

Abstract
Dianthus chinensis L. is used to treat various diseases including cancer; however, the molecular mechanism by which the ethanol extract of Dianthus chinensis L. (EDCL) induces apoptosis is unknown. In this study, the apoptotic effects of EDCL were investigated in human HepG2 hepatocellular carcinoma cells. Treatment with EDCL significantly inhibited cell growth in a concentration- and time-dependent manner by inducing apoptosis. This induction was associated with chromatin condensation, activation of caspases, and cleavage of poly (ADP-ribose) polymerase protein. However, apoptosis induced by EDCL was attenuated by caspase inhibitor, indicating an important role for caspases in EDCL responses. Furthermore, EDCL did not alter the expression of bax in HepG2 cells but did selectively downregulate the expression of bcl-2 and bcl-xl, resulting in an increase in the ratio of bax:bcl-2 and bax:bcl-xl. These results support a mechanism whereby EDCL induces apoptosis through the mitochondrial pathway and caspase activation in HepG2 cells.
1. Introduction
Hepatocellular carcinoma (HCC) is the fifth most commonly diagnosed cancer, with more than 1 million deaths reported annually worldwide [1]. Exposure to aflatoxin B1 and infection with hepatitis B virus and hepatitis C virus are high-risk factors for HCC [2–4]. The high prevalence and high death rate require novel strategies for the prevention and treatment of hepatic cancer. Natural products with antitumor activity are a promising approach to cancer prevention.
Plants are valuable sources of bioactive compounds and are used for medicinal purposes in Asia including Korea. Recently, oriental medicine has been the focus of scientific discovery efforts into novel drugs including anticancer agents [5–9]. Several herb-based components and extracts have been reported to reduce tumor growth and inhibit metastasis in the human HCC HepG2 model in vitro and in vivo [10, 11].
Dianthus chinensis L. (Caryophyllaceae, Rainbow pink) is commonly known as “Pae-raeng-ee-kot” in Korea. In Korea, this herb is used as a folk remedy for the treatment of menostasis, gonorrhea, cough, diuretic, and emmenagogue [12]. The chemical components of Dianthus chinensis L. are eugenol, phenylethylalcohol [13], melosides A and L [14] and dianchinenosides A, B [15], C, and D [16]. Hypotensive, anthelmintic, intestinal peristaltic, antitumor, and antioxidant activity was documented [12, 13, 17, 18]. However, apoptosis induction by this herb was never reported. The ethnomedical information described above formed the basis for the present study, which was conducted to evaluate the cytotoxic activity and mechanism of action of the ethanol extract of Dianthus chinensis L. in HepG2 HCC cells.
2. Materials and Methods
2.1. Plant and Preparation of Extracts
Dianthus chinensis L. was purchased as a dried herb from OmniHerb Co. (Yeongcheon, Korea) and authenticated based on microscopic and macroscopic characteristics by the Classification and Identification Committee of the Korea Institute of Oriental Medicine (KIOM). The dried herb (30.26 g) was extracted twice with 70% ethanol (with 2 h reflux) and the extract was then concentrated under reduced pressure. The decoction was filtered, lyophilized, and stored at 4°C. The yield of dried extract from starting crude material was approximately 18.57% (w/w). The lyophilized powder was dissolved in 10% dimethyl sulfoxide and then filtered through a 0.22 μm syringe filter to create a stock solution. EDCL denotes Dianthus chinensis L. ethanol extract. EDCL was diluted in culture medium to the final concentration indicated for each experiment.
2.2. Cell Culture
HepG2 human hepatocarcinoma cells were obtained from the American Type Culture Collection (Manassa, VA, USA). Cells were routinely maintained in Minimum Essential Medium with Earle's Balanced Salts and L-glutamine (MEM/EBSS, HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco BRL, Gaithersburg, MD, USA), 100 U/mL penicillin (Gibco BRL), and 100 μg/mL streptomycin (Gibco BRL) at 37°C in a humidified atmosphere of 5% CO2. The culture medium was replaced every 2 days.
2.3. Cell Viability Assay
Cells were seeded in 96-well culture plates at a density of 2 × 104 cells/well and allowed to adhere at 37°C for 12 h. The following day, several concentrations of EDCL were added and the cells were further incubated for 48 h. Then, cell viability was measured using the CCK-8 assay. 10 μL CCK-8 reagent was added to each well and incubated for 1 h at 37°C. Cell viability determination was based on the bioconversion of tetrazolium into formazan by intracellular dehydrogenase. Absorbance was measured at 450 nm using a Benchmark Plus Microplate Spectrophotometer (Bio-Rad, Hercules, CA, USA). Cytotoxicity was expressed as a percentage of the absorbance measured in control untreated cells.
2.4. Nuclear Staining with Hoechst 33342
Hoechst 33342 (Invitrogen, Eugene, Oregon, USA) staining was used to observe the apoptotic morphology of cells. Briefly, 5 × 105 cells/mL were seeded in six-well plates and incubated for 24 h. Then, the cells were exposed to different concentrations of EDCL (50–400 μg/mL) for 48 h. Next, the cells were collected and fixed with 3.7% formaldehyde in phosphate buffered saline (PBS) for 15 min and stained with Hoechst 33342 (10 μg/mL) at room temperature for 10 min. Finally, after the cells were washed with PBS, morphological changes, including a reduction in volume and nuclear chromatin condensation, were observed by fluorescence microscopy (Olympus Optical, Tokyo, Japan) and photographed at a 400x magnification.
2.5. Flow Cytometric Analysis for Measurement of Sub-G1 Phase
Cells were seeded in six-well plates at 1 × 106 cells/well and allowed to attach overnight. After exposure to EDCL, cells were collected, washed twice with ice-cold PBS buffer (pH 7.4), fixed with 80% ethanol at 4°C for 2 h, and then stained with PI/RNase Staining Buffer (BD PharMingen, San Diego, CA, USA) for 20 min in the dark at room temperature. Apoptotic cell analysis was conducted on a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA) and the data were analyzed using the CellQuest software.
2.6. Assay of Caspase-3/7, -8, and -9 Activity
Caspase activity was assayed using Caspase-Glo assay kits (Promega, Madison, WI, USA) according to manufacturer protocols. Briefly, cells were seeded at a density of 2 × 104 per well in triplicate wells onto 96-well plates and incubated for 24 h. Afterwards, the cells were exposed to several concentrations of EDCL (50–400 μg/mL) for 48 h or incubated with 180 μg/mL of EDCL for 6–48 h. After exposure to EDCL, culture supernatant (100 μL) was transferred into a white-walled 96-well plate. An equal volume of caspase substrate was added and samples were incubated at room temperature for 1 h. Culture medium was used as a blank control sample and luminescence was measured using an EnVision 2103 Multilabel Reader (PerkinElmer, Wellesley, MA, USA).
2.7. Protein Preparation and Western Blot Analysis
Cells were seeded in six-well plates at 1 × 106 cells/well and allowed to attach overnight. Afterwards, cells were exposed to different concentrations of EDCL for 48 h. Then, the cells were washed with ice-cold PBS twice and lysed with 1X RIPA lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 1 mM Protease Inhibitor Cocktail) for 30 min on ice. Lysates were cleared by centrifugation and supernatants were collected. The total protein content was quantified using the Bradford method. Proteins (30 μg) were mixed with 2X sample buffer, incubated at 95°C for 5 min, and loaded onto 12% polyacrylamide gels. Electrophoresis was performed using the Mini Protean 3 Cell (Bio-Rad). Proteins separated on the gels were transferred onto nitrocellulose membranes (Schueicher & Schell BioScience, Dassel, Germany). Membranes were blocked for 2 h using blocking buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20, 3% nonfat dry milk) and incubated at 4°C overnight with primary antibody (all antibodies were purchased from Cell Signaling Technology, Beverly, MA, USA). After washing with blocking buffer three times for 30 min, membranes were probed with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG) and anti-rabbit IgG (Cell Signaling Technology) for 2 h. The membranes were washed for 1 h (during which the wash buffer was changed three times) with Tris-buffered saline Tween 20 solution and developed with ECL Advance Western Blotting Detection Kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK) using a LAS-3000 luminescent image analyzer (Fuji Photo Film Co. Ltd., Kanagawa, Japan). Western blot signals were quantified and normalized to β-actin by densitometry analysis using the Multi-Gauge program of the LAS-3000 imaging system.
2.8. Statistical Analysis
Mean data values are presented with their deviation (mean ± SD) from three independent measurements. Statistical analyses were performed according to Prism 5 program (GraphPad, San Diego, USA). Analysis of variance (ANOVA) was followed by Dunett's test. A value of P < 0.05 was considered to be statistically significant.
3. Results
3.1. Effect of EDCL on HepG2 Cell Growth
The effect of EDCL on HepG2 cell growth was assessed using the CCK-8 assay. Figure 1 shows inhibition of HepG2 cell viability by several concentrations (50–400 μg/mL) of EDCL and over time (6–48 h). The results show concentration- and time-dependent inhibition, with IC50 values ranging from 314.98 μg/mL (24 h) to 186.64 μg/mL (48 h) (Figure 1).
Figure 1.
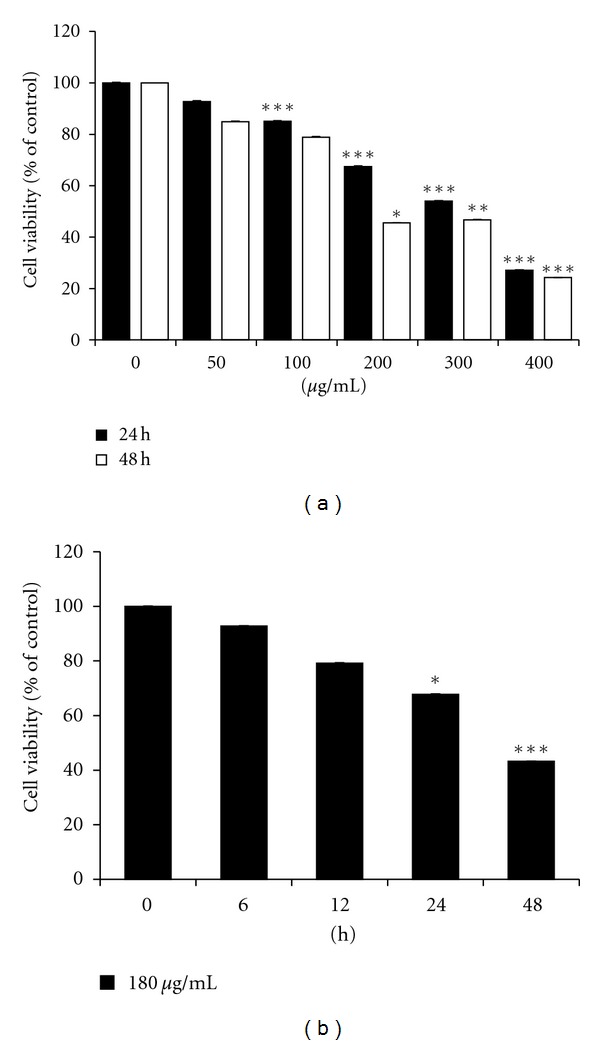
Exposure to EDCL induces growth inhibition in HepG2 cells. (a) Concentration response. Cells were incubated in the presence or absence of several concentrations of EDCL for 24 and 48 h. (b) Time course. Cells were exposed to 180 μg/mL EDCL over time (6–48 h). Cell viability was assessed by CCK-8 assay. The data are expressed as the means ± SD of triplicate samples. *P < 0.05, **P < 0.01, and ***P < 0.001 versus untreated EDCL.
3.2. Effect of EDCL on HepG2 Cell Apoptosis
To investigate the effect of EDCL on the morphology of apoptotic cells, Hoechst 33342 staining was conducted. Very few apoptotic cells were observed in the control culture, while the percentage of apoptotic cells in the presence of EDCL increased in an EDCL concentration-dependent manner (Figure 2(a)). The amount of sub-G1 DNA was analyzed to quantify the number of dead cells, since dead cells have a lower DNA content than cells in the G1 phase. Flow cytometric analysis indicated that exposure to EDCL markedly increased the number of sub-G1 phase cells in a concentration- and time-dependent manner (Figures 2(b) and 2(c)).
Figure 2.
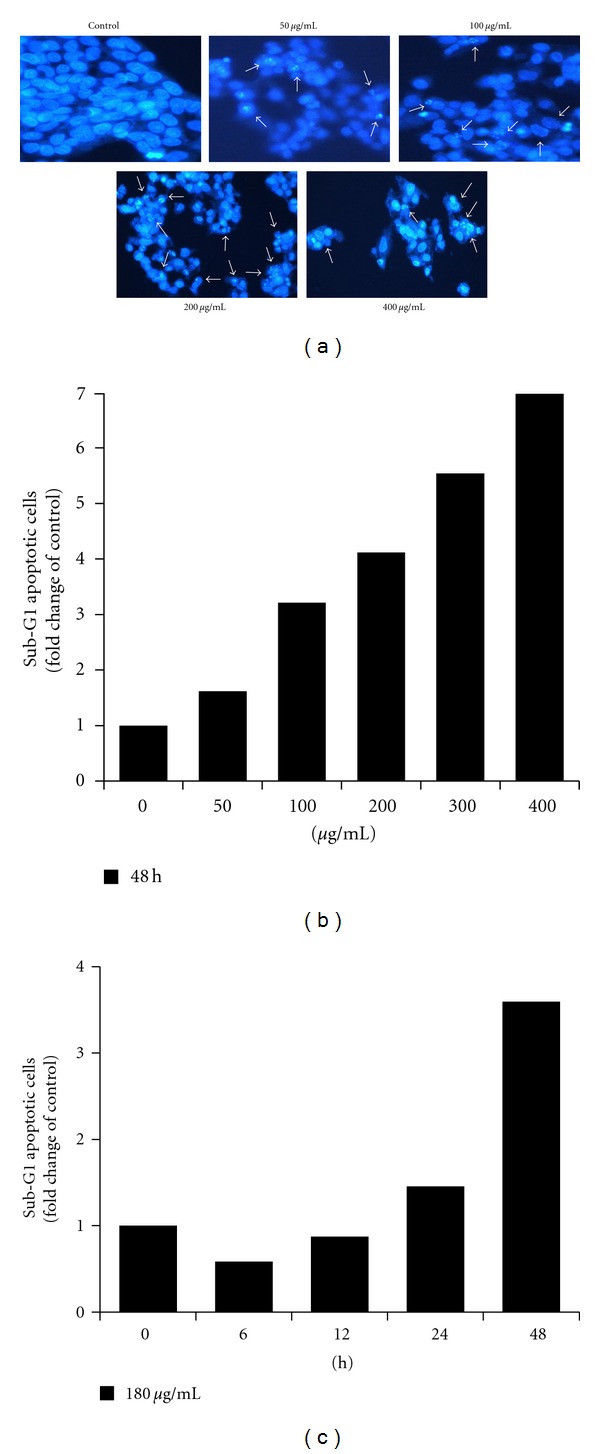
Exposure to EDCL induces apoptosis in HepG2 cells. (a) Cells were incubated in the presence or absence of several concentrations of EDCL for 48 h. Hoechst stain showed EDCL-induced chromatin condensation (arrow). Magnification, ×400. (b) Cells were exposed to several concentrations of EDCL for 48 h or (c) exposed to EDCL (180 μg/mL) over time. Apoptosis was measured using PI staining and flow cytometry.
3.3. Effect of EDCL on the Apoptotic Mitochondrial Pathway
The expression level of Bcl-2 family members interacting directly with mitochondria was studied. Western blotting (Figure 3(a)) revealed that the translational levels of bax expression, a proapoptotic protein, remained virtually unchanged in response to EDCL, whereas bcl-2, bcl-xl, and mcl-1, which are antiapoptotic proteins, were inhibited by exposure to EDCL. These data show that EDCL alters the bax:bcl-2 and bax:bcl-xl ratios in HepG2 cells in a concentration-dependent manner. Since proteins from the IAP family bind to caspases, leading to caspase inactivation in eukaryotic cells, the involvement of the IAP family in EDCL-induced apoptosis was further examined. The results indicated that the levels of IAP family members, such as cellular inhibitor-of-apoptosis protein (cIAP)-1, cIAP-2, and X-linked inhibitor of apoptosis protein (XIAP), were downregulated in HepG2 cells exposed to EDCL in a concentration-dependent manner (Figure 3(b)).
Figure 3.
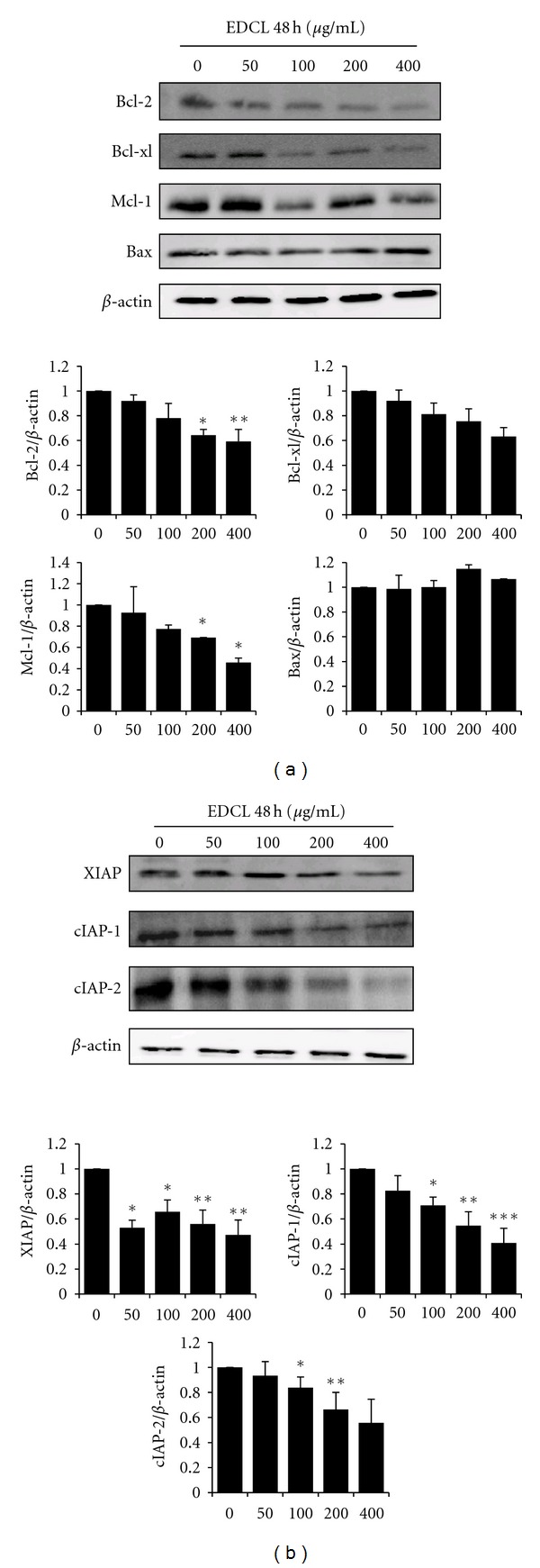
Exposure to EDCL downregulates the expression of Bcl-2 and IAP family members in HepG2 cells. Cells were exposed to several concentrations of EDCL for 48 h. Protein levels were monitored by Western blot analysis. Western blot signals were quantified and normalized to β-actin. Values are expressed as means ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001 versus untreated EDCL.
3.4. Effect of EDCL on Caspase Activity
To investigate the apoptotic cascade induced by EDCL, HepG2 cells were exposed to several concentrations of EDCL (50–400 μg/mL) for 48 h or incubated with 180 μg/mL of EDCL for 6–48 h, after which caspase-3/7, -8, and -9 activity was measured. The level of caspase activation in HepG2 cells exposed to EDCL was compared to that of control untreated cells arbitrarily set to 1.0. Results showed that EDCL markedly increased caspase-3/7, -8, and -9 activity, with maximum increase activity at 200 μg/mL. Results also showed that caspase activity increased over time in response to 180 μg/mL EDCL (Figures 4(a) and 4(b)). At the concentration of 200 μg/mL, the activity of caspase-3/7, caspase-8, and caspase-9 increased by 16.16-, 7.80-, and 14.17-fold, respectively. Furthermore, EDCL induced the degradation of poly (ADP-ribose) polymerase (PARP, 116 kDa), which is a protein substrate of caspase-3, and PARP cleavage fragments (89 kDa) increased over time (Figure 4(c)).
Figure 4.
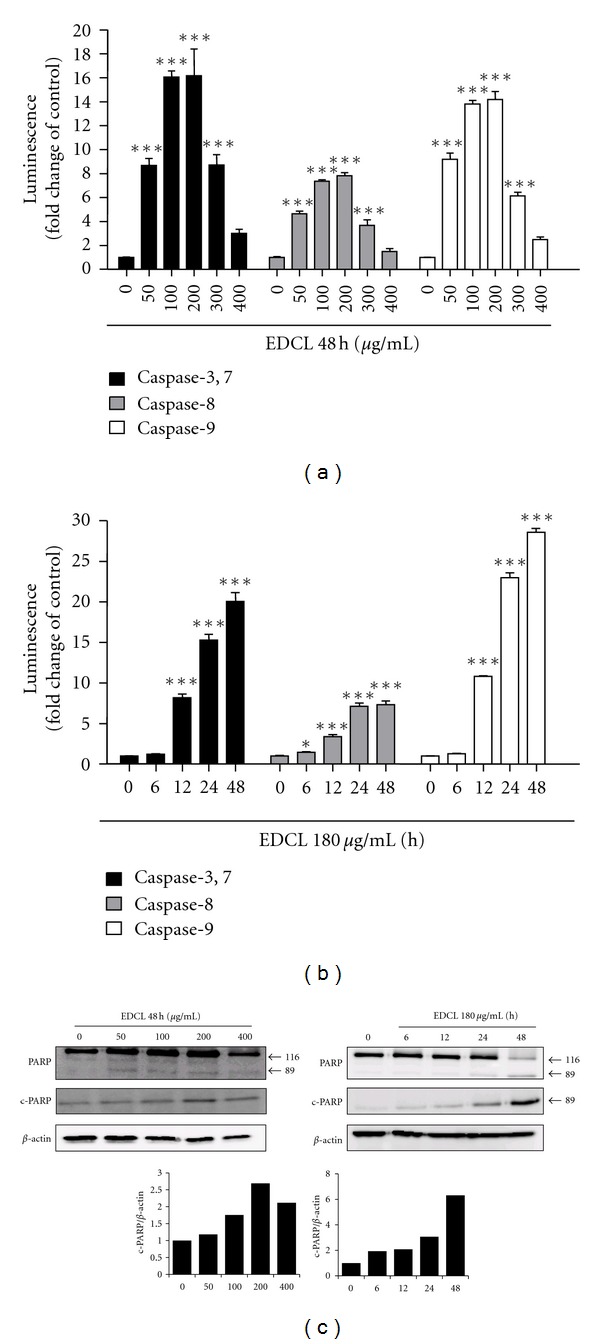
Exposure to EDCL shows activation of caspases and degradation of PARP protein in HepG2 cells. (a) Concentration response. Cells were incubated in the presence or absence of several concentrations of EDCL for 48 h. (b) Time course. Cells were incubated in the presence or absence of 180 μg/mL EDCL for different lengths of time. Upon completion of each exposure time, caspase activity was assessed using a Caspase-Glo assay kits assay, as described in Section 2. The data are expressed as the means ± SD of triplicate samples. *P < 0.05 and ***P < 0.001 versus untreated EDCL. (c) Cells were subjected to Western blot analysis using anti-PARP and anti-c-PARP antibodies. Western blot signals were quantified and normalized to β-actin.
3.5. Effect of Caspase Inhibitor on EDCL-Induced Apoptosis in HepG2 Cells
To confirm that caspase activation is a key step in EDCL-induced apoptosis, HepG2 cells were pretreated with z-vad-fmk (80 μM), a broad-spectrum caspase inhibitor, for 1 h, and then subsequently exposed to 180 μg/mL EDCL for 48 h. As shown in Figure 5(a), z-vad-fmk did not affect cell viability but inhibited the antiproliferative activity of EDCL. EDCL strongly stimulated caspase protease activity and pretreating cells with z-vad-vmk nearly abolished EDCL-induced caspase activity (Figure 5(b)). Furthermore, blockade of caspase activity by z-vad-vmk prevented EDCL-induced chromatin condensation (Figure 5(c)), PARP degradation (Figure 5(d)), and increase in sub-G1 population (Figure 5(e)). These results clearly show that EDCL-induced apoptosis is associated with caspase activation.
Figure 5.
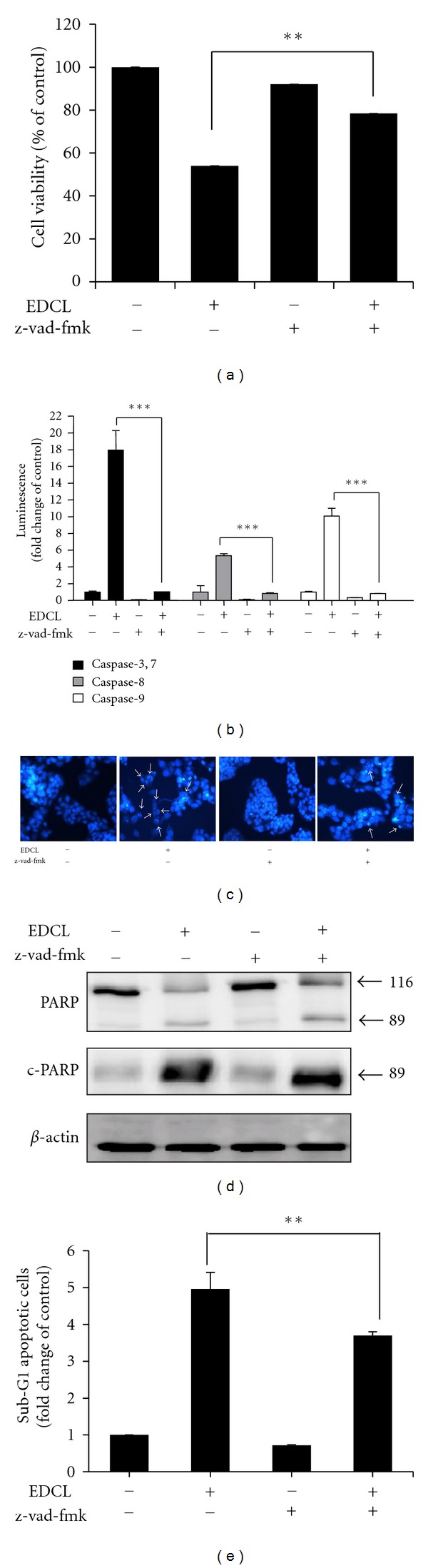
Caspase inhibition prevents EDCL-induced apoptosis in HepG2 cells. Cells were incubated in the presence or absence of z-vad-fmk for 1 h before being exposed to EDCL (180 μg/mL). (a) After 48 h of incubation with EDCL, cell viability was assessed using the CCK-8 assay and (b) caspase activity was measured. (c) Hoechst staining shows EDCL-induced chromatin condensation (arrow). Magnification, ×400. (d) Cells were subjected to Western blot analysis using anti-PARP and anti-c-PARP antibodies. (e) Cells were evaluated for sub-G1 DNA content by flow cytometry. The data are expressed as the means ± SD of triplicate samples. *P < 0.05, **P < 0.01, and ***P < 0.001 versus EDCL+z-vad-fmk.
4. Discussion
During the last decade, a considerable amount of research has focused on cancer cell apoptosis. Apoptosis, or programmed cell death, is the major control mechanism by which cells die if DNA damage is not repaired [19]. Apoptosis is also a critical protective mechanism against carcinogenesis, eliminating damaged cells or cells proliferating abnormally in response to carcinogens [20]. Therefore, induction of apoptotic cell death is a promising emerging strategy for the prevention and treatment of cancer [21]. The results of the present study clearly demonstrate that EDCL suppressed HepG2 cell viability by inducing apoptosis. After exposure to EDCL, chromatin condensation and apoptotic bodies were clearly observed. These results suggest that HepG2 cells exposed to EDCL undergo typical apoptosis. Furthermore, flow cytometric analysis after propidium iodide staining confirmed EDCL-induced apoptosis in HepG2 cells.
Members of the Bcl-2 family of proteins, such as bcl-2, bcl-xl, mcl-1, and bax, are the most prominent actors in controlling the release of cytochrome c and in mitochondria-mediated apoptosis [22]. Thus, it has been suggested that the ratio between the level of proapoptotic bax protein and the level of antiapoptotic bcl-2 protein determines whether a cell responds to an apoptotic signal [23]. In this study, EDCL did not alter the expression of bax in HepG2 cells but did selectively downregulate the expression of bcl-2 and bcl-xl, resulting in an increase in the ratio of bax:bcl-2 and bax:bcl-xl.
The execution of cellular demolition in apoptosis is also carried out by caspases [24]. The caspase family of proteins is one of the main executors of the apoptotic process. Caspases belong to a group of enzymes known as cysteine proteases and exist within the cell as inactive proforms or zymogens. These zymogens can be cleaved to form active enzymes following the induction of apoptosis. The IAP family of proteins blocks apoptosis by directly inhibiting at least two members of the caspase family of cell death proteases, caspase-3, and caspase-7. XIAP, cIAP-1, and cIAP-2 can prevent the proteolytic processing of procaspase-3, -6, and -7 by blocking the cytochrome c-induced activation of procaspase-9 [24, 25]. Studies have shown that exposure of HepG2 cells to EDCL caused proteolytic activation of caspases and down-regulation of XIAP, cIAP-1 and cIAP-2. The enzyme poly(ADP-ribose) polymerase, or PARP, was one of the first proteins identified as a substrate for caspases. PARP is involved in repair of DNA damage and functions by catalyzing the synthesis of poly (ADP-ribose) and by binding to DNA strand breaks and modifying nuclear proteins. PARP helps cells maintain viability, and the cleavage of PARP facilitates cellular disassembly and serves as a marker for cells undergoing apoptosis [26, 27]. In the present study, we examined whether the PARP protein, a substrate of caspase-3 [28], was cleaved in cells exposed to EDCL. As expected, PARP was clearly degraded in a concentration- and time-dependent manner that correlated with caspase activation. Under the same experimental conditions, z-vad-fmk prevented EDCL-induced apoptosis by blocking caspase activation. These data indicate that caspases are the key molecules mediating EDCL-induced apoptosis in HepG2 cells.
In conclusion, this study clearly demonstrates that EDCL strongly inhibits cell proliferation and induces apoptosis in HepG2 cells. EDCL induced apoptosis through the mitochondrial pathway, involving the activation of caspase-3/7, -8, and -9, the down-regulation of antiapoptotic proteins, and the degradation of PARP protein. Because induction of apoptosis is thought to be a suitable anticancer therapeutic mechanism, these results confirm the potential of EDCL as a chemotherapeutic agent in human hepatocellular carcinoma cells. In vivo studies are needed to fully establish the potential of EDCL as a chemopreventive and therapeutic agent in cancer.
Conflict of Interests
The authors have no conflict of interests to declare.
Acknowledgments
This work was supported by the project “Construction of the Basis for Practical Application of Herbal Resources” funded by the Ministry of Education, Science and Technology (MEST) of Korea to the Korea Institute of Oriental Medicine (KIOM). We thank the KIOM Classification and Identification Committee for critical authentication of plants and helpful discussions.
References
- 1.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. Ca-A Cancer Journal for Clinicians. 2005;55(1):10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Okuda K. Hepatocellular carcinoma: recent progress. Hepatology. 1992;15(5):948–963. doi: 10.1002/hep.1840150532. [DOI] [PubMed] [Google Scholar]
- 3.Seow TK, Liang RCMY, Leow CK, Chung MCM. Hepatocellular carcinoma: from bedside to proteomics. Proteomics. 2001;1(10):1249–1263. doi: 10.1002/1615-9861(200110)1:10<1249::AID-PROT1249>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 4.Kern MA, Breuhahn K, Schirmacher P. Molecular pathogenesis of human hepatocellular carcinoma. Advances in Cancer Research. 2002;86:67–112. doi: 10.1016/s0065-230x(02)86003-1. [DOI] [PubMed] [Google Scholar]
- 5.Hu H, Ahn NS, Yang X, Lee YS, Kang KS. Ganoderma lucidum extract induces cell cycle arrest and apoptosis in MCF-7 human breast cancer cell. International Journal of Cancer. 2002;102(3):250–253. doi: 10.1002/ijc.10707. [DOI] [PubMed] [Google Scholar]
- 6.Lee SMY, Li MLY, Tse YC, et al. Paeoniae Radix, a Chinese herbal extract, inhibit hepatoma cells growth by inducing apoptosis in a p53 independent pathway. Life Sciences. 2002;71(19):2267–2277. doi: 10.1016/s0024-3205(02)01962-8. [DOI] [PubMed] [Google Scholar]
- 7.Cheng YL, Lee SC, Lin SZ, et al. Anti-proliferative activity of Bupleurum scrozonerifolium in A549 human lung cancer cells in vitro and in vivo. Cancer Letters. 2005;222(2):183–193. doi: 10.1016/j.canlet.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 8.Park DI, Lee JH, Moon SK, et al. Induction of apoptosis and inhibition of telomerase activity by aqueous extract from Platycodon grandiflorum in human lung carcinoma cells. Pharmacological Research. 2005;51(5):437–443. doi: 10.1016/j.phrs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Tan ML, Sulaiman SF, Najimuddin N, Samian MR, Muhammad TST. Methanolic extract of Pereskia bleo (Kunth) DC. (Cactaceae) induces apoptosis in breast carcinoma, T47-D cell line. Journal of Ethnopharmacology. 2005;96(1-2):287–294. doi: 10.1016/j.jep.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 10.Chung TW, Moon SK, Chang YC, et al. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: complete regression of hepatoma growth and metastasis by dual mechanism. The FASEB Journal. 2004;18(14):1670–1681. doi: 10.1096/fj.04-2126com. [DOI] [PubMed] [Google Scholar]
- 11.Cheung CSF, Chung KKW, Lui JCK, et al. Leachianone A as a potential anti-cancer drug by induction of apoptosis in human hepatoma HepG2 cells. Cancer Letters. 2007;253(2):224–235. doi: 10.1016/j.canlet.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 12.WHO. Medicinal plants in the Republic of Korea. Manila, Philippines: World Health Organization, Regional Office for Western Pacific; 1998. [Google Scholar]
- 13.Hsu HY, Chen YP, Shen CS, Hsu CC, Chang HC. Oriental Materia Medica. Long Beach, Calif, USA: Oriental Healing Arts Institute; 1986. [Google Scholar]
- 14.Monties B, Bouillant ML, Chopin J. C-diholosylflavones dans les feuilles du melon (Cucumis melo) Phytochemistry. 1976;15(6):1053–1056. [Google Scholar]
- 15.Li HY, Koike K, Ohmoto T, Ikeda K. Dianchinenosides A and B, two new saponins from Dianthus chinensis. Journal of Natural Products. 1993;56(7):1065–1070. [Google Scholar]
- 16.Li Hong Yu, Koike K, Ohmoto T. Triterpenoid saponins from Dianthus chinensis. Phytochemistry. 1994;35(3):751–756. doi: 10.1016/s0031-9422(00)90599-5. [DOI] [PubMed] [Google Scholar]
- 17.Kosuge T, Yokota M, Sugiyama K. Studies on antitumor activities and antitumor principles of Chinese herbs. I. Antitumor activities of Chinese herbs. Yakugaku Zasshi. 1985;105(8):791–795. doi: 10.1248/yakushi1947.105.8_791. [DOI] [PubMed] [Google Scholar]
- 18.Kim SY, Kim JH, Kim SK, Oh MJ, Jung MY. Antioxidant activities of selected oriental herb extracts. Journal of the American Oil Chemists’ Society. 1994;71(6):633–640. [Google Scholar]
- 19.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21(3):485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 20.Schuchmann M, Galle PR. Sensitizing to apoptosis—sharpening the medical sword. Journal of Hepatology. 2004;40(2):335–336. doi: 10.1016/j.jhep.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 21.Hu W, Kavanagh JJ. Anticancer therapy targeting the apoptotic pathway. Lancet Oncology. 2003;4(12):721–729. doi: 10.1016/s1470-2045(03)01277-4. [DOI] [PubMed] [Google Scholar]
- 22.Borner C. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Molecular Immunology. 2003;39(11):615–647. doi: 10.1016/s0161-5890(02)00252-3. [DOI] [PubMed] [Google Scholar]
- 23.Salomons GS, Brady HJM, Verwijs-Janssen M, et al. The baxα:Bcl-2 ratio modulates the response to dexamethasone in leukaemic cells and is highly variable in childhood acute leukemia. International Journal of Cancer. 1997;71(6):959–965. doi: 10.1002/(sici)1097-0215(19970611)71:6<959::aid-ijc9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 24.Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO Journal. 1999;18(19):5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO Journal. 1997;16(23):6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker PR, Sikorska M. New aspects of the mechanism of DNA fragmentation in apoptosis. Biochemistry and Cell Biology. 1997;75(4):287–299. [PubMed] [Google Scholar]
- 27.Oliver FJ, De La Rubia G, Rolli V, Ruiz-Ruiz MC, De Murcia G, Ménissier-De Murcia J. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis: lesson from an uncleavable mutant. Journal of Biological Chemistry. 1998;273(50):33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 28.Tewari M, Quan LT, O’Rourke K, et al. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81(5):801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]