

Summary
Objectives
Obesity and genetic variation in aromatase and type 1 17-β hydroxysteroid dehydrogenase (HSD) could influence the E2 trajectory of decline during the menopause transition.
Design and participants
E2 trajectories during the menopause transition (phenotype) were identified using 5934 data points acquired annually from 681 women in Study of Women’s Health across the Nation (SWAN), a multiethnic study of the mid-life. E2 trajectories were related to CYP19 and type I 17-βHSD single-nucleotide polymorphisms (SNPs) and obesity.
Results
logE2 trajectories began to decline precipitously 2 years before the final menstrual period (FMP). The trajectory of the logE2 decline varied with genotypes and obesity. logE2 rates of decline were greater in nonobese women than in obese women, P < 0.05. Women with the CYP19rs936306 CT variant had logE2 rate of decline that was 54% as rapid as the rate of decline of women with the TT variant, P < 0.05. logE2 rate of decline in women with the CYP19rs749292 GG variant was two-thirds the rate of logE2 decline in women with the AG variant, P < 0.05. logRates of E2 decline with 17-βHSD SNPs (rs2830, rs592389, and rs615942) varied according to genotype within obesity groups. Within each obesity group, logE2 rate of decline was greater in heterozygous variants and much less in homozygotes (P < 0.05). Obese women with selected CYP19 and 17-β HSD gene variants had remarkably different E2 trajectories around the FMP, resulting in different postmenopausal E2 levels. The rate of the E2 decline and the subsequent postmenopausal E2 levels may be relevant to oestrogen-sensitive chronic diseases including cancers.
Introduction
While endogenous estradiol (E2) levels contribute to health and disease, E2 rates of decline during the menopause and subsequent E2 postmenopausal levels also influence disease states.1 E2 decline is the penultimate event of the menopause transition.2-8 It is now recognized that there are minimal changes in endogenous follicular phase E2 levels until about 2 years prior to the final menstrual period (FMP).4,8 and that this rapid decline is not predicated on the age of FMP.9 Factors that influence the E2 rate of decline and the ensuing postmenopausal E2 level are poorly elucidated.
Investigators hypothesized that E2 levels were sustained until just before the FMP because of increased ovarian aromatase activity in the late menopause transition10 which may, in part, be genetically related.11-13 Two genes considered include the aromatase gene (CYP19), controlling the rates of conversion of androgens (testosterone or androstenedione) to their parallel oestrogens (estradiol or estrone), and the type 1 17-β hydroxysteroid dehydrogenase (17HSD) gene that encodes the bidirectional enzyme converting estrone (E1) to E2 (Fig. 1)14,15 Earlier, we have previously reported that two CYP19 single-nucleotide polymorphisms (SNPs) were associated with variation in the endogenous serum testosterone-to-estradiol ratio, suggesting a functional role.16 Additionally, E1 can be converted to the more biologically active E2 by type 1 17HSD. The gradient in this bidirectional enzyme action is thought to favour the conversion of E1 to E2, so greater E1 levels in obese women could result in increased circulating E2. Moreover, the relative bidirectionality of type 1 17HSD depends on intracellular redox potential17 that is thought to be altered in obesity. Therefore, genetic variation in type 1 17HSD enzyme may alter the rate of conversion, particularly in the obese.18
Fig. 1.
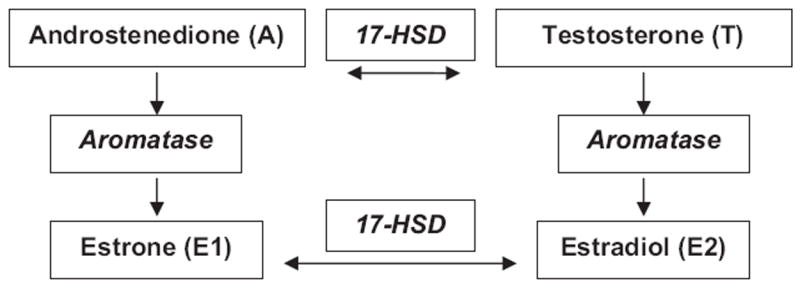
The aromatase enzyme converts androstenedione to estrone (E1) and testosterone to estradiol (E2). Estrone (E1) and E2 conversion by the type 1 17β-hydroxysteroid dehydrogenase enzyme is bidirectional.
Using E2 trajectories around the FMP as the phenotype, we evaluated the relation of aromatase (CYP19) and type 1 17-βHSD SNPs to the rates of change around the FMP. We hypothesized different rates of E2 decline in CYP 19 variants (rs936306 and rs749292) or type 1 17-βHSD variants (rs2830, 592389 or rs615942). Further, we hypothesized that relationships between the type 1 17-βHSD genotypes and the phenotype differed in obese vs nonobese women.
Methods and procedures
Sample population
The sample was 681 women with genotyping and a natural FMP from the Study of Women’s Health Across the Nation (SWAN), a multisite, community-based longitudinal study of women at mid-life.19 At the 1996/7 SWAN baseline, enrollees were aged 42–52 years and pre-or early perimenopausal. Enrollees had an intact uterus and ≥1 ovary, were not pregnant or lactating and were not using exogenous reproductive hormones. Each of the seven study sites enrolled Caucasian women plus women of one other racial/ethnic group, including African-American (Boston, Detroit area, Chicago and Pittsburgh), Japanese (Los Angeles), Chinese (Oakland, California) and Hispanic (New Jersey) women.
In 2001/2, 1988 women were eligible for the SWAN Genetics Study, and 88% provided blood or saliva.20,21 Further, 1538 women agreed to lymphocytes immortalization who subsequently became the source of extracted DNA. The DNA was genotyped for 26 SNPs associated with genes for oestrogen receptors or sex steroid metabolism pathway enzymes.19 Details about specimen collection and processing, SNP selection and genotyping have been reported.21 Genotyping was undertaken using a TaqMan (Roche Molecular Systems, Inc., Pleasanton, CA, USA) system with ABI 7900HT sequence detection technology.
Data from 681 women (with an FMP and 5934 annual hormone data points) were included of whom 198 (29%) were African-American, 327 (48%) were Caucasian, 77 (11%) were Chinese, and 79 (12%) were Japanese. Data were excluded from analyses for 598 women with genotype data but exogenous hormone use and 259 women without a natural FMP. The University of Michigan Institutional Review Board approved the protocol, and written informed consent was obtained from participants.
Hormone analyses
Fasting blood samples were collected at annually during days 2–5 of the menstrual cycle follicular phase and assayed for serum hormones. If phlebotomy could not be linked to menses, specimens were collected approximately 1 year apart. Aliquoted specimens were stored at −80 degrees Centigrade without thaw until assay. Serum E2 concentrations were measured in duplicate with a modified, offline ACS-180 (E2-6) immunoassay. Inter- and intra-assay coefficients of variation averaged 10.6% and 6.4%, respectively, over the assay range. The lower limit of detection was 1 pg/ml. Assay cross-reactivity was 0.75% for estrone, 0.28% for estriol and 0.00% for norethinedrone.22
Other measures
The FMP was defined following 12 months of amenorrhoea without alternative physiological explanations. A natural FMP was unobscured by hysterectomy, oophorectomy or exogenous hormone use with verification by medical record abstraction or visualizing packaging.
Annual measures of height (cm) and weight (kg) were used to calculate body mass index (BMI). Obesity was defined as a BMI > 30 kg/m2. Women reported their smoking behaviour and self-identified their race/ethnicity.
Data analyses
The data were examined for outliers and log-transformed to satisfy statistical assumptions of constant variance and normality in modelling. Frequencies and means were used to describe the SNPs, race/ethnic group, baseline smoking status, circulating E2 levels, age and BMI. Analysis of variance (ANOVA), contingency table analyses and analysis of covariance were used to compare characteristics of participants according to body size and estimate the group means for E2 across the transition period.
The assumption of Hardy–Weinberg equilibrium was tested using chi-square analyses; the likelihood of equality of allele frequencies across groups was evaluated with a test of equal proportions.
Analyses for E2 rates of change
logE2 rate of decline around the FMP was estimated using splines and nonparametric stochastic mixed modelling.8,23 logE2 rates of decline and accelerations were estimated as the first- and second-order derivatives of differential equations associated with the cubic spline function; curvature was approximated by integrating both rate of decline and acceleration/deceleration.8 Data from fitted models were organized into epochs by setting nodes for use in piecewise linear mixed modelling to describe the logE2 decline slope by genotype.24 Nodes were estimated at 2 years prior to the FMP, at the FMP and at 2 years after the FMP after which a new steady state was achieved. We expressed the regression betas of slopes as a proportion of the genotype with the fastest rate of decline. We used bootstrapping of 100 samples to build the 95% confidence bands for asymmetrical curves and their derivatives.25,26
Relating genotype to phenotype
Multiple variable regression modelling was used to relate logE2 rate of decline to BMI and the genotypes. We used orthogonal contrasts to test whether logE2 slopes were significantly different according to genotype. Interaction terms for time-to-FMP × obesity and time-to-FMP × genotype were included in models for each of the five SNPs. Additionally, models for the type 1 17β HSD genotypes included a BMI × genotype interaction term.
Statistical analyses were conducted using SAS™ (version 9.1; SAS Institute, Cary, NC, USA) SAS macro language, R™ (R Project, version 1.9.1, Free Software Foundation, Boston, MA, USA) and Matlab™ 7.0 (The MathWorks, Inc., Natick, MA, USA).
Results
At baseline, the mean (±SD) age of women was 47.0 ± 2.6 years. The mean (±SD) baseline E2 level was 55.7 ± 44.6 pg/ml. One-third of women were classified as obese (BMI > 30.0 kg/m2). The mean (±SD) age of FMP was 50.2 ± 2.2 years.
The phenotype was the precipitous change in the logE2 trajectory during the menopause transition. The rate of logE2 decline accelerated 2 years before the FMP and reached the maximum rate of change at the FMP (Fig. 2). Then, E2 began to decelerate and reached a new equilibrium about 2 years after the FMP. The rate of E2 decline was 50% greater in nonobese women compared to obese women (P < 0.001) (Fig. 2).
Fig. 2.
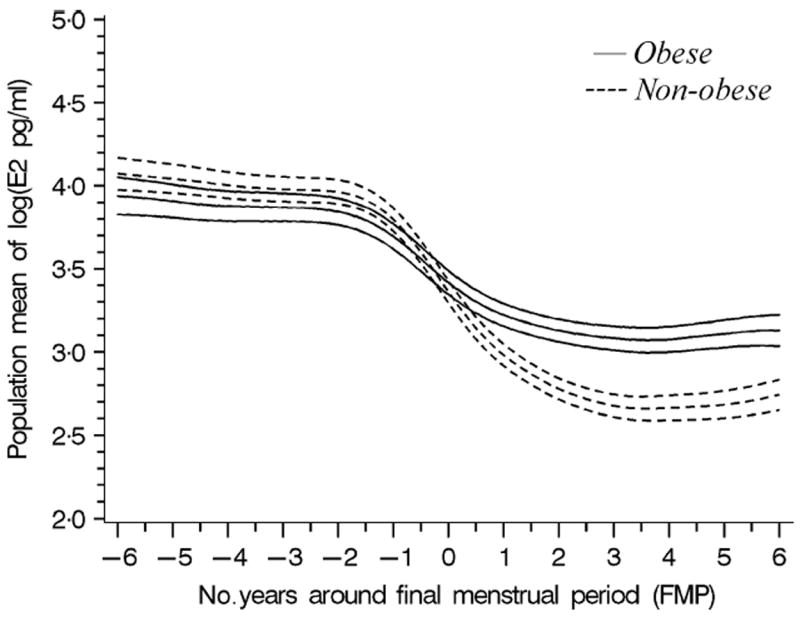
The modelled population mean change in logE2 (with 95% confidence bands) around the FMP in nonobese women (BMI<30, dashed lines) and obese women (BMI>30, solid lines).
Aromatase SNPs
CYP19 rs749292 AG variant was associated with the greatest rate of E2 decline (Table 1); the rate of E2 decline with the GG variant of rs749292 (β = −0.0337) was 67% of that of the AG variant (P < 0.0001). The rate of E2 decline with the CT variant (CYP19 rs936306) was 54% (β = −0.0318) of the TT variant decline (β = −0.0591, P < 0.0001). These relationships were similar in obese vs nonobese women (data not shown).
Table 1.
logEstradiol (E2) rates of change in the time from 2 years before final menstrual period (FMP) to FMP, by CYP19 rs749292 and CYP19 rs9363062
| Genotype | Number of women | Genotype slopes (β, pg/ml) | Slope as a proportion of the fastest rate of decline (%) |
|---|---|---|---|
| CYP19 rs749292 | |||
| AA | 143 | −0.0482 | 96 |
| AG | 328 | −0.0501 | 100 (referent) |
| GG | 200 | −0.0337* | 67 |
| CYP19 rs9363062 | |||
| CC | 356 | −0.0543 | 92 |
| CT | 230 | −0.0318* | 54 |
| TT | 89 | −0.0591 | 100 (referent) |
Genotype slope is statistically different than other genotype slopes, P < 0.05.
The type 1 17βHSD SNPs and obesity
The mean rate of E2 decline in obese women was at least 50% less than the E2 rate of decline of nonobese women within the three type 1 17HSD variants (Tables 2, 3 and 4). The rate of E2 decline in obese homozygous women was markedly less than the rate of decline in nonobese homozygous women.
Table 2.
logEstradiol (E2) rates of change as slopes according to obesity classification (BMI<30 vs BMI>30) and type 1 17β hydroxysteroid dehydrogenase (17β HSD) rs592389 genotypes
| Type 1 17β HSD rs592389
| ||||
|---|---|---|---|---|
| Number of women | Slopes (β, pg/ml) | Slope as a proportion of the fastest rate of decline (%) | Slope as a proportion of the rate of decline within obesity group (%) | |
| BMI < 30 kg/m2 | ||||
| AA | 99 | −0.0562* | 60 | 60 |
| AC | 222 | −0.0937*† | 100 (referent) | 100 (referent) |
| CC | 129 | −0.0589* | 63 | 6% |
| BMI ≥ 30 kg/m2 | ||||
| AA | 41 | −0.0072 | 8 | 16 |
| AC | 92 | −0.0446† | 47 | 100 (referent) |
| CC | 87 | −0.0099 | 11 | 22 |
Within genotype, BMI<30 group declines faster than BMI>30, P < 0.01.
Genotype slope is significantly different than other genotype slopes within the obesity group.
Table 3.
logEstradiol (E2) rates of change as slopes according to obesity classification (BMI<30 vs BMI>30) and type 1 17β hydroxysteroid dehydrogenase (17β HSD) rs615942 genotypes
| Type 1 17β HSD rs615942
| ||||
|---|---|---|---|---|
| Number of women | Slopes (β, pg/ml) | Slope as a proportion of the fastest rate of decline (%) | Slope as a proportion of the rate of decline within obesity group (%) | |
| BMI < 30 kg/m2 | ||||
| GG | 129 | −0.0619* | 71 | 71 |
| TG | 219 | −0.0866*† | 100 (referent) | 100 (referent) |
| TT | 106 | −0.0549* | 63 | 63 |
| BMI ≥ 30 kg/m2 | ||||
| GG | 65 | −0.0179† | 21 | 42 |
| TG | 98 | −0.0426† | 49 | 100 (referent) |
| TT | 59 | −0.0109 | 13 | 26 |
Within genotype, BMI<30 group declines faster than BMI>30, P < 0.01.
Genotype slope is significantly different than other genotype slopes within the obesity group.
Table 4.
logEstradiol (E2) rates of change as slopes according to obesity classification (BMI<30 vs BMI>30) and type 1 17β hydroxysteroid dehydrogenase (17β HSD) rs2830 genotypes
| Type 1 17β HSD rs2830
| ||||
|---|---|---|---|---|
| Number of women | Slopes (β, pg/ml) | Slope as a proportion of the fastest rate of decline (%) | Slope as a proportion of the rate of decline within obesity group (%) | |
| BMI < 30 kg/m2 | ||||
| AA | 132 | −0.0568* | 56 | 56 |
| AG | 220 | −0.1013† | 100 (referent) | 100 (referent) |
| GG | 99 | −0.0551* | 54 | 54 |
| BMI ≥ 30 kg/m2 | ||||
| AA | 87 | −0.0062 | 6 | 12 |
| AG | 93 | −0.0507† | 50 | 100 (referent) |
| GG | 40 | −0.0046 | 5 | 9 |
Within genotype, BMI<30 group declines faster than BMI>30, P < 0.01.
Genotype slope is significantly different than other genotype slopes within the obesity group.
In Table 2 showing type 1 17HSD rs592389, heterozygous (AC) obese women had a mean rate of E2 decline (β = −0.0446) that was 47% of the rate of E2 decline in nonobese women (β = −0.0937; P < 0.02). The rate of logE2 decline in nonobese women with the AA or CC genotypes was 60% or 63%, respectively, of the rate of logE2 decline in the nonobese women with the AC genotype. However, among the homozygous (AA and CC) obese women, the rate of logE2 decline was only 8% and 11% of the decline observed in the nonobese AC referent group.
Table 3 with type 1 17HSD rs615942 reveals that the rate of E2 declined 49% (β = −0.0426) in obese women less than in the nonobese women (β = −0.0866; P < 0.02). In nonobese women, the rate of logE2 decline in the GG and TT homozygotes was 71% and 63% of the decline observed in the heterozygous (GT) referent group (β = −0.0866). The women who were both obese and homozygous had much slower rates of decline (21% for GG and 13% for TT) compared to nonobese heterozygotes.
Table 4 indicates that the rate of logE2 decline was markedly greater in the nonobese women than in the obese women within each of the type 1 17HSD rs2830 genotypes. The rate of logE2 decline in the obese heterozygous women was 50% of the rate in the nonobese heterozygous women. In the nonobese women, the rates of decline in the two homozygous groups were about 50% of those seen in the heterozygous referent group. In the obese group, the rates of decline in the two homozygous groups were <10% of the decline in the referent nonobese heterozygous (AG) group.
Postmenopausal estradiol levels
Obesity and genotypes influenced resulting E2 levels at 2 years post-FMP. Women with slower E2 rates of decline had higher levels of endogenous E2 2 years following the FMP than did women with more rapid rates of decline. There was almost a 34–46% difference in postmenopausal E2 level according to obese vs nonobese and type 1 17HSD status.
Discussion
The rate of E2 decline around the FMP may contribute to the endocrine environment for subsequently occurring health-related conditions, including bone status and E2-sensitive cancers. Yet, there has been little study of the rates of E2 change around the FMP, and to our knowledge, this is the first report describing rates of change as a phenotype for genetic studies. We found obese women with selected CYP19 and 17HSD variants had modest E2 declines in the 4-year period around the FMP compared to more dramatic E2 declines in nonobese women and other selected CYP19 and 17HSD variants. Subsequently, women with slower rates of E2 decline had higher endogenous E2 following the FMP than women with more rapid rates of decline. The impact of variations in E2 decline at the FMP, and the ensuing E2 levels, awaits evaluation but may have immediate implications for presentation of hot flashes,27,28 bone loss at the menopause1 or risk of developing endometrial and breast cancer.13,29
Heterozygotes associated with type 1 17HSD SNPs had markedly different rates of E2 menopausal decline than did homozygotes. These findings are most consistent with the concept of overdominance in which apparent heterosis reflects superior biochemical, physiological, or evolutionary fitness of heterozygous genotypes over homozygous genotypes at a single locus.30 Overdominance can result from allele-specific gene expression and epigenetic regulation, as reported in circadian-mediated metabolic pathways.31 Further, our findings may result from epistasis, a model of genetic action in which there is an interaction between genotypes at two different gene loci accounting for modes of underdominance or overdominance.32-34 The selective advantages of over and underdominance has been well-documented in sickle cell anaemia, a condition determined by a single polymorphism that also confers some resistance to malaria. Because of selective pressures, homozygotes have little or no protection from malaria and a propensity to sickle cell anaemia whereas heterozygotes enjoy a partial resistance to both malaria and sickle cell anaemia. It would be useful to confirm the model of genetic action given the range of physiological actions associated with estradiol and consider how differences in the rate of decline in the 4-year period around the menopause transition might be permissive for the differential expression of pathologies or targeting of interventions.35
Obesity was prominently associated with the rate of E2 decline during the menopause transition. The mean rates of E2 decline in the obese were half those observed in the nonobese women. Slower rates of loss during the transition resulted in higher circulating E2 levels among obese women in the early postmenopause. It remains to be determined whether the attenuated decline in E2 around the FMP leading to greater E2 levels in the early postmenopause is important for the slower rate of bone loss among the obese in the menopause transition1 or the patterns of hot flash presentation with obesity.27
The interaction between type 1 β17HSD genotypes and obesity could be present because circulating androstenedione is converted to E1 by aromatase in adipose tissue, and in turn, E1 is then reduced to E2 by type 1 17HSD.18 The gradient in this bidirectional enzyme action is thought to favour the conversion of E1 to E2. Thus, greater E1 levels in obese postmenopausal women with increased fat mass and increased aromatase would be expected to result in higher circulating E2 in the obese compared to the nonobese.
We selected two aromatase variants for study because we had previously determined that baseline endogenous estradiol-to-testosterone (T) ratio was different in these two variants.16 We had reported that the TT genotype vs other genotypes of the CYP19 rs936306 polymorphism was associated with a significant difference in the T:E2 ratio (lower T and higher E2 levels), especially in African-American women.19 That these SNPs were also related to rate of decline, E2 decline adds credibility to a possible functional role.
This report includes notable strengths and limitations. These analyses included SNPs associated with genes that encode the enzymes that are biologically related to the process of interest, the rate of E2 decline around the FMP. The availability of data from a large number of women, followed through and observed natural menopause, provides a unique phenotype, the rate of E2 decline. The data analysis was designed to assess the rate of E2 decline using frequently acquired data across the menopause transition period. These findings are more likely to be generalizable to populations of women because the data were acquired from community rather than clinical samples of women. However, although the findings are intriguing, the five SNPs being evaluated represent a small number of possible variants; other variants and other genes might influence the rate of change in E2 levels,29 including other 17HSD genes14 as well as obesity genes. Until additional variants and genes are evaluated and their influence on the rate of E2 more fully elucidated, their role as intervention or therapeutic targets awaits further definition.
In summary, obesity and variation in five SNPs associated with the aromatase and type 1 β17HSD genes were related to the rate of log E2 decline in the critical transition period around the FMP. The rate of E2 decline in obese women was half that of nonobese women. With the three 17HSD variants, the interaction with body size suggested that the association with the rate of decline was determined by both body size and the gene that encodes the enzyme with bidirectional conversion of E1 and E2. The findings that type 1 β17HSD heterozygotes had markedly different rates of E2 menopausal decline than did homozygotes are most congruent with heterosis and overdominance genetic modes of action. To our knowledge, this represents the first opportunity to examine variables affecting the rates of E2 loss during the menopause transition. It is important to identify the factors and their genetic modes of action that influence rates of E2 decline as these contribute to subsequent circulating E2 levels in the postmenopause and, by extension, may contribute to health- and disease-related processes of the postmenopause, including bone loss and the development of oestrogen-related cancers.
Acknowledgments
The Study of Women’s Health Across the Nation (SWAN) has grant support from the National Institutes of Health, DHHS, through the National Institute on Aging (NIA), the National Institute of Nursing Research (NINR) and the NIH Office of Research on Women’s Health (ORWH) (Grants AG012505, AG012535, AG012531, AG012539, AG012546, AG012553, AG012554, AG012495; NR004061). The genotype data are from the SWAN Repository (AG17104). The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the NIA, NINR, ORWH or the NIH. This report is based on samples from the SWAN Core Repository. If scientists are interested in developing studies based on this resource, a description of the SWAN Core and DNA Repositories and how to obtain access to the resources can be found at http://www.swanrepository.org.
Footnotes
Authorship All authors had access to the data and participated in the writing of the manuscript.
Conflict of interest/financial disclosure
MFS, JFR, HZ, MJ, DM, SRK, CC & BN have no conflicts of interest or financial disclosure to this study.
References
- 1.Sowers MFR, Zheng H, Jannausch ML, et al. Amount of bone loss in relation to time around the final menstrual period and follicle-stimulating hormone staging of the transmenopause. The Journal of Clinical Endocrinology and Metabolism. 2010;95:2155–2162. doi: 10.1210/jc.2009-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherman BM, West JH, Korneman SG. The menopausal transition: analysis of LH, FSH, estradiol and progesterone concentrations during menstrual cycles of older women. The Journal of Clinical Endocrinology and Metabolism. 1976;42:629–636. doi: 10.1210/jcem-42-4-629. [DOI] [PubMed] [Google Scholar]
- 3.Metcalf MG, Donald RA, Livesey JH. Pituitary ovarian function in normal women during the menopause transition. Clinical Endocrinology. 1981;14:234–255. doi: 10.1111/j.1365-2265.1981.tb00193.x. [DOI] [PubMed] [Google Scholar]
- 4.Burger HG, Dudley EC, Hopper JL, et al. Prospectively measured levels of serum follicle-stimulating hormone, estradiol, and the dimeric inhibins during the menopausal transition in a population-based cohort of women. The Journal of Clinical Endocrinology and Metabolism. 1999;84:4025–4030. doi: 10.1210/jcem.84.11.6158. [DOI] [PubMed] [Google Scholar]
- 5.Overlie I, Moen MH, Morkrid L, et al. The endocrine transition around menopause – a five year prospective study with profiles of gonadotropins, estrogen, androgen and SHBG among healthy women. Acta Obstetricia et Gynecologica Scadinavica. 1999;10:642–647. [PubMed] [Google Scholar]
- 6.Burger HG, Dudley EC, Robertson DM, et al. Hormonal changes in the menopause transition. Recent Progress in Hormone Research. 2002;57:257–275. doi: 10.1210/rp.57.1.257. [DOI] [PubMed] [Google Scholar]
- 7.Randolph JF, Jr, Sowers M, Bondarenko IV, et al. Change in estradiol and follicle-stimulating hormone across the early menopausal transition: effects of ethnicity and age. The Journal of Clinical Endocrinology and Metabolism. 2004;89:1555–1561. doi: 10.1210/jc.2003-031183. [DOI] [PubMed] [Google Scholar]
- 8.Sowers MFR, Zheng H, McConnell D, et al. Estradiol rates of change in relation to the final menstrual period from a population-based cohort of women. The Journal of Clinical Endocrinology and Metabolism. 2008;93:3847–3852. doi: 10.1210/jc.2008-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Randolph JF, Crandall C, Gold EB, et al. Change in FSH and E2 across the menopausal transition: effect of age at the final menstrual period. Journal of Clinical Endocrinology and Metabolism. 2010 doi: 10.1210/jc.2010-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welt CK, Jimenez Y, Sluss PM, et al. Control of estradiol secretion in reproductive ageing. Human Reproduction. 2006;21:2189–2193. doi: 10.1093/humrep/del136. [DOI] [PubMed] [Google Scholar]
- 11.Dick IM, Devine A, Prince RL. Association of an aromatase TTTA repeat polymorphism with circulating estrogen, bone structure, and biochemistry in older women. American Journal of Physiology, Endocrinology and Metabolism. 2005;288:E989–E995. doi: 10.1152/ajpendo.00550.2004. [DOI] [PubMed] [Google Scholar]
- 12.Somner J, McLellan S, Cheung J, et al. Polymorphisms in the P450 c17 (17-hydroxylase/17, 20-Lyase) and P450 c19 (aromatase) genes: association with serum sex steroid concentrations and bone mineral density in postmenopausal women. The Journal of Clinical Endocrinology and Metabolism. 2004;89:344–351. doi: 10.1210/jc.2003-030164. [DOI] [PubMed] [Google Scholar]
- 13.Cai H, Shu XO, Egan KM, et al. Association of genetic polymorphisms in CYP19A1 and blood levels of sex hormones among postmenopausal Chinese women. Pharmacogenet and Genomics. 2008;8:657–664. doi: 10.1097/FPC.0b013e3282fe3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moeller G, Adamski J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Molecular and Cellular Endocrinology. 2009;301:7–19. doi: 10.1016/j.mce.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 15.Khan N, Sharma KK, Andersson S, et al. Human 17beta-hydroxysteroid dehydrogenases types 1, 2, and 3 catalyze bi-directional equilibrium reactions, rather than unidirectional metabolism, in HEK-293 cells. Archives of Biochemistry and Biophysics. 2004;429:50–59. doi: 10.1016/j.abb.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 16.Sowers MR, Wilson AL, Kardia SR, et al. Aromatase gene (CYP 19) polymorphisms and endogenous androgen concentrations in a multi-racial, multi-ethnic, multi-site study of women at midlife. The American Journal of Medicine. 2006;119(9A):S23–S30. doi: 10.1016/j.amjmed.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Agarwal AK, Auchus RJ. Minireview: cellular redox state regulates hydroxysteroid dehydrogenase activity and intracellular hormone potency. Endocrinology. 2005;146:2531–2538. doi: 10.1210/en.2005-0061. [DOI] [PubMed] [Google Scholar]
- 18.Brailly S, Gougeon A, Milgrom E, et al. Androgens and progestins in the human ovarian follicle: differences in the evolution of preovulatory, healthy nonovulatory, and atretic follicles. The Journal of Clinical Endocrinology and Metabolism. 1981;53:128–134. doi: 10.1210/jcem-53-1-128. [DOI] [PubMed] [Google Scholar]
- 19.Sowers MF, Crawford S, Sternfeld B, et al. Design, survey sampling and recruitment methods of SWAN: a multi-center, multi-ethnic, community-based cohort study of women and the menopausal transition. In: Lobos R, Marcus R, Kelsey JL, editors. Menopause. Academic Press; New York, NY: 2000. pp. 175–188. [Google Scholar]
- 20.Sowers MR, Wilson AL, Karvonen-Gutierrez CA, et al. Sex steroid hormone pathway genes and health-related measures in women of four races: the Study of Women’s Health Across the Nation (SWAN) The American Journal of Medicine. 2006;119(9A):S103–S110. doi: 10.1016/j.amjmed.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Kardia SR, Chu J, Sowers MR. Characterizing variation in sex steroid hormone pathway genes in women of 4 races/ethnicities: the Study of Women’s Health Across the Nation (SWAN) The American Journal of Medicine. 2006;119:S3–S15. doi: 10.1016/j.amjmed.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 22.England BG, Parsons GH, Possley RM, et al. Ultrasensitive semiautomated chemiluminescent immunoassay for estradiol. Clinical Chemistry. 2002;48:1584–1586. [PubMed] [Google Scholar]
- 23.Zhang D, Lin X, Sowers MF. Semiparametric stochastic mixed models for longitudinal hormone data. Journal of the American Statistical Association. 1998;93:710–719. [Google Scholar]
- 24.Neter J, Wasserman W, Kutner M. Applied Linear Statistical Models. 2. Homewood; Illinois: 1985. [Google Scholar]
- 25.Efron B, Tibshirani R. Bootstrap measures for standard errors, confidence intervals, and other measures of statistical accuracy. Statistical Science. 1986;1:54–77. [Google Scholar]
- 26.Claeskens G, Van Keilegom I. Bootstrap confidence bands for regression curves and their derivatives. Annals of Statistics. 2003;31:1852–1884. [Google Scholar]
- 27.Thurston RC, Sowers MF, Chang Y, et al. Adiposity and vasomotor symptom reporting among midlife women: the Study of Women’s Health Across the Nation (SWAN) American Journal of Epidemiology. 2008;167:78–85. doi: 10.1093/aje/kwm244. [DOI] [PubMed] [Google Scholar]
- 28.Schilling C, Gallicchio L, Miller SR, et al. Genetic polymorphisms, hormone levels, and hot flashes in midlife women. Maturitas. 2007;57:120–131. doi: 10.1016/j.maturitas.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunning AM, Dowsett M, Healey CS, et al. Polymorphisms associated with circulating sex hormone levels in postmenopausal women. Journal of the National Cancer Institute. 2004;96:936–945. doi: 10.1093/jnci/djh167. [DOI] [PubMed] [Google Scholar]
- 30.Comings DE, MacMurray JP. Molecular heterosis: a review. Molecular Genetics and Metabolism. 2000;71:19–31. doi: 10.1006/mgme.2000.3015. [DOI] [PubMed] [Google Scholar]
- 31.Chen ZJ. Molecular mechanisms of polyploidy and hybrid vigor. Trends in Plant Science. 2010;15:57–71. doi: 10.1016/j.tplants.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melchinger AE, Utz HF, Piepho HP, et al. The role of epistasis in the manifestation of heterosis: a systems-oriented approach. Genetics. 2007;177:1815–1825. doi: 10.1534/genetics.107.077537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Lu K, Chen Z, et al. Dominance, overdominance and epistasis condition the heterosis in two heterotic rice hybrids. Genetics. 2008;180:1725–1742. doi: 10.1534/genetics.108.091942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiévet JB, Dillmann C, de Vienne D. Systemic properties of metabolic networks lead to an epistasis-based model for heterosis. Theoretical and Applied Genetics. 2010;120:463–473. doi: 10.1007/s00122-009-1203-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Day JM, Foster PA, Tutill HJ, et al. 17 beta-hydroxysteroid dehydrogenase Type 1, and not Type 12, is a target for endocrine therapy of hormone-dependent breast cancer. International Journal of Cancer. 2008;122:1931–1940. doi: 10.1002/ijc.23350. [DOI] [PubMed] [Google Scholar]