

Abstract
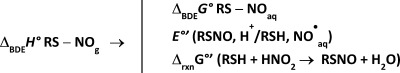
Nitrosothiols are powerful vasodilators. Although the mechanism of their formation near neutral pH is an area of intense research, neither the energetics nor the kinetics of this reaction or of subsequent reactions have been addressed. The following considerations may help to guide experiments. (1) The standard Gibbs energy for the homolysis reaction RSNO → RS• + NO•(aq) is +110 ± 5 kJ mol–1. (2) The electrode potential of the RSNO, H+/RSH, NO•(aq) couple is −0.20 ± 0.06 V at pH 7. (3) Thiol nitrosation by NO2– is favorable by 37 ± 5 kJ mol–1 at pH 7. (4) N2O3 is not involved in in vivo nitrosation mechanisms for thermodynamic — its formation from NO2– costs 59 kJ mol–1 — or kinetic — the reaction being second-order in NO2– — reasons. (5) Hemoglobin (Hb) cannot catalyze formation of N2O3, be it via the intermediacy of the reaction of Hb[FeNO2]2+ with NO• (+81 kJ mol–1) or reaction of Hb[FeNO]3+ with NO2– (+88 kJ mol–1). (6) Energetically and kinetically viable are nitrosations that involve HNO2 or NO• in the presence of an electron acceptor with an electrode potential higher than −0.20 V. These considerations are derived from existing thermochemical and kinetics data.
Short abstract
A value of 110 kJ mol−1 s−1 is derived for the Gibbs bond energy of RS−NO, which leads to −0.20 ± 0.06 for the electrode potential of the RSNO, H+/RSH, NO•(aq) couple at pH 7. These values allow a quantitative discussion of nitrosation mechanisms by HNO2 and NO•.
Introduction
How nitrogen monoxide can escape from blood to contribute to relaxation of blood vessels is an unsolved mystery. NO• in blood is rapidly consumed by binding to deoxyhemoglobin1,2 and reaction with oxyhemoglobin.3−5 Nitrosation of a thiol or formation of dinitrosyl iron complexes may be a way to preserve NO•, although reduction by one electron is necessary to set NO• free from a nitrosothiol. The energetics of these reactions have not been addressed. I show here that standard Gibbs energies and electrode potentials and the rate constants derived from these are easily calculated. The results allow one to eliminate reaction mechanisms and thereby to focus on possible pathways.
Nitrosation by NO2–
Standard Gibbs Bond Dissociation Energy of RS–NO
The energetics of nitrosation of thiols require knowledge of the Gibbs energy of reaction 1, in which RSNO represents a nitrosated cysteine as in S-nitrosoglutathione
| 1 |
For CH3CH2SNO, Bartberger et al. calculated a bond dissociation energy of 134 kJ mol–1 and a Gibbs dissociation energy of 89.5 kJ mol–1 in the gas phase.6 With the same ab initio technique, Baciu and Gauld reproduced this value and calculated a slightly higher bond dissociation energy of 139 kJ mol–1 for nitrosocysteine.7 Assuming that the –TΔS terms for both nitroso compounds are the same, −44.5 kJ mol–1, one arrives at a gas-phase Gibbs bond dissociation energy of nitrosocysteine of 94.5 kJ mol–1. To derive a Gibbs bond dissociation energy that is valid in water, we must dissolve nitrosocysteine, cysteine, and NO•. To a first approximation, the hydration energies of cysteine and nitrosocysteine are assumed to be the same. Additionally, NO• needs to be dissolved, which costs 15 kJ mol–1 (Table 1). Thus, in water, ΔrxnG°1 = +110 kJ mol–1 with an estimated error of 5 kJ mol–1, which reflects the uncertainty in the ab initio calculations and the fact the hydration energies do not fully cancel because R–SNO is more polar than R–SH.8,9
Table 1. Thermodynamic and Kinetic Quantitiesa.
| property | molecule, reaction | value at 25 °C | ref |
|---|---|---|---|
| ΔfG° | kJ·mol–1 | ||
| NO2– | –38 | (41) | |
| H2O (l) | –237.13 | (42) | |
| NO•(g) | +86.5 | (42) | |
| NO2•(g) | +51.3 | (42) | |
| N2O3(g) | +139.5 | (42) | |
| Henry constant | M/100 kPa | ||
| NO• | 1.93 × 10–3 | (43,44) | |
| NO2• | 1.1 × 10–2 | (45,46) | |
| N2O3 | 0.70 | (47) | |
| ΔfG° | kJ·mol–1 | ||
| NO•(aq) | +102 | b | |
| ONOO•(aq) | +117 ± 10 | (32) | |
| NO2•(aq) | +62.5 | b | |
| N2O3(aq) | +140 | b | |
| E° | V vs NHE | ||
| E°′2 | RSNO, H+/RSH, NO•(aq) | –0.20 ± 0.06 (pH 7) | c |
| E°′3 | RS•, H+/RSH | +0.94 ± 0.03 (pH 7) | (15) |
| E°4 | HNO2, H+/NO•(aq), H2O | +0.81 (pH 0) | b |
| E°′4 | +0.18 (pH 7) | ||
| E°8 | NO2•(aq)/NO2– | +1.04 | (41) |
| E°′–15 | HbFe3+/HbFe2+ | +0.122 (pH 7.1, 0.2 M Cl–) | (51) |
| E°′18 | CytcFe3+/CytcFe2+ | +0.26 V (pH 7) | (52) |
| E° | HbFe3+, NO•(aq) /Hb[FeNO]2+ | +0.71 (T
state) +0.80 (R state) |
c c |
| E° | Hb[FeNO]3+/Hb[FeNO]2+ | +0.47 (T
state) +0.54 (R state) |
c c |
| E°′ | S•–, H+/HS– | +0.92 V (pH 7) | (16) |
| S•–, 2H+, H2S | +0.92 V (pH 7) | c | |
| ΔrxnG°, K, k | |||
| ΔrxnG°1 | RSNO → RS• + NO• | +110 ± 5 kJ·mol–1 | c |
| ΔrxnG°5 | RSH + NO2– + 2H+ → RSNO + H2O | –37 ± 5 kJ·mol–1 | c |
| ΔrxnG°6 | 2NO2– + 2H+ → N2O3(aq) + H2O | –21 ± 3 kJ·mol–1 | b |
| ΔrxnG°′6(pH 7) | +59 ± 3 kJ·mol–1 | b | |
| K6 (pH 7) | 4.5 × 10–11 M–1 d | b | |
| k6, k–6 (pH 7) | k6 = 2.4 × 10–8 M–1 s–1, k–6 = 5.3 × 102 s–1 | (48) | |
| ΔrxnG°7 | N2O3(aq) → NO•(aq) + NO2•(aq) | +24.0 kJ/mol | b |
| K7 | 6.1 × 10–5 M–1 | (48,49) | |
| k7, k–7 | k7 = 8.0 × 104 s–1, k–7 = 1.1 × 109 M–1s–1 | (48) | |
| ΔrxnG°′10 | HbFe2+ + NO2– + 2H+ → HbFe3+ + NO•(aq) + H2O | –6 kJ mol–1 (pH 7) | c |
| k10 | 1.0 M–1 s–1 | (31) | |
| ΔrxnG°11 | HbFe3+ + NO2– → Hb[FeNO2]2+ | –16 to –19 kJ mol–1 | (21,50) |
| K11 | Hb-Fe3+ + NO2– → Hb[FeNO2]2+ | 2.0 × 103 M–1 (37 °C, pH 7.4), 0.56 × 103 M–1 (pH 7.4) | (21,50) |
| ΔrxnG°12 | Hb[FeNO2]2++ NO•(aq) → HbFe2++ N2O3(aq) | +81 kJ mol–1 | c |
| K13 | HbFe3+ + NO•(aq) → Hb[FeNO]3+ | 1.3 × 104 M–1 | (25) |
| ΔrxnG°14 | Hb[FeNO]3+ + NO2– → HbFe2+ +N2O3 | +88 kJ mol–1 | c |
| ΔrxnG°′16 | Hb[FeNO]3+ + H2O → HbFe2+ + NO2– + 2H+ | +22 kJ mol–1 (pH 7) | c |
| HbFe2+ + NO•(aq) → Hb[FeNO]2+ | 8.7 × 109 M–1 (T state), 1.7 × 1011 M–1 (R state) | (25) |
Errors, where indicated, are estimates. Numerical subscripts refer to equations in the text.
The value follows directly from the literature values quoted in the table.
The value is derived in the text.
Value corrected for pH and the standard state of water.
How easily is RSNO reduced by one electron to liberate NO•? The electrode potential of the RSNO, H+/RSH, NO•(aq) couple, reaction 2, follows from addition of reactions 1 and 3 (Table 1) and is −0.20 ± 0.06 V at pH 7 vs the normal hydrogen electrode.
| 2 |
| 3 |
Monohydrogenascorbate, with E°′(asc•–, H+/Hasc–) = +0.28 V,10 should thus not reduce RSNO, as observed. On the basis of this observation and that dithionite did reduce RSNO, Bohle and co-workers concluded that the electrode potential was less than 0 V,11 in agreement with the present estimate. A value of −0.20 V implies that, in the presence of redox couples with electrode potentials larger than that value, generation of NO•(aq) is uphill. On the other hand, redox couples with such potentials would help formation of RSNO from RSH and NO•. Indeed, iron is known to help in formation of nitrosothiols.12
Energetics of Nitrosation
The energetics of nitrosation
by HNO2 are now calculated by addition of reactions −1,
−3, and 4 (Table 1), in which RSH stands
for glutathione and represents thiols in general. Here and below frequent
use is made of the equalities ΔrxnG° = −RT lnK = −nFΔE° in which R is the gas constant, n the number of electrons
in the reaction equation, and F the Faraday constant. 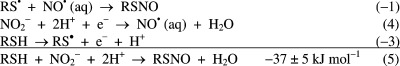 Nitrosation of RSH by HNO2 is thus favorable
by −37 ± 5 kJ mol–1 at pH 7, which compares
well with the −33 kJ mol–1 derived from the
equilibrium constant of 6 × 105 M–1 listed by Williams13 for cysteine. What
do these Gibbs energies mean? Given a concentration of 0.5 μM
nitrite, the ratio of RSNO to RSH should be between 0.5:1 and 1:1.
Thus, given millimolar concentrations of thiols, one would also expect
millimolar concentrations of nitrosothiols. This is not found, which
may show that production is rate limiting.
Nitrosation of RSH by HNO2 is thus favorable
by −37 ± 5 kJ mol–1 at pH 7, which compares
well with the −33 kJ mol–1 derived from the
equilibrium constant of 6 × 105 M–1 listed by Williams13 for cysteine. What
do these Gibbs energies mean? Given a concentration of 0.5 μM
nitrite, the ratio of RSNO to RSH should be between 0.5:1 and 1:1.
Thus, given millimolar concentrations of thiols, one would also expect
millimolar concentrations of nitrosothiols. This is not found, which
may show that production is rate limiting.
Nitrosation may also
involve two NO2– and N2O3 as an intermediate according to the following set of reactions
(see Table 1), but the overall energetics are
those of reaction 5, −37 kJ mol–1.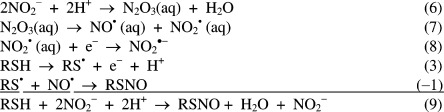 Nitrosation by the simplest of nitrosothiols, HSNO, is
ca. 10 kJ mol–1 less favorable, as is easily calculated
from the minor difference between the bond strengths, that of RS–NO
being ca. 12 kJ stronger6 than that of
HS–NO,14 and between the electrode
potentials, E°′3(RS•,H+/RSH) = +0.94 V15 and E°′(S•–,H+/HS–) = +0.92 V,16 at
pH 7. Given a pK1 of H2S of 7.1, this value also applies to E°′(S•–,2H+/H2S) at pH 7. Consequently, transnitrosation of RSH by HSNO is downhill by
the same amount.
Nitrosation by the simplest of nitrosothiols, HSNO, is
ca. 10 kJ mol–1 less favorable, as is easily calculated
from the minor difference between the bond strengths, that of RS–NO
being ca. 12 kJ stronger6 than that of
HS–NO,14 and between the electrode
potentials, E°′3(RS•,H+/RSH) = +0.94 V15 and E°′(S•–,H+/HS–) = +0.92 V,16 at
pH 7. Given a pK1 of H2S of 7.1, this value also applies to E°′(S•–,2H+/H2S) at pH 7. Consequently, transnitrosation of RSH by HSNO is downhill by
the same amount.
Nitrosation by N2O3
One or Two NO2–?
Given that nitrosation is possible with one or two NO2–, we now ask which reaction is more likely. An interesting observation, published in 2003,17 is that a concentration of ca. 2.5 μM in blood causes some vasodilation with a much larger effect observed at about 200 μM. How can one produce NO•, or RSNO, from NO2– and deliver the former to the endothelial cell from where it can diffuse into the muscle layer surrounding the blood vessel? Basu et al.18 followed up on a proposal by Robinson and Lancaster19 that deoxyhemoglobin catalyze formation of N2O3 from NO2– according to reactions 10–12. The advantage of N2O3 is that it does not interact with Fe2+ in hemoglobin.
| 10 |
| 11 |
| 12 |
Addition of Reactions 10–12 results in reaction 6; thus, hemoglobin is thought to act as a catalyst. As shown in Table 1, ΔrxnG°′6 = +59 kJ mol–1 at pH 7, which, given a plasma concentration of 2.5 μM NO2–,17 results in an equilibrium concentration of 2.8 × 10–22 M N2O3. Given that the rate of hydrolysis, k–6, is known (Table 1), k6 is 2.4 × 10–8 M–1 s–1. Half-lives of Reactions 6 and −6 can now be calculated. The t1/2 of hydrolysis of N2O3 (reaction −6) is (ln 2)/k–6 or 0.693/530 s–1 = 1.3 ms. The t1/2 of reaction 6 is given by 1/(k6[NO2–]) or 1.7 × 1013 s or slightly more than 500 000 years. At a concentration of 200 μM NO2–, these numbers are different but still do not support formation of N2O3.
Another reaction that ought to be taken into account is reaction 7, dissociation of N2O3. If N2O3 were formed it would, at that dilute concentration, completely dissociate into NO• and NO2• before it hydrolyzes: the t1/2 of reaction 7 (Table 1) is 0.693/8.0 × 104 s–1 = 8.7 μs. Thus, formation of N2O3 is thermodynamically and kinetically unlikely.
Catalysis by Hemoglobin
Can hemoglobin act as a catalyst as proposed?18 Formation of N2O3 must be fast or NO• disappears by binding to hemoglobin or reaction with oxyhemoglobin. Thus, given a t1/2 of 1.7 × 1013 s, N2O3 needs to be produced on the second time scale, ca. 1013 times faster. If that were feasible, it would not help because the rate of hydrolysis would increase by the same factor. Are these results very sensitive to the precise values of the Gibbs energies? The answer is no: to be physiologically relevant, nanomolar concentrations of N2O3 need to be produced. To achieve a 1 nM concentration of N2O3 at equilibrium, the Gibbs energy of reaction 6 has to change by 69 kJ/mol to become −10 kJ mol–1. Selective use of the R and T states of hemoglobin, if possible, may change the energetics favorably by ca. 10 kJ/mol,20 which still does not make formation of N2O3 possible. It needs to be pointed out, pro forma, that if R and T states are involved then hemoglobin is not acting as a true catalyst.
The energetics of reactions 10 and 11 are easily calculated from the data collected in Table 1: these are −6 and −16 kJ/mol, respectively. Given that ΔrxnG°′6 = +59 kJ/mol at pH 7, the Gibbs energy change of reaction 12 at pH 7 is +81 kJ/mol. Were one to use the binding constant of the methemoglobin–nitrite complex determined by Goetz et al.,21 then the Gibbs energy change of reaction 12 is +84 kJ mol–1. This shows that tighter binding of NO2– to methemoglobin does not help the formation of N2O3.
Following a study by Fernandez and Ford,22 Hopmann et al.23 also considered an alternative mechanism for formation of N2O3
| 13 |
| 14 |
| 15 |
| 4 |
Reactions 4 and 13–15 also add up to reaction 6; thus, ΔrxnG°′14 = +88 kJ mol–1 at pH 7.
Comparison with DFT Calculations
The energetics of reactions 12 and 14 have been estimated by density functional theory calculations. Hopmann et al.23 report that reaction 12 is uphill by 71–84 kJ mol–1, in good agreement with the value of +81 kJ mol–1. However, Berto and Lehnert,24 using a more refined model of the active site, claim that reaction 12 is slightly exothermic, between −4 and −13 kJ mol–1, which is incorrect by 85–95 kJ mol–1. For reaction 14, Hopmann et al.23 conclude that electron transfer from NO2– to Hb[FeNO]3+ is favorable by 29 kJ mol–1, which is off by 117 kJ mol–1. In spite of the large difference in Gibbs energies for reactions 12 and 14, Hopmann et al.23 conclude that both reactions are energetically reasonable. However, electron transfer from NO2– to Hb[FeNO]3+ is not exothermic: from the equilibria between NO• and HbFe2+ and NO• and HbFe3+ 25 one calculates (Table 1) that E°(Hb[FeNO]3+/Hb[FeNO]2+) is +0.47 V for T-state hemoglobin and +0.54 V for R-state hemoglobin. Combined with the electrode potential of the NO2•/NO2– couple, 1.04 V (Table 1), electron transfer is unfavorable by at least 48 kJ/mol. It is truly regrettable that these ab initio calculations provide neither consistent nor proper estimates of Gibbs energies because it implies that any proposed intermediates and transition states are similarly compromised. Gibbs energies can be correctly and rapidly calculated per manum.
Are reactions 12 and 14 possible if we let N2O3 hydrolyze? We deduct the Gibbs energy of reaction 6 and obtain +22 and +29 kJ mol–1, respectively. These numbers are small enough to let the reactions proceed if products are removed. The Gibbs energy of +29 kJ mol–1 also applies to reaction 16
| 16 |
Reaction 11 results in Hb[FeNO2]2+, in which NO2– is thought18 to be partially oxidized by Fe3+. Given a binding energy of only 16 kJ mol–1 (reaction 10) and the difference in electrode potential between the couples HbFe3+/HbFe2+ and NO2•(aq)/NO2– of 0.9 V (Table 1), such a partial electron transfer is unlikely, as was recognized by Berto and Lehnert.24
The conclusion is that N2O3 cannot play a role in the preservation of NO•. Furthermore, given the low physiological concentration of NO2–, any mechanism that relies on two NO2– to occur on a second time scale is kinetically doomed.
Nitrosation by HNO2, by NO• and an Electron Acceptor, and by ONOO•
Returning to the original observation, which is that injection of 0.40 mM NO2– into the brachial artery of the upper arm resulted in a final concentration of ca. 2.5 μM as measured in the ipsolateral antecubital vein, led to noticeable vasodilation17 and having shown that the N2O3 pathway is most unlikely, one can ask whether HNO2, present under these conditions at a concentration of ca. 0.25 nM, is the agent responsible. Like N2O3, HNO2 is neutral and could penetrate endothelial cells. Nitrosation is thermodynamically possible, but is it fast enough? The rate of nitrosation is given by
| 17 |
in which k = 4.6 × 105 M–2 s–1.13 It is of course not correct to use eq 17 if the thiol of interest, for instance, hemoglobin β-chain cysteine 93, is not homogeneously distributed. The following considerations, therefore, result in only a rough estimate. Given a concentration inside the red blood cell of 5 mM hemoglobin, and thus of 10 mM β-chain cysteine 93, and of a HNO2 concentration of 0.25 × 10–9 M, then the rate of nitrosothiol formation is 1× 10–13 M s–1, which would appear to be too slow. However, the concentration of NO2– at the site of injection was much higher. It may thus be possible that the small extent of vasodilation was caused by HNO2.
If nitrosation does not involve NO2– but NO• then an electron
acceptor with an electrode potential larger than −0.20 V (reaction 2) is required. Iron(III) cytochrome c is such an electron acceptor for the nitrosation of glutathione,26 and the overall reaction is favorable: 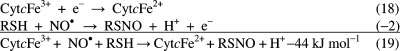 Mechanistically, reaction 19 requires three reactants to
be present at the same time in close proximity, which makes it kinetically
unlikely. However, this problem is obviated by binding of glutathione
to cytochrome c prior to reaction with NO•. A similar mechanism can be written for methemoglobin with an overall
Gibbs energy change of −26 kJ mol–1. Dioxygen
may also act as an electron acceptor;27 given an E°(O2/O2•–) of −0.35 V,28 the reaction is uphill but pulled through by the diffusion-controlled
reaction of the product, O2•–,
with another NO•.29 As
experimentally observed, this nitrosation reaction is second order
in NO•.27
Mechanistically, reaction 19 requires three reactants to
be present at the same time in close proximity, which makes it kinetically
unlikely. However, this problem is obviated by binding of glutathione
to cytochrome c prior to reaction with NO•. A similar mechanism can be written for methemoglobin with an overall
Gibbs energy change of −26 kJ mol–1. Dioxygen
may also act as an electron acceptor;27 given an E°(O2/O2•–) of −0.35 V,28 the reaction is uphill but pulled through by the diffusion-controlled
reaction of the product, O2•–,
with another NO•.29 As
experimentally observed, this nitrosation reaction is second order
in NO•.27
Alternatively,
NO• may first bind to iron(III)
followed by the nitrosation reaction. In the case of methemoglobin
this process is less favorable but still possible: 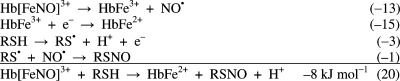 Experimental evidence for this pathway exists.30 However, in vivo, this reaction pathway seems
unlikely as the concentration of methemoglobin is small and because
NO• is more likely to react with oxy- and deoxy-hemoglobin.
Experimental evidence for this pathway exists.30 However, in vivo, this reaction pathway seems
unlikely as the concentration of methemoglobin is small and because
NO• is more likely to react with oxy- and deoxy-hemoglobin.
A modification that involves NO2– and
hemoglobin as a catalyst allows the following kinetically and thermodynamically
feasible reactions: ![]() The only assumption made is that reaction of Hb[FeNO]3+ with RSH takes place before NO• relocates
to HbFe2+ or reacts with oxyhemoglobin. Indeed, if dissociation
of NO• from HbFe3+ takes place then we
have reaction 10, which is slow, 1 M–1 s–1 at pH 7.5.31
The only assumption made is that reaction of Hb[FeNO]3+ with RSH takes place before NO• relocates
to HbFe2+ or reacts with oxyhemoglobin. Indeed, if dissociation
of NO• from HbFe3+ takes place then we
have reaction 10, which is slow, 1 M–1 s–1 at pH 7.5.31
We found recently evidence for ONOO• as an intermediate in the oxidation of NO• to NO2•.32 Can it play a role in nitrosation? Given an electrode potential of +0.51 V33 for E°(ONOO•/ONOO–), oxidation of RSH (E°′3 = +0.94 V, Table 1) is uphill, but overall the reaction would be favorable, as the oxidation is followed by reaction of NO• with RS•, reaction −1. Now there are several possibilities in theory, but none of them is practical: Dissociation of ONOO– could yield the necessary NO•, but this reaction is slow, 0.02 s–1.34 More likely is protonation of ONOO– to yield ONOOH, which oxidizes or nitrates other molecules in the vicinity. Formation of RSNO with a second NO• is kinetically unlikely. Oxidation of RSH by NO2• is similarly unrealistic, not in the least because formation of the latter from NO• under in vivo conditions is extremely slow.35
Conclusion
Nitrosation by N2O3 and ONOO• can be excluded, by HNO2 may be possible, and reactions that involve NO• require a suitable electron acceptor. The mechanism proposed in reactions −16 and 20 needs to be investigated further.
The equations and energetics provided here can be used as LEGO blocks to build a reaction mechanism. Once an energetically favorable mechanism has been established, one must ask the question whether the kinetics are fast enough. It is important to keep in mind that the reactions used to calculate a Gibbs energy, such as reactions −1, 3, −13, and −15 above, do not necessarily take place: they serve to produce the Gibbs energy of reaction 20. In particular, given that the RS• radical is in equilibrium with R•SH, where R• stands for a carbon-centered radical elsewhere in the molecule,36,37 one would do well to avoid RS• in mechanisms of nitrosation.
The approach used here is not new,38−40 requires only pencil and paper, and may help in defining the reaction one has an interest in prior to embarking on possibly elaborate, expensive, and technically difficult laboratory experiments or in silico calculations.
Acknowledgments
Discussions with Dr. P. L. Bounds, Dr. T. Nauser, Dr. R. Kissner, and Prof. I. Ivanovich are gratefully acknowledged. I am indebted to Dr. N. Hogg and Dr. M. Gladwin for clarifying various issues related to the proposed anhydratase function of haemoglobin.
The authors declares no competing financial interest.
References
- Cassoly R.; Gibson Q. H. Conformation, co-operativity and ligand binding in human hemoglobin. J. Mol. Biol. 1975, 91, 301–313. [DOI] [PubMed] [Google Scholar]
- Moore E. G.; Gibson Q. H. Cooperativity in the dissociation of nitric oxide from hemoglobin. J. Biol. Chem. 1976, 251, 2788–2794. [PubMed] [Google Scholar]
- Doyle M. P.; Pickering R. A.; Cook B. R. Oxidation of oxymyoglobin by nitric oxide through dissociation from cobalt nitrosyls. J. Inorg. Biochem. 1983, 19, 329–338. [Google Scholar]
- Eich R. F.; Li T.; Lemon D. D.; Doherty D. H.; Curry S. R.; Aitken J. F.; Mathews A. J.; Johnson K. A.; Smith R. D.; Phillips G. N.; Olson J. S. Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 1996, 35, 6976–6983. [DOI] [PubMed] [Google Scholar]
- Herold S. Kinetic and spectroscopic characterization of an intermediate peroxynitrite complex in the nitrogen monoxide induced oxidation of oxyhemoglobin. FEBS Lett. 1998, 439, 85–88. [DOI] [PubMed] [Google Scholar]
- Bartberger M. D.; Mannion J. D.; Powell S. C.; Stamler J. S.; Houk K. N.; Toone E. J. S–N Dissociation energies of S-nitrosothiols: On the origins of nitrosothiol decomposition rates. J. Am. Chem. Soc. 2001, 123, 8868–8869. [DOI] [PubMed] [Google Scholar]
- Baciu C.; Gauld J. W. An assessment of theoretical methods for the calculation of accurate structures and S–N bond dissociation energies of S-nitrosothiols (RSNO). J. Phys. Chem. A 2003, 107, 9946–9952. [Google Scholar]
- Li H. Z.; Tao W.; Gao T.; Li H.; Lu Y. H.; Su Z. M. Improving the accuracy of density functional theory (DFT) calculation for homolysis bond dissociation energies of Y-NO bond: Generalized regression neural network based on Grey relational analysis and principal component analysis. Int. J. Mol. Sci. 2011, 12, 2242–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timerghazin Q. K.; English A. M.; Peslherbe G. H. On the multireference character of S-nitrosothiols: A theoretical study of HSNO. Chem. Phys. Lett. 2008, 454, 24–29. [Google Scholar]
- Williams M. H.; Yandell J. K. Outer-sphere electron transfer reactions of ascorbate anions. Aust. J. Chem. 1982, 35, 1133–1144. [Google Scholar]
- Arulsamy N.; Bohle D. S.; Butt J. A.; Irvine G. J.; Jordan P. A.; Sagan E. Interrelationships between conformational dynamics and the redox chemistry of S-nitrosothiols. J. Am. Chem. Soc. 1999, 121, 7115–7123. [Google Scholar]
- Vanin A. F.; Malenkova I. V. Iron is a catalyst of cysteine and glutathione S-nitrosation on contact with nitric oxide in aqueous solutions at neutral pH. Biochemistry (Moscow) 1996, 61, 374–379. [Google Scholar]
- Williams D. L. H.Nitrosation Reactions and the Chemistry of Nitric Oxide; Elsevier B.V.: 2004. [Google Scholar]
- Timerghazin Q.; Peslherbe G. H.; English A. M. Structure and stability of HSNO, the simplest S-nitrosothiol. Phys. Chem. Chem. Phys. 2008, 10, 1532–1539. [DOI] [PubMed] [Google Scholar]
- Folkes L. K.; Trujillo M.; Bartesaghi S.; Radi R.; Wardman P. Kinetics of reduction of tyrosine phenoxyl radicals by glutathione. Arch. Biochem. Biophys. 2011, 506, 242–249. [DOI] [PubMed] [Google Scholar]
- Das T. N.; Huie R. E.; Neta P.; Padmaja S. Reduction potential of the sulfhydryl radical: Pulse radiolysis and laser flash photolysis studies of the formation and reactions of .SH and HSSH.– in aqueous solutions. J. Phys. Chem. A 1999, 103, 5221–5226. [Google Scholar]
- Cosby K.; Partovi K. S.; Crawford J. H.; Patel R. P.; Reiter C. D.; Martyr S.; Yang B. K.; Waclawiw M. A.; Zalos G.; Xu X.; Huang K. T.; Shields H.; Kim-Shapiro D. B.; Schechter A. N.; Cannon R. O. III; Gladwin M. T. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 2003, 9, 1498–1505. [DOI] [PubMed] [Google Scholar]
- Basu S.; Grubina R.; Huang J.; Conradie J.; Huang Z.; Jeffers A.; Jiang A.; He X.; Azarov I.; Seibert R.; Mehta A.; Patel R.; King S. B.; Hogg N.; Ghosh A.; Gladwin M. T.; Kim-Shapiro D. B. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat. Chem. Biol. 2007, 3, 785–794. [DOI] [PubMed] [Google Scholar]
- Robinson J. M.; Lancaster J. R. Hemoglobin-mediated, hypoxia-induced vasodilation via nitric oxide. Am. J. Respir. Cell Mol. Biol. 2005, 32, 257–261. [DOI] [PubMed] [Google Scholar]
- Holt J. M.; Klinger A. L.; Yarian C. S.; Keelara V.; Ackers G. K. Asymmetric distribution of cooperativity in the binding cascade of normal human hemoglobin. 1. Cooperative and noncooperative oxygen binding in Zn-substituted hemoglobin. Biochemistry 2005, 44, 11925–11938. [DOI] [PubMed] [Google Scholar]
- Goetz B. I.; Shields H. W.; Basu S.; Wang P.; King S. B.; Hogg N.; Gladwin M. T.; Kim-Shapiro D. B. An electron paramagnetic resonance study of the affinity of nitrite for methemoglobin. Nitric Oxide 2010, 22, 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez B. O.; Ford P. C. Nitrite catalyzes ferriheme protein reductive nitrosylation. J. Am. Chem. Soc. 2003, 125, 10510–10511. [DOI] [PubMed] [Google Scholar]
- Hopmann K. H.; Cardey B.; Gladwin M. T.; Kim-Shapiro D. B.; Ghosh A. Hemoglobin as a nitrite anhydrase: Modeling methemoglobin-mediated N2O3 formation. Chem.—Eur. J. 2011, 17, 6348–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berto T. C.; Lehnert N.. Density functional theory model of the proposed anhydrase funtion of hemoglobin in hypoxia sensing. Inorg. Chem. 2011, 50, 7361−7663. [DOI] [PubMed] [Google Scholar]
- Ford P. C.; Laverman L. E.; Lorkovic I. M.. Reaction mechanisms of nitric oxide with biologically relevant metal centers. In Advances in Inorganic Chemistry. Solvent Exchange on Metal Ions; Academic Press: New York, 2003; Vol. 54, pp 203–257. [Google Scholar]
- Basu S.; Keszler A.; Azarova N. A.; Nwanze N.; Perlegas A.; Shiva S.; Broniowska K. A.; Hogg N.; Kim-Shapiro D. B. A novel role for cytochrome c: Efficient catalysis of S-nitrosothiol formation. Free Radical Biol. Med. 2010, 48, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A. J.; Buerk D. G.; Ischiropoulos H. A novel reaction mechanism for the formation of S-nitrosothiol in vivo. J. Biol. Chem. 1997, 272, 2841–2845. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H.; Stanbury D. M.; Bounds P. L. Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radical Biol. Med. 2010, 49, 317–322. [DOI] [PubMed] [Google Scholar]
- Botti H.; Möller M.; Steinmann D.; Nauser T.; Koppenol W. H.; Denicola A.; Radi R. Distance-dependent diffusion-controlled reaction of •NO and O2•– at chemical equilibrium with ONOO–. J. Phys. Chem. B 2010, 114, 16584–16593. [DOI] [PubMed] [Google Scholar]
- Herold S.; Röck G. Reactions of deoxy-, oxy-, and methemoglobin with nitrogen monoxide - Mechanistic studies of the S-nitrosothiol formation under different mixing conditions. J. Biol. Chem. 2003, 278, 6623–6634. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Pickering R. A.; DeWeert T. M.; Hoekstra J. W.; Pater D. Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J. Biol. Chem. 1981, 256, 12393–12398. [PubMed] [Google Scholar]
- Galliker B.; Kissner R.; Nauser T.; Koppenol W. H. Intermediates in the autoxidation of nitrogen monoxide. Chem.—Eur. J. 2009, 15, 6161–6168. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Arbault S.; Bruce D.; de Oliveira P.; Erard M.; Vuillaume M. Characterization of the electrochemical oxidation of peroxynitrite: Relevance to oxidative stress bursts measured at the single cell level. Chem.—Eur. J. 2001, 7, 4171–4179. [DOI] [PubMed] [Google Scholar]
- Sturzbecher M.; Kissner R.; Nauser T.; Koppenol W. H. Homolysis of the peroxynitrite anion detected with permanganate. Inorg. Chem. 2007, 46, 10655–10658. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H.The chemistry and biochemistry of oxidants derived from nitric oxide. In Nitric Oxide and Radicals in the Pulmonary Vasculature; Weir E. K., Archer S. L., Reeves J. T., Eds.; Futura Publishing Co., Inc.: Armonk, NY, 1996; pp 355–362. [Google Scholar]
- Nauser T.; Casi G.; Koppenol W. H.; Schöneich C. Reversible intramolecular hydrogen transfer between cysteine thiyl radicals and glycine and alanine in model peptides: Absolute rate constants derived from pulse radiolysis and laser flash photolysis. J. Phys. Chem. B 2008, 112, 15034–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstetter D.; Nauser T.; Koppenol W. H. Hydrogen exchange equilibria in glutathione radicals - Rate constants. Chem. Res. Toxicol. 2010, 23, 1596–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol W. H. Reactions involving singlet oxygen and the superoxide anion. Nature 1976, 262, 420–421. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H. Oxygen activation by cytochrome P450: A thermodynamic analysis. J. Am. Chem. Soc. 2007, 129, 9686–9690. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H. Oxyradical reactions: From bond-dissociation energies to reduction potentials. FEBS Lett. 1990, 264, 165–167. [DOI] [PubMed] [Google Scholar]
- Stanbury D. M. Reduction potentials involving inorganic free radicals in aqueous solution. Adv. Inorg. Chem. 1989, 33, 69–138. [Google Scholar]
- Wagman D. D.; Evans W. H.; Parker V. B.; Schumm R. H.; Halow I.; Bailey S. M.; Churney K. L.; Nuttal R. L. Selected values for inorganic and C1 and C2 organic substances in SI units. J. Phys. Chem. Ref. Data 1982, 11 (Suppl. 2), 37–38. [Google Scholar]
- Wilhelm E.; Battino R.; Wilcock R. J. Low-pressure solubility of gases in liquid water. Chem. Rev. 1977, 77, 219–262. [Google Scholar]
- Zacharia I. G.; Deen W. M. Diffusivity and solubility of nitric oxide in water and saline. Ann. Biomed. Eng. 2005, 33, 214–222. [DOI] [PubMed] [Google Scholar]
- Schwartz S. E.; White W. H. Kinetics of reactive dissolution of nitrogen dioxides into aqueous solution. Adv. Environ. Sci. Technol. 1983, 12, 1–116. [Google Scholar]
- Cheung J. L.; Li Y. Q.; Boniface J.; Shi Q.; Davidovits P.; Worsnop D. R.; Jayne J. T.; Kolb C. E. Heterogeneous interactions of NO2 with aqueous surfaces. J. Phys. Chem. A 2000, 104, 2655–2662. [Google Scholar]
- Markovits G. Y.; Schwartz S. E.; Newman L. Hydrolysis equilibrium of dinitrogen trioxide in dilute acid solution. Inorg. Chem. 1981, 20, 445–450. [Google Scholar]
- Grätzel M.; Taniguchi S.; Henglein A. Pulsradiolytische Untersuchung der NO-Oxydation und des Gleichgewichts N2O3 ≤=> NO + NO2 in wässriger Lösung. Ber. Bunsen Ges. Phys. Chem. 1970, 74, 488–492. [Google Scholar]
- Treinin A.; Hayon E. Absorption spectra and reaction kinetics of NO2, N2O3, and N2O4 in aqueous solution. J. Am. Chem. Soc. 1970, 92, 5821–5828. [DOI] [PubMed] [Google Scholar]
- Schwab D. E.; Stamler J. S.; Singel D. J. Nitrite-methemoglobin inadequate for hypoxic vasodilation. Nat. Chem. Biol. 2009, 3, 366. [DOI] [PubMed] [Google Scholar]
- Taboy C. H.; Faulkner K. M.; Kraiter D.; Bonaventura C.; Crumbliss A. L. Concentration-dependent effects of anions on the anaerobic oxidation of hemoglobin and myoglobin. J. Biol. Chem. 2000, 275, 39048–39054. [DOI] [PubMed] [Google Scholar]
- Schejter A.; Aviram I.; Goldkorn T.. The contribution of electrostatic factors to the oxidation reduction potentials of c-type cytochromes. In Electron transport and oxygen utilization; Ho C., Ed.; Elsevier North Holland, Inc.: 1982; pp 95–99. [Google Scholar]