

The indole alkaloids of the terrestrial blue-green algae[1] have elicited a broad response in the form of new chemical methods and preparations for these structurally diverse natural products. This enterprise is driven, in part, by the diverse biological activity that is often associated with metabolites of their resident cyanobacteria.[2] Beginning with Natsume's syntheses of (nonchlorinated) hapalindoles J & M,[3] members of the fischerindole,[4] hapalindole,[3, 5] welwitindolinone,[4, 6] and ambiguine[7] natural products have been accessed by total chemical synthesis.[8] The chlorinated congeners further increase the functional and stereochemical diversity of this natural product class, but solutions for their synthesis are few in number. Fukuyama reported the first synthesis of a hapalindole containing a chiral secondary alkyl chloride (hapalindole G, 3).[5g] The syn relationship between the chlorine and methyl groups was established by cyclopropane ring opening with lithium chloride. In 2005, Baran and coworkers reported syntheses of fischerindoles I and G. The neopentyl chloride of these linear tetracycles is diastereomeric relative to that in 3, and was also constructed through a series of stereospecific transformations, leading with an epoxide ring opening reaction.[4] The same neopentyl chloride is contained within welwitindolinone A, and Wood's solution to this involved a chloronium-induced [1,2]-methyl shift to establish the anti-relationship between the methyl and chlorine. Equally elegant was the Baran group's preparation of (+)-welwitindolinone A from (−)-fischerindole I.[4, 7]
We now report the first total synthesis of hapalindoles K and A, and a formal synthesis of hapalindole G (Figure 1). In so doing, use of i) a two-fold electrophilic aromatic substitution (EAS) reaction between indole and α-methyl tiglic acid chloride, ii) a demanding intermolecular [4+2] cycloaddition, and iii) a late stage Ritter reaction provided the needed convergency and functional group installation to deliver each target in 15 steps or less.
Figure 1.
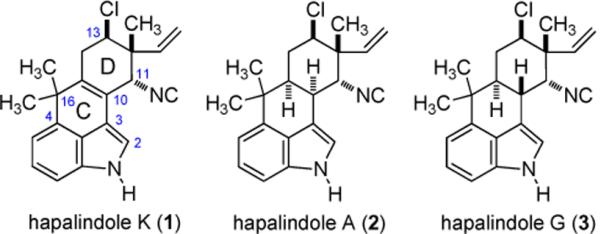
Structures of Hapalindole K, A, and G
In an earlier report, we applied a rhodium(II) catalyzed cyclohexannulation to construct the ABC ring system of ambiguine G which bears a substituent at C1.[9] The needs of hapalindoles 1–3 are more directly addressed by construction of the tricyclic ring system through an acylation/alkylation protocol in two steps (Scheme 1). Et2AlCl mediated Friedel-Crafts acylation of indole with α-methyl tiglic acid chloride provided adduct 4 in excellent yield.[10] An extensive investigation of Lewis acids to effect the desired C4–C16 bond formation was unsuccessful, with retro-Friedel-Crafts acylation to return indole predominating in most cases. However, we discovered that the desired tricyclic ketone (6) could be obtained by treatment with a molten AlCl3-NaCl solution.[11] Temperature is very critical to the regioselective outcome of this cyclization step, since optimal temperatures (117–120 °C) provided 6 as the major product, but elevated temperatures led predominantly to regioisomer 5. On larger scale, purification was reserved until after sulfonamide formation since undesired 5 (fischerindole backbone) was unreactive under these conditions. Ketone 7 was subsequently converted to its enol triflate[12] and treated with Zn(CN)2 and Pd(PPh3)4 to give the α,β-unsaturated nitrile in excellent yield.[13] Nitrile reduction with DIBAL was followed by treatment of the resulting enal (8) with TBSOTf and Et3N. This sequence furnished the requisite diene (9) with the desired geometry.
Scheme 1.
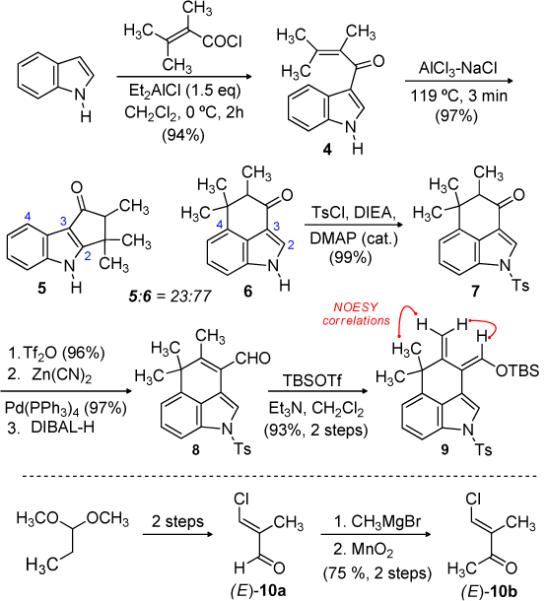
Diene and Dienophile Synthesis
With diene 9 and dienophile 10a in hand, different strategies were evaluated to effect the desired intermolecular Diels-Alder cycloaddition.[14] Attempts to effect the cycloaddition under thermal conditions led to slow desilylation of 9 to form the corresponding enal (8), as well as an undefined decomposition pathway for the dienophile. Lewis acid-promoted Diels-Alder cycloaddition between diene 9 and β-chloro methacrolein 10a[15] was also investigated, but the Mukaiyama aldol product predominated in all attempts.
In order to slow the 1,2-addition pathway, we evaluated dienophile 10b as a substitute for β-chloro-α-methyl acrolein (Scheme 1). A range of Lewis acids were examined to promote the cycloaddition, as partially outlined in Table 1.
Table 1.
Lewis Acid Promoted Intermolecular Diels-Alder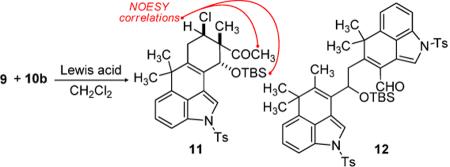
| Entry | Lewis acid | temp, time | 11:12:8[a] | yield (11)[b] |
|---|---|---|---|---|
| 1 | TMSOTf | −78 °C, 2 h | 1:0:1 | 18% |
| 2 | TiCl4 | −20 °C, 0.5 h | 1:3:1 | 24% |
| 3 | Ti(iOPr)4 | 0 °C, 12 h | 0:0:1 | – |
| 4 | Et2AlCl | 0 °C, 12 h | 0:0:1 | – |
| 5 | (CH3)3Al | 0 °C, 12 h | 0:0:1 | – |
| 6 | EtAlCl2 | −20 °C, 1 h | 3:1:0 | 36% |
| 7 | EtAlCl2[c] | −78 → −20 °C, 3 h | 13:1:1 | 59% |
Determined by analysis of the crude reaction mixture by 1H NMR.
Isolated yield.
Substitution of CH2Cl2 for toluene as the solvent provided 11 in 54% yield.
Trimethylsilyl triflate provided the first evidence of cycloaddition, but suffered from low conversion and formation of enal 8 (Table 1, entry 1). It appeared, however, that a single regioand diastereomer of the desired Diels-Alder adduct 11 was formed. Titanium tetrachloride improved conversion to products of C-C bond formation, but Mukaiyama aldol product 12 was formed as the major product. It should be noted that 12 could be recycled to enal 8 upon treatment with TiCl4, with little overall loss of material. Many titanium and aluminum Lewis acids favored the formation of enal 8 (Table 1, entries 3–5). Ethyl aluminum dichloride, however, successfully improved selectivity and conversion to 11 (Table 1, entry 6), and could be further optimized to obtain the Diels-Alder cycloadduct in 59% isolated yield (Table 1, entry 7). The isolated yield, combined with analysis of the crude reaction mixture by 1H NMR, suggest a high degree of regio- and diastereoselection, and an additional amount (15%) of 8 and 12 could be isolated.
With the tetracyclic core of the hapalindoles in place, our attention focused on elaboration of the cyclohexene ring. Reduction of the ketone, and treatment of the alcohol in situ with triflic anhydride and pyridine provided alkene 13 in 56% yield (Scheme 2). Evidence for production of a Grob fragmentation product was noted when using the purified alcohol, and may have contributed to the slightly depressed yield. Desilylation using TBAF led to allylic alcohol 14.
Scheme 2.
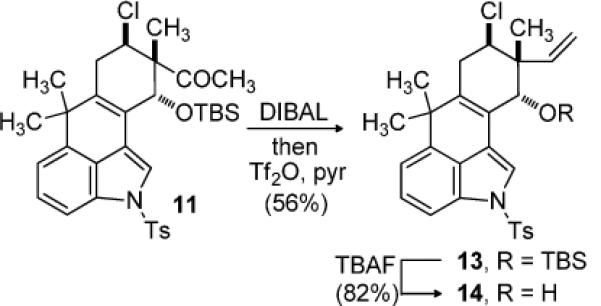
Completion of the Neopentyl Chloride Subunit
At this stage, efforts to construct the C11-N bond using an established five step protocol were unsuccessful.[4, 5g] In this case decomposition of the starting material was observed, perhaps influenced by the presence of the chloride and C10–C15 unsaturation. However, exposure of 14 to H2SO4–AcOH provided acetate 15a in excellent yield and good diastereoselection (dr = 7:1) (Scheme 3). The relative stereochemistry was assigned by chemical correlation of 15a to alcohol 14. Furthermore, the largely retention of configuration is consistent with approach of acetic acid to the intermediate allylic cation along an axial trajectory. The coupling constants associated with the chloro-substituted axial methine (J = 9 & 5.4 Hz) support assigment of the acetate to an axial position. Application of a Ritter reaction to 14 using TMSCN as a nitrogen source provided formamide 15b as a single diastereomer.[16] Formation of an allylic cation and axial approach of the nucleophile again rationalizes the diastereoselection, which was further confirmed by the observation of a NOESY crosspeak as shown in Scheme 4. A second possible source of stereocontrol is the close proximity of the equatorial position to C2 of the indole ring, resulting in an unfavorable steric interaction. Further investigation revealed that silyl ether 13 was a suitable Ritter substrate, providing 15b in comparable yield. Subsequently, the synthesis of hapalindole K could be accomplished in two straightforward synthetic operations which included tosyl deprotection and isonitrile formation (Scheme 4).
Scheme 3.
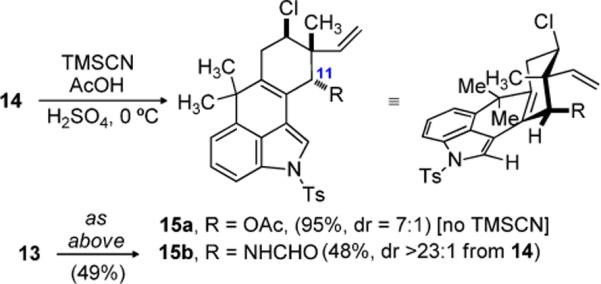
The Key Ritter Reaction to Set C11-N Bond
Scheme 4.

Preparation of Hapalindoles K (1), A (2)
Hapalindole A exhibits two additional chiral centers at C10 and C15 in the perhaps higher energy cis-decalin ring system (rings C and D). The potential complication of the neopentyl chloride notwithstanding, we elected to examine Natsume's reductive approach in this context.[17] Exposure of formamide 15b to lithium aluminum hydride provided the desired cis-fused ring system as formamide 18 (Scheme 4). Additionally, alcohol 17 and the detosylated indole 16 could be isolated. Treatment of formamide 18 with phosgene then provided hapalindole A (2).
Modification of this sequence can also provide access to the trans-fused hapalindoles, such as hapalindole G (3). Reduction of allylic alcohol 14, again using the Natsume protocol, afforded alcohol 19 (Scheme 5). Dess-Martin periodinane-mediated oxidation[18] of alcohol 19 was followed by protection of the indole nitrogen as an allyl carbamate (21). Epimerization at C10 was effected by triethylamine, leading to trans-decalin 22. This intermediate was prepared by Fukuyama in his total synthesis of (−)-hapalindole G.[5g]
Scheme 5.
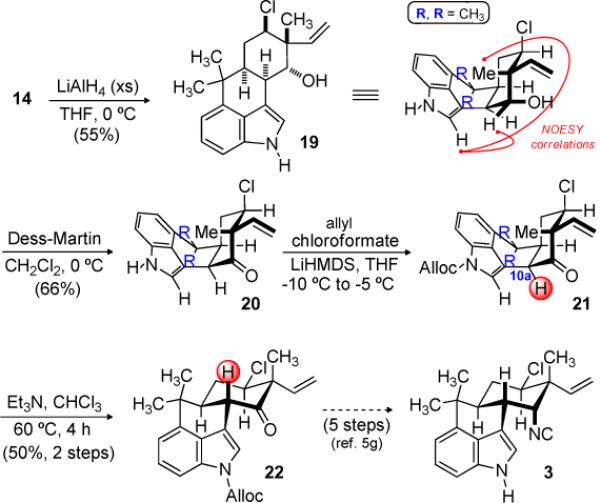
Preparation of Hapalindole G
In summary, we describe a 12 step total synthesis of hapalindoles A and K respectively, and a formal synthesis of hapalindole G. Each case highlights the value of a key progenitor – neopentyl chloride 11 – using a convergent, stereocontrolled approach. A second new development in this overall approach was the use of a Ritter reaction to establish the C10–N bond stereoselectively. This aspect was particularly key to the brevity of each synthesis.
Supplementary Material
Acknowledgments
[**] This work was supported by the NIH (GM 063557).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.((Please delete if not appropriate))
References
- [1].a) Moore RE, Cheuk C, Patterson GML. J. Am. Chem. Soc. 1984;106:6456–6457. [Google Scholar]; b) Moore RE, Cheuk C, Yang XQG, Patterson GML, Bonjouklian R, Smitka TA, Mynderse JS, Foster RS, Jones ND. J. Org. Chem. 1987;52:1036–1043. [Google Scholar]; c) Stratmann K, Moore RE, Bonjouklian R, Deeter JB, Patterson GML, Shaffer S, Smith CD, Smitka TA. J. Am. Chem. Soc. 1994;116:9935–9942. [Google Scholar]
- [2].a) Burja AM, Banaigs B, Abou-Mansour E, Grant Burgess J, Wright PC. Tetrahedron. 2001;57:9347–9377. [Google Scholar]; b) Mo S, Krunic A, Chlipala G, Orjala J. J. Nat. Prod. 2009;72:894–899. doi: 10.1021/np800751j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Muratake H, Natsume M. Tetrahedron. 1990;46:6331–6342. [Google Scholar]
- [4].Baran PS, Richter JM. J. Am. Chem. Soc. 2005;127:15394–15396. doi: 10.1021/ja056171r. [DOI] [PubMed] [Google Scholar]
- [5].a) Muratake H, Kumagami H, Natsume M. Tetrahedron. 1990;46:6351–6360. [Google Scholar]; b) Sakagami M, Muratake H, Natsume M. Chem. Pharm. Bull. 1994;42:1393–1398. [Google Scholar]; c) Vaillancourt V, Albizati KF. J. Am. Chem. Soc. 1993;115:3499–3502. [Google Scholar]; d) Kinsman AC, Kerr MA. Org. Lett. 2001;3:3189–3191. doi: 10.1021/ol0165138. [DOI] [PubMed] [Google Scholar]; e) Kinsman AC, Kerr MA. J. Am. Chem. Soc. 2003;125:14120–14125. doi: 10.1021/ja036191y. [DOI] [PubMed] [Google Scholar]; f) Baran PS, Richter JM. J. Am. Chem. Soc. 2004;126:7450–7451. doi: 10.1021/ja047874w. [DOI] [PubMed] [Google Scholar]; g) Fukuyama T, Chen X. J. Am. Chem. Soc. 1994;116:3125–3126. [Google Scholar]
- [6].a) Reisman SE, Ready JM, Hasuoka A, Smith CJ, Wood JL. J. Am. Chem. Soc. 2006;128:1448–1449. doi: 10.1021/ja057640s. [DOI] [PubMed] [Google Scholar]; b) Reisman SE, Ready JM, Weiss MM, Hasuoka A, Hirata M, Tamaki K, Ovaska TV, Smith CJ, Wood JL. J. Am. Chem. Soc. 2008;130:2087–2100. doi: 10.1021/ja076663z. [DOI] [PubMed] [Google Scholar]; c) Bhat V, Allan KM, Rawal VH. J. Am. Chem. Soc. 2011:5798–5801. doi: 10.1021/ja201834u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Baran PS, Maimone TJ, Richter JM. Nature. 2007;446:404–408. doi: 10.1038/nature05569. [DOI] [PubMed] [Google Scholar]
- [8].See the Supporting Information for Structural Formulas
- [9].Chandra A, Viswanathan R, Johnston JN. Org. Lett. 2007;9:5027–5029. doi: 10.1021/ol702247a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Okauchi T, Itonaga M, Minami T, Owa T, Kitoh K, Yoshino H. Org. Lett. 2000;2:1485–1487. doi: 10.1021/ol005841p. [DOI] [PubMed] [Google Scholar]
- [11].Bergman J, Venemalm L, Gogoll A. Tetrahedron. 1990;46:6067–6084. [Google Scholar]
- [12].Padwa A, Bur SK, Zhang H. J. Org. Chem. 2005;70:6833–6841. doi: 10.1021/jo0508797. [DOI] [PubMed] [Google Scholar]
- [13].Lucas S, Heim R, Ries C, Schewe KE, Birk B, Hartmann RW. J. Med. Chem. 2008;51:8077–8087. doi: 10.1021/jm800888q. [DOI] [PubMed] [Google Scholar]
- [14].Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew. Chem. Int. Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- [15].a) Arnold Z, Zemlicka J. Collect. Czech. Chem. Commun. 1959;24:2378–2384. [Google Scholar]; b) Arnold Z, Zemlicka J. Collect. Czech. Chem. Commun. 1959;24:2385–2392. [Google Scholar]; c) Williard PG, Grab LA, De Laszlo SE. J. Org. Chem. 1983;48:1123–1125. [Google Scholar]; d) Mooridian A, Cloke JB. J. Am. Chem. Soc. 1946;68:785–789. [Google Scholar]; e) Viswanathan R. Indiana University; Bloomington: 2005. [Google Scholar]
- [16].Jirgensons A, Kauss V, Mishnev AF, Kalvinsh I. J. Chem. Soc., Perkin Trans. 1999;1:3527–3530. [Google Scholar]
- [17].Muratake H, Natsume M. Tetrahedron. 1990;46:6343–6350. [Google Scholar]
- [18].Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.