

Abstract
The preparation and reactivity of steroidal vinyldiazo compounds is reported, providing a convenient, substituent tolerant, chemo- and stereoselective entry into 4- and 6-substituted androgen analogues from a common precursor. Under dirhodium catalysis, O—H insertion occurs at the carbenoid site, leading to 4-substituted steroids, but under silver catalysis, O—H insertion occurs at the vinylogous position, leading to 6-substituted steroids.
The steroid skeleton is one of Nature’s privileged motifs.1 The ubiquity of this structure throughout a variety of human biochemical pathways has made it a target of considerable interest to a range of drug discovery programs.2 These efforts have revolved around either total syntheses of specific analogues3 or hemisynthesis as a means of accessing the tetracyclic core.4
Aromatase cytochrome P450 is the enzyme responsible for the oxidation of androgens to estrogens. It is the only mammalian enzyme capable of converting an aliphatic six-membered ring to an aromatic ring. As such it is essential to the modulation of steroidal biogenesis. However, it has been found to be over-expressed in >75% of postmenopause breast cancer cells.5 Consequently, it has been a significant clinical target.6 Until recently, the mechanism of aromatization was not completely understood, and many current medications have been designed without the benefit of access to a detailed structural analysis of the active site of the enzyme.7
The first X-ray crystal structure of aromatase P450 was solved by Ghosh et al. in 2009.8 As part of a collaborative program with the Ghosh group we have sought to construct a series of androgen analogues to futher probe the active site and ultimately develop, through informed design, highly selective aromatase P450 inhibitors. In order to achieve this goal, we required ready access to 4- and 6-functionalized alkoxy analogues. This paper describes a carbene approach that allows selective 4- or 6-functionalization by exploiting the complementary dirhodium(II)- or silver(I)-catalyzed donor/acceptor carbene O—H insertion chemistry that has recently been developed.9
Our investigations began by developing an efficient method for the installation of the diazo functionality at the 4-position (Scheme 1). The synthesis of the diazo compounds 2 and 4 was achieved through a three-step process. The synthesis began from the commercially available 1,4-androstadienedione 1 and 1,4-androstenedione 3. Radical bromination10 followed by zinc promoted de-bromination and concommitent de-conjugation11 afforded the diazo precursors. Use of a water-soluble diazo-transfer reagent12 was essential in the preparation of the steroidal diazo compounds 2 and 4 as removal of the water-soluble by-products greatly facilitated their isolation.
Scheme 1.

Synthesis of vinyldiazo steroids 2 and 4
We then examined the optimization of the O—H insertion of the rhodium carbenes derived from these diazo compounds (Table 1). Formation of the rhodium carbene from 2 and subsequent insertion into the O—H bond of ethanol afforded the 4-ethoxy substituted steroid 5a, the product of direct OH insertion. The product was found to exist as a mixture of tautomers, predominantly favoring the enol form, likely due to internal H-bonding stabilization. Low yields were obtained when the alcohol was used as the solvent (entry 1). When hexane was used low conversion was observed (entry 2), presumeably due to solubility issues. However, effective OH insertion was observed when the reaction was conducted in trifluorotoluene (TFT) with ten equivalents of alcohol (entry 3 vs 4). The reaction was found to be catalyzed by a range of dirhodium(II) catalysts, though Rh2(S-DOSP)4 proved to be the most efficient (entry 4).
Table 1.
Optimization of the catalyst and solvent
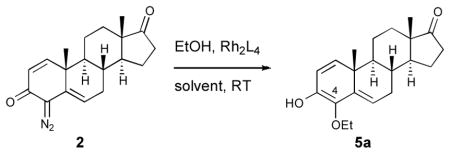 | ||||
|---|---|---|---|---|
| entry | Rh2L4a | solventb | equiv. EtOH | yield (%) |
| 1 | Rh2(S-DOSP)4 | neat | 10 | 12 |
| 2 | Rh2(S-DOSP)4 | hexanes | 10 | 8 |
| 3 | Rh2(S-DOSP)4 | TFT | 5 | 51 |
| 4 | Rh2(S-DOSP)4 | TFT | 10 | 61 |
| 5 | Rh2(Ooct)4 | TFT | 10 | 48 |
| 6 | Rh2(S-PTAD)4 | TFT | 10 | 38 |
| 7 | Rh2(OAc)4 | TFT | 10 | 50 |
In all reactions 1 mol% of catalyst was employed. The reaction was allowed to react for 4h after addition of the solution of 2.
All reactions were run at a concentration of 0.15 M with respect to 2.
With conditions established for the efficient O—H insertion, our focus turned to the scope of this transformation. The reaction with a range of alcohols and acids is described in Table 2. The O—H insertion of the rhodium carbene derived from diazoketone 2 proved to be general, with a range of aliphatic and aromatic 4-substituted ethers and esters 5a-f afforded in moderate to good yield (41–68%). Sterically and electronically diverse alcohols are suitable for this reaction system. The O—H insertion reaction proceeds with excellent regioselectivity. The 6-substituted androgens, which arise from vinylogous reaction of the rhodium carbene, were generally observed in only trace amounts.
Table 2.
Rhodium-catalyzed O—H insertion
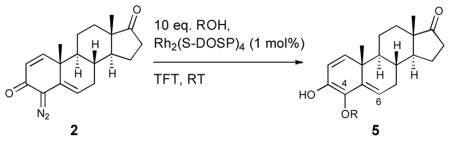 | |||||
|---|---|---|---|---|---|
| entry | R | product | time (h) | C-4:C-6a | yield (%)b |
| 1 | -CH2CH3 | 5a | 6 | >95:5 | 61 |
| 2 | -CH3 | 5b | 6 | 76:24 | 49 |
| 3 | -CH2Ph | 5c | 12 | >95:5 | 52 |
| 4 | -(CO)CH3 | 5d | 8 | 92:8 | 68 |
| 5 | -(CO)CH2CH3 | 5e | 6 | 88:12 | 55 |
| 6 | -(CO)CH2Ph | 5f | 16 | >95:5 | 41 |
Ratio determined by 1H NMR spectroscopy of the crude reaction mixture. All reactions were run at a concentration of 0.15 M with respect to 2.
Isolated yield of the C-4 regioisomer
The rhodium carbene arising from the decomposition of 4 was also found to be capable of O—H bond insertion reactions (Table 3). The 4-substituted ethers 6a-c were prepared in moderate yield (entries 1–3; 44–53%). The regioselectivity of the reaction was high, with only trace amounts of the 6-substituted analogues detected.
Table 3.
Rhodium-catalyzed O—H insertion of 4
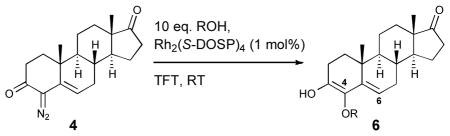 | |||||
|---|---|---|---|---|---|
| entry | R | product | time (h) | C-4:C-6a | yield (%)b |
| 1 | -CH2CH3 | 6a | 8 | 90:10 | 53 |
| 2 | -CH3 | 6b | 10 | 93:7 | 44 |
| 3 | -CH2Ph | 6c | 10 | >95:5 | 45 |
Ratio determined by 1H NMR spectroscopy of the crude reaction mixture. All reactions were run at a concentration of 0.15 M with respect to 2.
Isolated yield of the C-4 regioisomer
The nature of the metal catalyst employed to form the stabilized donor/acceptor carbene has been established to have a profound effect on the reaction outcome.9 Previous studies on the donor/acceptor vinyldiazoacetate functionality have demonstrated that while rhodium carbenes typically react at the carbene carbon, the silver(I) salts promote reactions predominantly at the vinylogous position via a carbene intermediate.9 We chose to exploit this to provide an expedient entry into 6-substituted androgens from the same vinylcarbenoid precursors 2 and 4. Indeed, by employing 10 mol% of silver triflate, reaction of the carbene derived from 2 afforded the 6-substituted analogues 7a-g, arising from vinylogous insertion (Table 4). The scope of the reaction is general, affording both the 6-substituted ethers and esters in moderate to good yield (39–71%). The insertion proceeds with excellent diastereoselectvity, favoring the 6-β-isomer, and regioselectivity, with the 4-substituted analogues generally present in only trace amounts.
Table 4.
Silver-catalyzed O—H insertion of 2
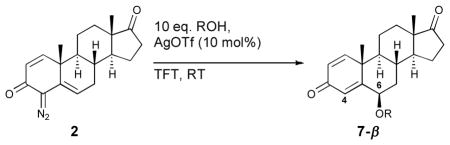 | ||||||
|---|---|---|---|---|---|---|
| entry | R | product | time (h) | C-4:C-6a | α:βa | yield (%)b |
| 1 | -CH2CH3 | 7a | 4 | 10:90 | 15:85 | 41 |
| 2 | -CH3 | 7b | 4 | 8:92 | 12:88 | 46 |
| 3 | -CH2Ph | 7c | 6 | 5:>95 | 22:78 | 39 |
| 4 | -C(CH3) | 7d | 3 | 5:>95 | 8:92 | 70 |
| 5 | -(CO)CH3 | 7e | 4 | 5:>95 | 5:>95 | 66 |
| 6 | -(CO)CH2CH3 | 7f | 10 | 5:>95 | 11:89 | 71 |
| 7 | -(CO)CH2Ph | 7g | 6 | 5:>95 | 5:>95 | 38 |
Ratio determined by 1H NMR spectroscopy of the crude reaction mixture. All reactions were run at a concentration of 0.15 M with respect to 2.
Isolated yield of the 7-β diastereomer
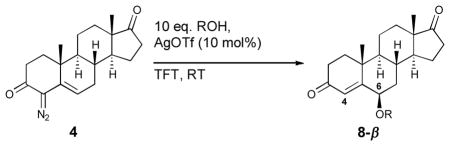
The silver triflate promoted vinylogous reaction was also efficient in the saturated system. The O–H insertion of the silver carbene derived from 4 led to essentially exclusive formation of the corresponding 6-substituted ethers 8a-d with only trace amounts of the 6-substituted analogues detected (Table 5). The ethers were prepared in moderate yield (39–61%), with high levels of both regio- and diastereoselectivity. The 6-β-androgens again were found to be highly favored. Attempts at preparing the acetate derivative 8e were unsuccessful as the compound appeared to be unstable and prone to elimination.
Table 5.
Silver-catalyzed O—H insertion of 4
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | product | time (h) | C-4:C-6a | α:βa | yield (%) |
| 1 | -CH2CH3 | 8a | 6 | 5:>95 | 12:88 | 42 |
| 2 | -CH3 | 8b | 8 | 8:92 | 10:90 | 39 |
| 3 | -CH2Ph | 8c | 12 | 5:>95 | 5:>95 | 61 |
| 4 | -C(CH3) | 8d | 10 | 5:>95 | 5:>95 | 46 |
| 5 | -(CO)CH3 | 8e | 16 | - | - | <5 |
Ratio determined by 1H NMR spectroscopy of the crude reaction mixture. All reactions were run at a concentration of 0.15 M with respect to 2.
Isolated yield of the 8-β diastereomer
The remarkable diastereoselectivity of the O—H insertion reaction of the silver carbenes derived from 2 and 4, deserves further comment. In all instances the 6-β-androgen product was strongly favored. The stereochemical assignment was initially made on the basis of the distinctive coupling constants for the C-6 proton, which indicated that the alkoxy group was in the 6-β-position. This assignment was then confirmed by the single crystal X-ray analysis of 7d (Figure 1).13
Fig. 1.

Single crystal X-ray analysis of 6-β androgen 7d
An inspection of the parent diazo structures might suggest that substrate-controlled stereoselection would be dictated by the C-19 β-methyl groups; this would direct the reaction to the α-face. We can account for the observed stereoselectivity if we consider the transition state (9) (Figure 2). While pathway b reduces the steric clash with the C–19 methyl group, pathway a is favored on stereoelectronic grounds, representing axial attack to form the conformationally preferred chair product.
Fig. 2.

Stereoelectronic rationalization of the formation of the 6-β substituted ethers and esters
In summary, we have developed a convenient procedure to regioselectively functionalize the steroid skeleton at the 4-and 6-positions, with a broad range of analogues accessible via common intermediates. The regioselectivity of the reaction was controlled effectively through choice of metal catalyst employed. Rhodium-catalyzed reactions result in selective reactions at the carbenoid site, whereas silver catalyzed reactions preferentially functionalize the vinylogous position of the carbenoid. Substrate-controlled diastereoselectivity was observed in the vinylogous reaction mediated by silver triflate, allowing the preparation of 6-β-substituted analogues. These compounds are currently being used to probe the active site of the aromatase P450 enzyme, the results of which will be disclosed in due course.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (R01GM086893). We thank Dr. Kenneth I. Hardcastle for the X-ray crystallographic structure determination.
Footnotes
Electronic Supplementary Information (ESI) available: [Experimental details and spectral data]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Beillmann J-F. Chem Rev. 2003;105(5):2019. [Google Scholar]
- 2.(a) Chapelon AS, Moraleda D, Rodriguez R, Olivier C, Santelli M. Tetrahedron. 2007;63:11511. [Google Scholar]; (b) Giroux S, Corey EJ. J Am Chem Soc. 2007;129:9866. doi: 10.1021/ja074306i. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hu QY, Rege PD, Corey EJ. J Am Chem Soc. 2004;126:5984. doi: 10.1021/ja048808x. [DOI] [PubMed] [Google Scholar]; (b) Canales E, Corey EJ. Org Lett. 2008;10:3271. doi: 10.1021/ol8011502. [DOI] [PubMed] [Google Scholar]
- 4.(a) Shi J, Shigehisa H, Guerrero C, Shenvi RA, Li CC, Baran PS. Angew Chem Int Ed. 2009;48:4328. doi: 10.1002/anie.200901116. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Van den Heuvel MJ, VanBokhoven CW, De Jongh HP, Zeelan FJ. Recl Trav Chim Pays-Bas. 1988;107:331. [Google Scholar]
- 5.(a) O’Neill SJ, Elton RA, Miller WR. Br Med J (Clin Res Ed) 1988;296:741. doi: 10.1136/bmj.296.6624.741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blankenstein MA, Szymczak J, Daroszewski J, Milewicz A, Thijssen JH. Gynecol Endocrinol. 1992;6:13. doi: 10.3109/09513599209081001. [DOI] [PubMed] [Google Scholar]
- 6.Eisen A, Trudeau M, Shelley W, Messersmith H, Pritchard KI. Cancer Treat Rev. 2008;34:157. doi: 10.1016/j.ctrv.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Destra Z, Nguyen A, Flockhart D, Skaar T, Fletcher R, Weinshilboum R, Berlin DS, Klein TE, Altman RB. Pharmacogenet Genomics. 2009;19:554. doi: 10.1097/FPC.0b013e32832e0ec1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Ghosh D, Griswold J, Erman M, Pangborn W. Nature. 2009;457:219. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh D, Griswold J, Erman M, Pangborn W. J Ster Biochem Mol Bio. 2010;118:197. doi: 10.1016/j.jsbmb.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Yue Y, Wang Y, Hu W. Tetrahedron Lett. 2007;48:3975. [Google Scholar]; (b) Hansen JH, Davies HML. Chem Sci. 2011;2:457. [Google Scholar]
- 10.Kaufmann S, Pataki J, Rosenkranz G, Romo J, Djerassi C. J Am Chem Soc. 1950;72:4531. [Google Scholar]
- 11.Nussbaum AL, Topliss GB, Popper TL, Oliveto EP. J Am Chem Soc. 1959;81:4574. [Google Scholar]
- 12.Moreau RL, Sorenson EJ. Tetrahedron. 2007;63:6446. [Google Scholar]
- 13.The crystal structure of 8d has been deposited at the Cambridge Crystallographic Data Centre under deposition number CCDC 829542A.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.