

Abstract
The intronic GABRG2 mutation, IVS6+2T→G, was identified in an Australian family with childhood absence epilepsy and febrile seizures (Kananura et al., 2002). The GABRG2 intron 6 splice donor site was found to be mutated from GT to GG. We generated wild-type and mutant γ2 subunit bacterial artificial chromosomes (BACs) driven by a CMV promoter and expressed them in HEK293T cells and expressed wild-type and mutant γ2 subunit BACs containing the endogenous hGABRG2 promoter in transgenic mice. Wild-type and mutant GABRG2 mRNA splicing patterns were determined in both BAC-transfected HEK293T cells and transgenic mouse brain, and in both, the mutation abolished intron 6 splicing at the donor site, activated a cryptic splice site, generated partial intron 6 retention, and produced a frameshift in exon 7 that created a premature translation termination codon (PTC). The resultant mutant mRNA was either degraded partially by nonsense-mediated mRNA decay or translated to a stable, truncated subunit (the γ2-PTC subunit) containing the first six GABRG2 exons and a novel frameshifted 29 aa C-terminal tail. The γ2-PTC subunit was homologous to the mollusk AChBP (acetylcholine binding protein) but was not secreted from cells. It was retained in the ER and not expressed on the surface membrane, but it did oligomerize with α1 and β2 subunits. These results suggested that the GABRG2 mutation, IVS6+2T→G, reduced surface αβγ2 receptor levels, thus reducing GABAergic inhibition, by reducing GABRG2 transcript level and producing a stable, nonfunctional truncated subunit that had a dominant-negative effect on αβγ2 receptor assembly.
Introduction
Epilepsy is one of the most common neurological disorders, affecting up to 3% of the general population. One-third to one-half of all epilepsy syndromes have a genetic basis (Berkovic et al., 2006), and patients with genetic epilepsy syndromes have absence, myoclonic, and/or generalized tonic-clonic seizures (Beghi et al., 2006). Most mutations associated with genetic epilepsies have been identified in genes encoding voltage- or ligand-gated ion channels (Noebels, 2003).
GABAA receptors mediate the majority of inhibitory neurotransmission in the CNS. Epilepsy mutations have been identified in GABAA receptor subunit genes (GABRs) GABRA1, GABRB3, and GABRG2 (Kang and Macdonald, 2009), but most of the mutations are in GABRG2. The α1, β2, and γ2 subunits form the most abundant GABAA receptor subtype in the CNS (Sieghart and Sperk, 2002; Whiting, 2003; Farrant and Nusser, 2005), and the γ2 subunit plays a critical role in brain function. In mouse brain, ∼75–80% of GABAA receptors contain the γ2 subunit (Olsen and Sieghart, 2008). Mice lacking γ2 subunits (γ2−/− mice) died shortly after birth (Günther et al., 1995). These γ2−/− mice lost 94% of their benzodiazepine binding sites, but GABA binding sites were only decreased slightly. The γ2 subunit is required for maintaining postsynaptic GABAA receptor clustering (Essrich et al., 1998). Heterozygous γ2+/− mice had significantly decreased benzodiazepine binding sites and increased extrasynaptic GABAA receptor radioligand binding sites in the CNS, but unchanged muscimol binding sites (Sinkkonen et al., 2004), and these animals had decreased GABAA receptor clustering in hippocampus and cerebral cortex (Crestani et al., 1999). The γ2+/− mice had increased anxiety (Crestani et al., 1999), a behavior recapitulated in γ2 subunit knockdown mice (Chandra et al., 2005). Epilepsy, however, has not been reported in γ2−/− or γ2+/− mice.
GABR mutations have been associated with seizures ranging from relatively benign absence and/or febrile seizures to severe myoclonic seizures (Macdonald et al., 2006). The most well characterized γ2 subunit missense mutation is GABRG2(R82Q) associated with childhood absence epilepsy and febrile seizures (Wallace et al., 2001). This mutation impaired α1β2γ2 receptor assembly, retained mutant γ2 subunits in the endoplasmic reticulum, and reduced receptor surface trafficking (Bianchi et al., 2002; Hales et al., 2005; Eugène et al., 2007). Knock-in mice harboring the GABRG2(R82Q) mutation had reduced cell surface γ2 subunit expression and reduced cortical inhibition, even in heterozygous animals (Tan et al., 2007). Mice heterozygous for the mutation also had absence seizures.
GABRG2(IVS6+2T→G) is a mutation of the intron 6 splice donor site from GT to GG identified in an Australian family with childhood absence epilepsy and febrile seizures (Kananura et al., 2002). The basis for the epilepsy in this family results from the specific alteration in splicing of GABRG2(IVS6+2T→G) mRNA and on subsequent translation of protein. To determine this splicing pattern, we generated wild-type and mutant GABRG2(IVS6+2T→G) bacterial artificial chromosomes (BACs) and determined how the IVS6+2T→G mutation altered intron 6 splicing and γ2 subunit expression in HEK293T cells and transgenic mouse brain. We then characterized the biogenesis and function of the translated mutant γ2 subunit.
Materials and Methods
Expression vectors with GABAA receptor subunits.
The coding sequences of human α1, β2, γ2S, and γ2L GABAA receptor subunits from the translation initiation codon ATG to the stop codon were cloned into pcDNA3.1 expression vectors (Invitrogen) or pLVX-IRES-ZsGreen1 vectors (Clontech) as previously described (Gallagher et al., 2005). The cDNA encoding the HA peptide, YPYDVPDYA, was introduced between the fourth and fifth amino acids of mature γ2S and γ2L subunits, which has been reported to be a functionally silent position (Connolly et al., 1996). In recent studies, the position of the mutant and variant amino acids in α1, β3, and δ subunits have been specified in the immature peptide that includes the signal peptide, but mutations in γ2 subunit have been reported in the mature peptide, excluding the signal peptide. For consistency, in this study, the positions of γ2 subunit mutations were designated also in the immature peptide.
The BAC clone number RP11-1035I20 (BACPAC Resources; http://bacpac.chori.org) contains a human chromosome 5 fragment that included the wild-type human GABRG2 gene genomic sequence (and thus a complete intron 6) and 20 kb upstream and 40 kb downstream human chromosome 5 sequences (see Fig. 1A). The BAC sequence was confirmed by restriction enzyme digestion and direct DNA sequencing. The BAC clone was recombined with the pEHHG vector (Wade-Martins et al., 2001), which contained the eGFP reporter gene driven by the HSV early gene promoter. In target cells expressing these BAC vectors, eGFP fluorescence was detected. In this BAC clone, hGABRG2 was predicted to be driven by the promoter sequence in the 20 kb upstream human chromosome sequence, while the eGFP was driven by a separate HSV promoter, and thus, expression of eGFP was independent of expression of hGABRG2. To introduce the point mutation in hGABRG2 at the IVS6+2 position, we used galK-facilitated recombineering (Warming et al., 2005; Chandler et al., 2007). The galK gene encodes galactose kinase and provides both positive and negative selection factors in this technique. Using galK-facilitated BAC recombineering, the human chromosome sequence upstream of the GABRG2 translation initiation sequence ATG was replaced with a CMV promoter (see Fig. 1A). Unless otherwise specified, wt and mutant hGABRG2 BACs were driven by the CMV promoter and contained the eGFP reporter gene. The oligo sequences for BAC recombineering are available upon request.
Figure 1.
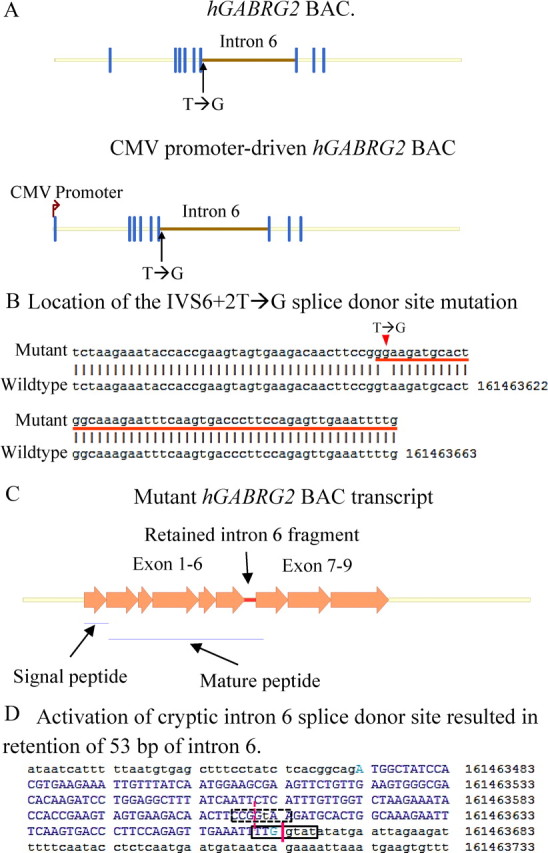
The GABRG2(IVS6+2T→G) BAC transcript retained 53 bp of intron 6 sequence. A, Structures of the human GABRG2 genomic sequence used in this study. The genomic sequence of GABRG2 in the BAC used in this study (RP11-1035I20) and the CMV promoter-driven GABRG2 BAC are shown. The yellow horizontal lines represent introns and 3′- and 5′-UTRs. The brown horizontal line represents intron 6. The vertical dark blue lines represent exons. The black arrow points to the mutation. The brown arrow at the left end represents the CMV promoter. The length of each line is proportional to the molecular size of the represented region (nucleic acid number). B, The mutant mRNA was cloned into the TOPO cloning vector (top) and human genomic mRNA (bottom) sequences were aligned. The last 91 bp of the mutant exon 6 alignment result including the retained intron 6 sequence are presented. The number on the bottom line shows the position of each nucleotide in human chromosome 5. The red arrow points to the IVS6+2T→G mutation, and the red line underlines the retained 53 bp intron 6 sequence. C, The structure of the GABRG2(IVS6+2T→G) BAC transcript is shown. The arrows represent exons 1–9. The red line represents the retained intron 6 fragment. The yellow lines represent 3′- and 5′-UTRs. The blue lines underline the signal and mature peptides. The length of each arrow and line is proportional to the molecular size of the represented region (nucleic acid number). D, The sequence of the mutant intron 6 splice donor site is shown. In the full mutant exon 6 sequence, a “t” in lowercase is shown at the T→G mutation site. The dotted black box shows the wild-type intron 6 splice site conservative sequences, and the dotted red line marks the wild-type splice site. The solid black box encloses the seven nucleic acids at the mutant donor site sequence, and the solid red line marks the splice site. The nucleotides in uppercase are exon 6 sequences, and the nucleotides in lowercase are intron sequences.
Cell culture, transfection, and RNAi.
Human embryonic kidney cells (HEK293T) (ATCC; CRL-11268) and HeLa cells (ATCC; CCL-2) were incubated at 37°C in humidified 5% CO2, 95% air and grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Cells were transfected with cDNAs using the FuGENE 6 transfection reagent (Roche Applied Science) or Lipofectamine 2000 (Invitrogen) at a DNA/transfection reagent ratio of 1:3 according to the manufacturer's instructions. The transfected cells were harvested after 36 h in culture for the following experimental protocols.
Sprague Dawley rat cortex was dissected from E18 embryos and dissociated using 0.25% trypsin and mild trituration (Banker and Goslin, 1998). Neurons were plated on poly-l-ornithine-coated coverslips in DMEM (Invitrogen) supplemented with 10% horse serum, 2 mm glutamine, and 1 mm Na-pyruvate. After 4 h, medium was replaced by 1 ml of serum-free culture medium containing Neurobasal with B27 supplement, glutamine (2 mm), and penicillin/streptomycin. Cultures were maintained at 36°C in a humidified CO2 incubator for up to 4 weeks and fed once a week. Cultured neurons were transfected with Lipofectamine 2000 (Invitrogen) at 7 DIV according to the manufacturer's instructions. A mixture of 1 μg of DNA and 3 μl of Lipofectamine in 60 μl of Opti-MEM (Invitrogen) was added to the well. One hour after incubation, the culture medium containing the Lipofectamine/DNA complex was completely replaced with fresh serum-free Neurobasal/B27 culture medium. Neurons were immunostained 7 d after transfection.
Nonsense-mediated mRNA decay (NMD) efficiency was decreased by knocking down the essential factor UPF1. Silencer select predesigned and validated siRNA (Ambion; siRNA ID s11926) was transfected to cells using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's manual. Twenty-four hours later, the same cells were transfected again with the wild-type or mutant BAC constructs and harvested 2 d later for RT-PCR. The efficiency of UPF1 knockdown was confirmed by Western blot.
RNA extraction, RT-PCR, and TaqMan real-time qPCR.
Total RNAs were extracted from transfected HEK293T cells by using the PerfectPure RNA Cultured Cell kit (5 PRIME) following the manufacturer's protocol, total RNA in mouse brain tissue was expressed by TRIzol reagent (Invitrogen) and PureLink RNA mini kit (Invitrogen) according to manufacturer's manual, and human total brain RNA was obtained from Ambion. Two hundred nanograms of total RNA of each sample was reverse transcribed to cDNA in a 10 μl volume using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). The transcribed cDNA was used as a template to perform regular PCR using Expand High Fidelity PCR Kit (Roche Applied Science) following the manufacturer's manual. One microliter of the 50× diluted transcribed cDNA was mixed with TaqMan Universal PCR Master Mix (Applied Biosystems) and TaqMan probes in a total volume of 5 μl for the TaqMan qPCR experiments. TaqMan probe detecting human GABRG2 gene mRNA, human GAPDH gene, 18S rRNA, or eGFP (part number 4331348; Custom TaqMan Gene Expression Assay Service) were used. The catalog number of each probe is available upon request. Each sample was run in triplicate, and the average threshold cycle (Ct) value of each sample was calculated by the Sequence Detection System, version 2.3, Standard Edition (Applied Biosystems). The average Ct values of GABRG2 gene mRNA were normalized to the endogenous human GAPDH, 18S rDNA, or eGFP amount, and the normalized Ct values of samples were compared to get the relative RNA abundance.
Generation and maintenance of hGABRG2 BAC transgenic mice.
The cesium chloride density centrifugation purified BAC DNAs were microinjected into the male pronucleus of C57BL/6J F1 fertilized mouse embryos and implanted into pseudopregnant ICR surrogate mice by the Vanderbilt Transgenic/ES Cell Shared Resource Facility. Founder mice were bred to C57BL/6J mice to establish transgenic lines. All animals used in these studies were handled in strict compliance with the guidelines of the American Association for Laboratory Animal Science and the Vanderbilt University Institutional Animal Care and Use Committee Protection of Research Subjects.
Transgenic mouse genotyping PCR.
Mouse tail samples collected at P14–P21 were extracted using red Extract-N-AMP tissue PCR kit (Sigma-Aldrich) according to the manufacturer's manual. Forward primers binding to either HA tag (primer sequence: TACCCCTACGACGTGCCCGACTACGCC) or intron 1/exon 2 border (GTAATCTATGTGTTTTTTGACCAATATGTTTTTTCTTAGCTTCACTAGCCAGAAATCTG) and reverse primer binding to intron 2 (CACCTCTCCCACTCATAGGCCTGAATG) were used for genotyping. PCR cycling conditions were as follows: 95°C, 5 min initial denature step; 95°C, 1 min/68°C, 1 min/72°C, 1 min (30 cycles); 72°C, 5 min final step.
Immunohistochemistry.
Brains were removed from CO2-killed mice, fresh frozen in powdered dry ice, and stored at −80°C until sectioned. Parasagittal sections, 20 μm thick, were prepared with a cryostat (CM1950; Leica Microsystems) and stored at −80°C until immunostaining (Schneider Gasser et al., 2006). Brain slices were fixed and permeabilized with 2% paraformaldehyde (Sigma-Aldrich) in PBS for 2 min, and washed with PBS. Slices were incubated overnight in rabbit monoclonal anti-HA epitope-tagged antibodies (1:500; clone C29F4; Cell Signaling) in PBS with 0.2% Triton X (Sigma-Aldrich) to detect HA epitope-tagged γ2 subunits, followed by 2 h incubation in IRDye800-conjugated donkey anti-rabbit IgG secondary antibodies at 1:1000 dilution in PBS with 0.2% Triton X. Immunolabeled slices were scanned with Odyssey imaging system (LI-COR) after air dry. Scan parameters were as follows: resolution, 21 μm; quality, highest; focus offset, 0.0 mm; intensity, 5.0 in both 700 and 800 channels. The 700 channel fluorescence signal was scanned to show autofluorescence of the brain sections. Scanned images were analyzed with Odyssey, version 3.0 (LI-COR).
Immunocytochemistry and confocal microscopy.
HEK293T cells were plated on poly-l-ornithine-coated, glass-bottom imaging dishes at a density of 3 × 105 cells/dish and cotransfected with 0.5 μg each of human subunit plasmid. Cells were fixed with 1% paraformaldehyde for 15 min to stain surface proteins, or permeabilized with CytoPerm (BD Biosciences) for 15 min to stain total proteins. The fixed/permeabilized cells were stained with rabbit polyclonal BIP antibodies (Abcam) for an hour, then a mixture of Alexa 568-conjugated donkey anti-rabbit secondary antibodies, Alexa 488-conjugated mouse monoclonal HA antibodies, and Alexa 647-conjugated mouse monoclonal α1 subunit antibodies (Millipore) for an hour. BIP protein (GRP78) is an ER specific marker. BIP antibodies visualized ER in total staining and showed membrane integrity in surface staining.
Neurons were fixed with 4% paraformaldehyde/4% glucose in PBS for 15 min to stain surface proteins, or permeabilized with CytoPerm (BD Biosciences) for 15 min to stain total proteins. Coverslips were then blocked for 1 h with 10% BSA in PBS, and incubated in mouse monoclonal antibody against the HA epitope tag (Covance) and rabbit polyclonal antibodies against ER marker BIP (Abcam) for 2 h, followed by Alexa 568-conjugated donkey anti-mouse IgG antibodies (Invitrogen) and Alexa 647-conjugated donkey anti-rabbit IgG antibodies (Invitrogen) for 1 h. Antibodies were diluted in 4% BSA in PBS for surface staining, or in 4% BSA in PBS containing 0.2% Triton X-100 for total staining. Coverslips were mounted with 5% n-propyl gallate (Sigma-Aldrich) in PBS/glycerol. The ZsGreen translated from the pLVX-IRES-ZsGreen1 vector (Clontech) was a marker for transfected neurons.
Confocal experiments were performed in part using the Vanderbilt University Medical Center Cell Imaging Shared Resource. Images were obtained using a Zeiss LSM 510 META inverted confocal microscope. Stained HEK293T cells or cultured neurons were excited with the 488 nm laser for the Alexa 488 fluorophore or ZsGreen signal, 543 nm laser for the Alexa 568 fluorophore signal, and 633 nm laser for the Alexa 647 fluorophore signal. We adjusted the pinhole of all channels to obtain 1 μm sections from HEK293T cells, or 2 μm sections from cultured neurons. In each experiment, we adjusted the laser intensity and detector sensitivity to use the full linear range of detection. Images were obtained with 8 bit, 1024 × 1024 pixel resolution, and an average of four scans was taken to decrease the background noise.
Flow cytometry.
To collect cells for flow cytometry analysis, monolayer cultures of HEK293T cells were dissociated by 37°C trypsin (Invitrogen) for 2 min, and then isolated to single cell in 4°C PBS containing 2% fetal bovine serum and 0.05% sodium azide (FACS buffer) by pipette up and down 10 times. Surface levels of each subunit were also quantified in 2 mm EDTA dissociated cells compared with trypsinized cells. The relative surface levels were not affected by trypsinization (data not shown). To evaluate total subunit levels, cells were permeabilized with CytoPerm (BD Biosciences) for 15 min, and washed with CytoWash (BD Biosciences).
Following washes with FACS buffer for surface staining or CytoWash for total staining, cells were incubated with anti-HA epitope-tagged antibodies (clone 16B12; Covance) conjugated to the Alexa 647 fluorophore (Invitrogen) for 1 h. Cells were then washed three times and fixed with 2% paraformaldehyde. Flow cytometry experiments were performed in the Vanderbilt Medical Center Flow Cytometry Shared Resource, which is supported by the Vanderbilt Ingram Cancer Center and the Vanderbilt Digestive Disease Research Center. Data were acquired using FACSDiva 6.0 (BD Biosciences) and analyzed off-line using FlowJo 7.5 (Tree Star). The mean fluorescence intensity of each sample was evaluated and normalized to the 100% control (α1β2γ2LHA or α1β2γ2SHA as noted in each figure legend). The normalized mean fluorescence intensity was represented as a percentage of the 100% control.
Immunoblotting.
Transgenic mouse brain tissue samples or cultured HEK293T cells were sonicated in radioimmunoprecipitation assay (RIPA) buffers (Pierce) and a protease inhibitor mixture (Sigma-Aldrich). Total tissue or cell lysates were cleaned by centrifugation at 20,000 × g for 30 min in 4°C. The supernatants were mixed with Nupage LDS sample buffer (Invitrogen), and then subjected to SDS-PAGE. Proteins in gels were transferred to Millipore Immobilon FL PVDF membrane (Millipore). Nonspecific binding on the membrane was blocked with the Odyssey blocking buffer (LI-COR). Rabbit polyclonal anti-GABAA receptor γ2 subunit antibodies (final concentration, 2 μg/ml; Alomone) and monoclonal anti-HA epitope-tagged antibodies (0.2 μg/ml; clone 16B12; Covance) were used to detect endogenous mouse γ2 subunits and HA epitope-tagged γ2 subunits, respectively. Monoclonal anti-GABAA receptor α1 subunit antibodies (final concentration, 5 μg/ml; clone BD24; Millipore Bioscience Research Reagents) and monoclonal anti-GABAA receptor β2/3 antibodies (4 μg/ml; clone 62-3G1; Millipore) were used to detect wild-type human α1 and β2 subunits, respectively. The polyclonal anti-human Upf-1 (hUpf-1) antibodies (Abgent; AP1905c) were used at a final concentration of 125 ng/ml. Anti-Na+/K+-ATPase antibodies (0.2 μg/ml; clone ab7671; Abcam) were used to check loading variability. Following incubation with primary antibodies, IRDye secondary antibodies were used at a 1:10,000× dilution (LI-COR) for visualization of specific bands with the Odyssey imaging system (LI-COR). The band intensities of scanned images were quantified with the Odyssey analysis software (LI-COR).
Glycosidase digestion.
Whole-cell lysates obtained from 10 mm Tris RIPA buffer (10 mm Tris-HCl, 150 mm NaCl, 1.0 mm EDTA, 1% Nonidet P-40, and 0.25% sodium deoxycholate) extraction were subjected to Endo H and peptide N-glycosidase-F digestion (New England Biolabs) following the manufacturer's recommended protocol. The digestion reactions were performed at 37°C for 3 h and terminated by addition of sample buffer.
Immunoprecipitation.
Protein complexes containing HA-tagged GABAA receptor subunits were immunoprecipitated using EZview Red anti-HA M2 beads (Sigma-Aldrich) for 30 min at room temperature following the manufacturer's manual. After three washes with extracting RIPA buffer, protein complexes were eluted with 100 μg/ml HA peptide (Sigma-Aldrich).
Electrophysiology.
Lifted whole-cell recordings were obtained from transfected HEK293T cells as previously described (Bianchi et al., 2002). Briefly, cells were bathed in an external solution consisting of the following (in mm): 142 NaCl, 8 KCl, 6 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose, pH 7.4, 325 mOsm. Electrodes were fire-polished to resistances of 0.8–1.5 MΩ and filled with an internal solution consisting of the following (in mm): 153 KCl, 1 MgCl2, 2 MgATP, 10 HEPES, 5 EGTA, pH 7.3, 300 mOsm. The combination of internal and external solutions produced a chloride equilibrium potential of ∼0 mV. For all recordings, cells were voltage clamped at −20 mV. GABA (1 mm) was applied to cells for 4 s, and cells were then washed with external solution for 40 s. Zn2+ (10 μm) was then preapplied for 10 s followed by coapplication of GABA (1 mm) and Zn2+ (10 μm) for 4 s. Finally, cells were washed with external solution for 10 s followed by application of GABA (1 mm) for 4 s. Whole-cell currents were low-pass filtered at 2–5 kHz and digitized at 10 kHz, and peak current amplitudes were quantified using the pClamp9 software suite (Molecular Devices).
Statistical analysis.
Data are presented as means ± SEM. We used Student's t test for two group comparisons, and one-way or two-way ANOVA with Bonferroni's multiple-comparison test for multiple comparisons. Data were plotted and analyzed with GraphPad Prism 5 (GraphPad Software).
Results
The GABRG2(IVS6+2T→G) mutation generated a mutant hGABRG2(IVS6+2T→G) BAC transcript that retained a 53 bp intron 6 fragment
The GABRG2(IVS6+2T→G) mutation altered the GABRG2 intron 6 splice donor site sequence. As a result, it was proposed that intron 6 is spliced out either with the donor site from another intron, resulting in exon skipping, or with an alternative donor site downstream of the wild-type site, resulting in cryptic splice donor site activation and partial intron 6 retention in the mutant mature mRNA (Kananura et al., 2002). However, the actual splice pattern of the mutant mRNA is unknown, and patient tissues or RNA samples are not available. Our approach to determine the splicing pattern of the mutant gene was to study splicing in vitro and in vivo of a BAC construct that contained hGABRG2 genomic sequence, and thus a full-length intron 6 (Fig. 1A) (for construct details, see Materials and Methods). Intron splicing is cell type specific, and the optimal approach to study splicing of GABRG2 is to do so in cells with endogenous GABRG2 expression. We used Lipofectamine 2000 to transfect either wild-type or mutant hGABRG2 BACs containing their native promoter and the eGFP reporter gene into PC12 cells, which have been reported to have endogenous GABRG2 expression (Tyndale et al., 1994). Although GFP expression was observed in BAC-transfected PC12 cells, using RT-PCR we were unable to demonstrate hγ2 subunit mRNA. As an alternative strategy, we replaced the hGABRG2 promoter with a CMV promoter and expressed the CMV promoter-driven hGABRG2 BAC in HEK293T cells (Fig. 1A) (for construct details, see Materials and Methods), and using RT-PCR, we were able to demonstrate hγ2 subunit mRNA expression. Thus, unless otherwise specified, all hGABRG2 BAC constructs in the remainder of the in vitro studies used the CMV promoter.
To determine wild-type and mutant hGABRG2 splicing patterns, we expressed both wild-type hGABRG2 BAC and control γ2S cDNA constructs in HEK293T cells and collected total RNA. DNA sequencing of hGABRG2 BAC RT-PCR products using primers binding to exons 5 and 9 of the GABRG2 coding sequence showed that the intervening introns 6, 7, and 8 were completely spliced out, and only γ2S subunit mRNA was transcribed from the hGABRG2 BAC (data not shown). This was consistent with the finding that the γ2S subunit splice variant is the default splicing product and that generation of the γ2L subunit splice variant requires positive regulation such as the function of neuron-specific RNA binding protein Nova-1 (Zhang et al., 1996, 1999; Dredge and Darnell, 2003).
The mutant hGABRG2(IVS6+2T→G) BAC transcript was expressed in HEK293T cells and cloned and sequenced (Fig. 1B). The mutant hGABRG2(IVS6+2T→G) BAC intron 6 used a cryptic splice donor site 53 bp downstream of the wild-type splice donor site, and thus the mutant transcript retained a 53 bp intron 6 fragment (Fig. 1C,D). None of the intron splice donor site prediction models that we used detected this site, suggesting that its sequence did not comply with general splice donor site rules. The mutant splice donor site was predicted to be much weaker than the wild-type site (Carmel et al., 2004), having less hydrogen bonding with the splice machinery, and hence forming a less stable mRNA–protein complex. These are all common properties of mutant splice donor sites (Buratti et al., 2007).
The mutant GABRG2(IVS6+2T→G) mRNA should be translated to a truncated subunit containing the signal peptide and N-terminal 217 aa of the wild-type γ2 subunit
In silico translation, using Vector NTi (Invitrogen), showed that the mutant transcript should be translated to a polypeptide containing the signal peptide and N-terminal 217 aa of the wild-type γ2 subunit. The retained 53 bp intron 6 fragment caused a frameshift in exon 7, which generated a stop codon 33 bp from the end of the fragment. The retained intron 6 fragment and the exon 7 frameshift sequence are predicted to be translated to a novel 29 aa peptide tail at the C terminus of the mutant protein (Fig. 2A), so the mutant protein contained the N terminus of the wild-type subunit and the novel C-terminal tail [γ2-premature translation termination codon (PTC) subunit] (Fig. 2B). The hydrophobicity of the 29 aa tail was evaluated by ProtScale at Expasy.org (Wilkins et al., 1999) and was found to be hydrophilic at the N terminus and hydrophobic at the C terminus (Fig. 2C). The maximum hydrophobic region was the C terminus, where the calculated maximum hydrophobicity was 1.43 (Abraham and Leo, 1987). This was very close to the maximum hydrophobicity of the wild-type γ2S subunit, which was 1.75 when evaluated by the same model. The structure of the mutant γ2-PTC subunit was unknown, but bioinformatics models did not predict that any secondary structure formed in this fragment.
Figure 2.

The mutant BAC transcript was predicted to encode a truncated protein containing most of the γ2 subunit N terminus and a novel hydrophobic C-terminal tail translated from the retained intron 6 fragment and the exon 7 frameshift product. A, The predicted sequence of the C-terminal tail of the GABRG2(IVS6+2T→G) BAC transcript (γ2-PTC subunit) is shown. The blue background shows the retained 53 bp intron 6 fragment, and the red octagon shows the position of the PTC in exon 7. Sequences of both DNA strands are shown. The predicted amino acids are shown in blue above the DNA sequences. B, The predicted membrane topology of the γ2-PTC subunit is shown. The gray circles represent the wild-type γ2 subunit N terminus 217 aa peptide. The dark blue circles represent the 29 aa novel C terminus translated from retained intron 6 and exon 7 frameshift product. C, The hydrophobicity of the novel C-terminal tail translated from retained intron 6 and exon 7 frameshift product was determined based on an amino acid scale (Abraham and Leo, 1987). The average hydrophobicities of five adjacent amino acids (y-axis) are plotted against the amino acid positions in the peptide (x-axis). D, Peptide sequence alignment by ClustalW2 showing the γ2-PTC subunit is homologous to AChBP identified from different species. Ac-AChBP, Aplysia californica; Bt-AChBP, Bulinus truncatusi; Ls-AChBP, Lymnaea stagnalis.
This truncated subunit was reminiscent of the soluble acetylcholine-binding proteins (AChBPs) found in mollusk glial cells (Smit et al., 2001; Hansen et al., 2004; Celie et al., 2005). AChBP sequences are homologous to the N-terminal extracellular domains of cys-loop family ligand-gated ion channel (LGIC) subunits, and the crystal structure and protein function are similar to the ligand-binding domain of the nicotinic acetylcholine receptor α subunit. GABAA receptors also belong to the cys-loop LGIC family. AChBPs oligomerize to form homopentamers containing binding sites for agonists and antagonists including acetylcholine. Upon acetylcholine release, AChBPs are released from glia cells into synaptic gaps and inhibit cholinergic neurotransmission by binding free acetylcholine molecules (Smit et al., 2001). Sequence alignment showed that the γ2-PTC subunit has the highest homology with the Aplysia californica AChBP (Ac-AChBP) (Fig. 2D). ClustalW alignment showed that the γ2-PTC subunit had a 21–29% sequence identity with AChBPs (data not shown), which was even higher than the 13–25% sequence identity between AChBPs and LIGC subunit N-terminal extracellular domains (Celie et al., 2005). The GABRG2(IVS6+2T→G) mutation might generate a mutant protein, the γ2-PTC subunit, that structurally resembles AChBPs and interferes with GABAergic neurotransmission in a similar way.
The expression pattern of hGABRG2HA BACs in transgenic mouse brain was similar to that of the endogenous GABRG2
The transcription product of the CMV-driven hGABRG2(IVS6+2T→G) BAC in HEK293T cells, the γ2-PTC subunit mRNA, retained a 53 bp intron 6 fragment in the mutant exon 6. However, it has been reported that promoter usage can affect intron splicing pattern (Kornblihtt, 2005), and thus the mutant hGABRG2(IVS6+2T→G) BAC might be spliced to another mRNA when driven by its endogenous promoter. The intron splicing pattern is also cell type dependent. To minimize possible artifacts, we studied intron splicing of the hGABRG2(IVS6+2T→G) BAC with its endogenous promoter region in transgenic mouse brain (Fig. 3). We first expressed an HA-tagged hGABRG2 BAC in C57BL/6J mice, which is the C57BL/6-Tg(hGABRG2HA)RLM mouse line [according to The Jackson Laboratory mouse nomenclature, Tg(hGABRG2HA mice)]. The HA tag was introduced to a functionally silent position in the wild-type subunit coding sequence. The HA-tagged γ2 subunits were not recognized by polyclonal γ2 subunit antibodies (data not shown). These antibodies recognized a strong nonspecific band below 50 kDa and weak γ2 subunit-specific bands below the nonspecific band (Fig. 3A). The Western blot on adult mouse total brain tissue lysate showed that both transgenic mice (Fig. 3Aa, lanes 4, 5) and wild-type littermates (Fig. 3Aa, lanes 1–3) had endogenous mouse γ2 subunits, but only transgenic Tg(hGABRG2HA) mice had both endogenous mouse and HA-tagged human γ2 subunits (Fig. 3Ab, lanes 4, 5). The merged image showed that the HA band molecular mass was similar to the endogenous mouse γ2 subunit band (Fig. 3Ac). We repeated this Western blot four times with 15 adult Tg(hGABRG2HA) mouse brain samples and detected the same HA tag band in addition to the endogenous mouse γ2 subunit band. Although the HA-tagged hGABRG2 BAC construct had human GABRG2 gene genomic sequence and transcription regulatory elements, it was translated to protein in the transgenic mouse brain.
Figure 3.
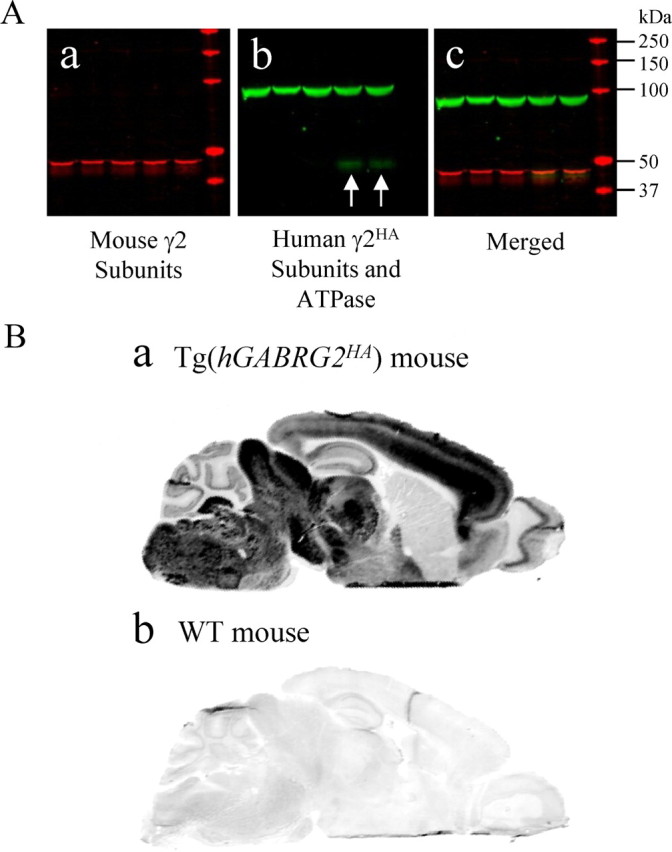
The wild-type human hGABRG2HA BAC in transgenic mouse brain had the same expression pattern as the endogenous mouse mGABRG2. A, Western blot on brain total lysate of Tg(hGABRG2HA) transgenic mice showing transgenic mice expressed both endogenous mouse γ2 subunits and HA-tagged human γ2 subunits (n = 4). a, Endogenous mouse γ2 subunits were labeled in the red channel. b, ATPase and HA-tagged proteins were labeled in the green channel. c, The merged image showed the molecular size of HA-tagged human γ2 subunits and endogenous mouse γ2 subunits were similar. The white arrows in b point to the HA bands. B, The expression pattern of γ2HA subunits in the Tg(hGABRG2HA) mouse brain was similar to that of γ2 subunits in wt mouse brain. HA antibodies stained parasagittal sections of adult Tg(hGABRG2HA) BAC transgenic mouse brain (a) or adult wild-type littermate brain (b). Sections were scanned in Odyssey scanner as one image after immunolabeling, which was presented in grayscale. The signal in the Tg(hGABRG2HA) section was oversaturated in some regions because the setting was chosen to visualize the nonspecific binding in the wild-type littermate section (n = 3).
We then collected brain samples from Tg(hGABRG2HA) mice and wild-type littermates, cryosectioned them to 20 μm sections, stained the sections with HA antibodies and IRDye800-conjugated donkey anti-mouse IgG secondary antibodies, and scanned the immunolabeled sections with the Odyssey imaging system (Fig. 3Ba,Bb). Wild-type mouse brain sections only showed weak fluorescence signal in the 800 channel (Fig. 3Bb). Its pattern was similar to the pattern of its autofluorescence scanned in the 700 channel, which did not receive any antibody labeling (data not shown). The Tg(hGABRG2HA) mouse brain section had enhanced 800 channel fluorescence signal primarily in olfactory bulb, cortex, hippocampus, thalamus, midbrain, pons, and cerebellum. The expression pattern of HA-tagged human γ2 subunits in the Tg(hGABRG2HA) mouse brain was similar to endogenous mouse γ2 subunits as reported previously (Fritschy et al., 1994; Fritschy and Mohler, 1995). The human GABRG2 gene promoter in hGABRG2HA BACs and the endogenous mouse GABRG2 gene promoter functioned similarly.
The γ2-PTC subunit was expressed as a stable protein in HEK293T cells and Tg(hGABRG2IVS6+2T→G) mouse brain
Having confirmed the expression pattern of hGABRG2 BACs, we next determined the effect of the mutation on mRNA splicing in the transgenic mouse brain. We introduced the IVS6+2T→G mutation into the BAC without the HA tag, expressed the mutant BAC in C57BL/6-Tg(hGABRG2IVS6+2T→G)RLM mice [Tg(hGABRG2IVS6+2T→G) mice], collected transgenic mouse and wild-type littermate brain total RNAs, and converted them to total cDNAs. To determine the effect of the mutation on mRNA splicing pattern in transgenic mice, we performed RT-PCR using primers binding to exon 5–7 of the γ2 subunit cDNA. The amplified fragment from wild-type human or mouse γ2 subunit cDNA was 320 bp, and the fragment amplified from the CMV-driven BAC transcript in HEK293T cell, the γ2-PTC subunit, was 373 bp. The primers amplified only one band from wild-type littermate total cDNAs but two bands that were almost overlapping from mutant Tg(hGABRG2IVS6+2T→G) mouse total cDNAs (data not shown). The IVS6+2T→G mutation generated an NciI restriction enzyme site in the middle of the γ2-PTC subunit cDNA. NciI should cut the amplified mutant exon 5–7 fragment into two fragments of 204 and 169 bp, thus allowing more separation on the gel between the amplified mouse fragment (320 bp) and the digested mutant transgene products (204 and 169 bp). We repeated the exon 5–7 RT-PCR in mutant transgenic mouse brain total RNAs and in wild-type human γ2S subunit or γ2-PTC subunit cDNA-transfected cell total RNAs. We then digested the RT-PCR products with NciI and separated digested products in ethidium bromide-stained agarose gel (Fig. 4A). The human γ2S subunit PCR product was undigested and remained ∼320 bp as expected (Fig. 4A, lane a), but the γ2-PTC subunit PCR product was digested to a broad ∼200 bp band, consistent with two 204 and 169 bp products (Fig. 4A, lane b). The Tg(hGABRG2IVS6+2T→G) mouse total RNA RT-PCR product showed two bands of ∼320 and 200 bp (again consistent with two 204 and 169 bp products) after digestion (Fig. 4A, lane 5), while their wild-type littermate RT-PCR fragments had only one 320 bp fragment after digestion (Fig. 4A, lanes 6–8). Direct DNA sequencing of the cloned RT-PCR products showed that the Tg(hGABRG2IVS6+2T→G) mouse brain had human γ2-PTC subunit cDNA identical with the transcription product of CMV-hGABRG2(IVS6+2T→G) BAC in HEK293T cells, as well as endogenous mouse γ2 subunit cDNA. This RT-PCR was repeated in seven Tg(hGABRG2IVS6+2T→G) mouse brain total RNA samples at different ages (three at P0, four at P35), and the same mutant human BAC transcript was detected in these animals. Thus, the splicing pattern of the mutant hGABRG2(IVS6+2T→G) BAC intron 6 in mouse brain was the same as the splicing pattern of the mutant CMV-driven BAC intron 6 in transfected HEK293T cells.
Figure 4.

The γ2-PTC subunit was expressed as a stable protein in HEK293T cells and Tg(hGABRG2IVS6+2T→G) mouse brain. A, Mutant γ2-PTC subunit mRNA was detected in Tg(hGABRG2IVS6+2T→G) mouse brain total RNA. RT-PCR experiment amplifying exons 5–7 of γ2 subunit, followed by NciI digestion, showed that the IVS6+2T→G mutant γ2 subunit mRNAs were expressed in Tg(hGABRG2IVS6+2T→G) mutant BAC transgenic mice brain total RNA. Ba, The γ2S or γ2-PTC subunit cDNAs were transfected in HEK293T cells at different amounts, total protein levels were evaluated by Western blot using antibodies against the epitope at the γ2 subunit mature peptide N terminus. Bb, Western blot on the Tg(hGABRG2IVS6+2T→G) mouse and wild-type littermates total brain lysates show that mutant γ2-PTC subunits were expressed in Tg(hGABRG2IVS6+2T→G) mouse brain. The numbers below each lane represent the mouse numbers. The experiment was repeated three times, and seven Tg(hGABRG2IVS6+2T→G) mice were studied.
Although mutant GABRG2(IVS6+2T→G) BAC γ2 subunit mRNAs were susceptible to degradation by NMD, we still detected γ2-PTC subunit mRNAs in transfected HEK293T cells and Tg(hGABRG2IVS6+2T→G) mouse brain. In silico translation predicted that the γ2-PTC subunit retained most of the wild-type γ2 subunit N terminus. We transfected wild-type γ2S subunit cDNA and γ2-PTC subunit cDNA in HEK293T cells, separated total cell lysates using a 4–12% gradient NuPage Novex Bis-Tris gel (Invitrogen), and ran Western blots using polyclonal γ2 subunit antibodies (Fig. 4Ba). All samples had a faint nonspecific band slightly smaller than 50 kDa and a specific γ2 subunit band of wild-type γ2 subunit that was ∼40 kDa (Fig. 4Ba, lanes 1, 2, top arrow). The γ2-PTC subunit-transfected cells had the same nonspecific band and showed a γ2 subunit-specific doublet band smaller than 37 kDa (Fig. 4Ba, lanes 3, 4, bottom arrow). The faint bottom band in the doublet was visible only at the higher γ2-PTC subunit amount and was obvious in 3 μg of cDNA-transfected cells (Fig. 4Ba, lane 3) but not in the 1 μg of cDNA transfected (Fig. 4Ba, lane 4). It was probably generated by a different pattern of subunit glycosylation (Lo et al., 2010). The predicted γ2-PTC subunit encodes a ∼33 kDa protein containing a signal peptide of 4 kDa and mature peptide of 28 kDa. The molecular mass of this protein band in SDS-PAGE gel was larger than predicted for the mature peptide, probably because of posttranslational modifications. Thus, the γ2-PTC subunit was translated as a stable protein in HEK293T cells.
We then collected Tg(hGABRG2IVS6+2T→G) mouse brain total lysate and blotted with the same antibodies (Fig. 4Bb). All mouse samples had the γ2 subunit-specific band at the same size as the wild-type γ2S subunit in HEK293T cells, which were endogenous mouse γ2 subunits (Fig. 4Ba, lanes 5–8, top arrow). The Tg(hGABRG2IVS6+2T→G) mouse brain sample (from mouse no. 5), however, had an extra doublet band at the same size as the γ2-PTC subunit in HEK293T cells (Fig. 4Ba, lane 1, bottom arrow). We evaluated 12 Tg(hGABRG2IVS6+2T→G) mouse brain samples at ages varying from P0 to P80 and detected the same staining pattern. We also introduced the hGABRG2(IVS6+2T→G) BAC to the B6D2F1/J mouse and made a B6D2-Tg(hGABRG2IVS6+2T→G) mouse. Western blot on four B6D2-Tg(hGABRG2IVS6+2T→G) mouse brain samples at P30 showed the same mouse γ2 and human γ2-PTC subunit band migration pattern as the C57BL/6-Tg(hGABRG2IVS6+2T→G) mouse brain samples. The mutant human GABRG2(IVS6+2T→G) BAC transgene was detected as stable γ2-PTC subunits in the Tg(hGABRG2IVS6+2T→G) mouse brain. These mice expressed both endogenous mouse γ2 and human γ2-PTC subunits in brain. Therefore, both Tg(hGABRG2IVS6+2T→G) mice and transfected HEK293T cells can be used to study the function of the γ2-PTC subunit.
NMD decreased mutant γ2-PTC subunit mRNA levels
The GABRG2 mutation, IVS6+2T→G, generated a PTC in exon 7. The mature mutant γ2-PTC subunit mRNA, therefore, should be degraded by the NMD machinery, since it contains a PTC that is >55 bp upstream of an exon–exon junction complex. In contrast, the GABRG2(Q390X) mutation is an autosomal dominant mutation associated with Dravet syndrome (Harkin et al., 2002). The mutation generates a PTC in the last exon, so the mature mutant GABRG2(Q390X) mRNA should not be degraded by NMD and should be translated to truncated γ2(Q390X) subunits (Kang et al., 2009b). To determine whether NMD was activated by these PTCs, wild-type hGABRG2, NMD-susceptible mutant hGABRG2(IVS6+2T→G), and NMD-resistant mutant hGABRG2(Q350X) BACs were expressed in HEK293T cells expressing siRNAs against the NMD essential factor UPF1, and γ2 subunit mRNA levels were quantified using the TaqMan real-time PCR assay. BACs were also transfected into HEK293T cells expressing siRNAs without cellular function according to the manufacturer's manual. The TaqMan probe was designed to bind to the borders of exons 4 and 5. The levels of BAC-derived γ2 subunit mRNAs were compared with GFP mRNA levels for each condition, and then the γ2 subunit mRNA levels of UPF1 siRNA-transfected cells were compared with negative control siRNA-transfected cells for each BAC construct. Western blot showed that the UPF1 protein level was unchanged in negative control siRNA-transfected HEK293T cells compared with untransfected control cells but was decreased to ∼20% in UPF1 siRNA-transfected cells (data not shown). Real-time PCR results showed that the wild-type γ2 subunit mRNA levels were not changed (1.16 ± 0.14-fold; n = 6) after UPF1 knockdown (Fig. 5A). The γ2-PTC subunit mRNA level, however, was increased 2.14 ± 0.52-fold after UPF1 knockdown (p < 0.05, n = 6), while as expected the γ2(Q390X) subunit mRNA level was not changed (1.19 ± 0.20-fold; not significant; n = 6). We also evaluated γ2 subunit mRNA level after blocking another NMD essential factor, SMG6, and obtained similar results (data not shown). Thus, γ2-PTC, but not γ2 or γ2(Q390X), subunit mRNA was subject to degradation by NMD.
Figure 5.

The mutant γ2-PTC subunit mRNA level was decreased by NMD, while the undegraded mRNA was translated to the immature γ2-PTC subunit with an ER glycosylation pattern. A, The mRNA level of the γ2-PTC subunit was increased after UPF1 knockdown (n = 6). RNA levels were evaluated by TaqMan quantitative real-time PCR as described in Materials and Methods. *p < 0.05, one-way ANOVA with Bonferroni's multiple-comparison test. B, The protein level of the γ2-PTC subunit was increased also after UPF1 knockdown (n = 4). **p < 0.01, one-way ANOVA Bonferroni's multiple-comparison test. Error bars indicate SEM. C, In HEK293T cells, the γ2-PTC subunit is a stable protein that was not secreted into the culture medium. HA-tagged wild-type γ2S subunit cDNA, γ2-PTC subunit cDNA, and pcDNA empty vector were expressed in HEK293T cells. Culture media of each cell and total cell lysate were both collected and incubated with HA beads to pull down HA-tagged proteins. Pull-down proteins were eluted with HA peptide, separated by SDS-PAGE, and blotted with HA antibodies. The experiment was repeated three times, and a representative gel was shown. D, The γ2-PTC subunit had an ER glycosylation pattern. HA-tagged wild-type γ2L and γ2-PTC subunits were expressed in HEK293T cells as either single subunits or were coexpressed with α1β2 subunits (Receptor). Total cell lysates from each condition were collected and digested with endoglycosidase Endo H or PNGase F. Digested and undigested proteins were blotted with HA antibodies. U, Undigested; H, Endo H digested; F, PNGase F digested. The experiment was repeated four times, and a representative gel was shown.
NMD decreased mutant γ2-PTC subunit levels
We collected the BAC-transfected cell lysates and blotted proteins using endogenous γ2 subunit antibodies (data not shown). We quantified the γ2 subunit band intensity of each lane normalized to the ATPase band intensity of the same lane, and compared the normalized γ2 subunit band intensity between cells expressing UPF1 siRNA and cells expressing negative control siRNA (Fig. 5B). The wild-type band intensity was unchanged (87.2 ± 18.9%; n = 4), the GABRG2(IVS6+2T→G) BAC protein band intensity was increased to 232.6 ± 31.5% (p < 0.01; n = 4), while the GABRG2(Q390X) BAC band intensity was unchanged (116.0 ± 20.5%; p = 0.095; n = 4) relative to the wild-type band intensity. The increased amount of GABRG2(IVS6+2T→G) BAC protein was consistent with the increased mRNA level. The wild-type and mutant γ2 subunit protein level increases after blocking SMG6 had the same trend (data not shown).
These data demonstrated that the amount of the GABRG2(IVS6+2T→G) BAC translation product, the γ2-PTC subunit, was increased by decreasing NMD efficiency in cells. The γ2-PTC subunit protein levels in the brain of the Tg(hGABRG2IVS6+2T→G) mouse, and presumably human patients, would be determined by both mutant GABRG2(IVS6+2T→G) gene transcription level and NMD efficiency.
The γ2-PTC subunit was not secreted into the culture medium
As noted above, the γ2-PTC subunit is homologous to the AChBPs (Brejc et al., 2001), which are secreted into the extracellular space by glial cells where they bind acetylcholine to terminate synaptic transmission. When Ac-AChBPs were expressed in HEK293T cells, homopentameric Ac-AChBPs were secreted into the culture media (Hansen et al., 2004). Thus, by analogy, it is possible that γ2-PTC subunits are folded correctly, form pentamers, and are secreted from cells. However, due to the increased hydrophobicity at the C-terminal tail, it is also possible that the subunit has a transmembrane segment that folds and assembles as a membrane bound protein and is not secreted. Epitope-tagged γ2-PTCHA subunits were used to determine the cellular fate of the truncated γ2-PTC subunits. The HA tag was added to the same site that was functionally silent in wild-type subunits. When wild-type γ2SHA or γ2-PTCHA subunits were expressed in HEK293T cells, both were stable proteins (Fig. 5C). As the γ2 subunit antibodies showed in Tg(hGABRG2IVS6+2T→G) mouse brain samples, HA-tagged mutant γ2-PTCHA subunits (lane 2) were smaller than wild-type γ2SHA subunits (Fig. 5C, lane 1). The HA beads successfully pulled down HA-tagged wild-type γ2SHA or mutant γ2-PTCHA subunits from total cell lysate (Fig. 5C, lanes 4, 5) but pulled down nothing from the cell culture medium (Fig. 5C, lanes 7, 8). We collected ∼15 ml of culture media from each sample. If the γ2-PTCHA subunit was secreted from cells at the same efficiency as Ac-AChBP (1 - 3 mg/L) (Hansen et al., 2004), there would be ∼15–45 μg of γ2-PTC subunit protein in 15 ml of culture media. We used the Odyssey quantitative Western blot system to detect the γ2-PTCHA subunit. According to the manufacturer's (LI-COR) document, even if the amount of γ2-PTCHA subunit was a hundred times less than 15–45 μg, it should still be sufficient for detection by our Western blot. Although the γ2-PTC subunit is highly homologous to the secreted Ac-AChBP, γ2-PTCHA subunits were present, but not secreted into the culture medium. In Tg(hGABRG2IVS6+2T→G) mouse brain or human patients, the mutant allele would be translated to γ2-PTC subunits, which are likely to be also expressed inside of the neurons and not secreted to extrasynaptic spaces.
The γ2-PTC subunit attained altered ER-associated glycosylation
While not secreted, γ2-PTC subunits could still form homo-oligomers or hetero-oligomers that are trafficked to the surface membrane. During the process of subunit maturation, immature N-linked mannose-rich oligosaccharides attached in the ER are replaced by mature glycans that are attached in the trans-Golgi region. Wild-type γ2L subunits show only low levels of membrane trafficking when expressed alone, which increased substantially with coexpression of α1 and β2 subunits (Connolly et al., 1999a). To determine whether the γ2-PTC subunits had mature glycosylation consistent with surface membrane trafficking, we compared the glycosylation patterns of γ2L and γ2-PTC subunits without or with cotransfection of α1 and β2 subunits.
Endo H cleaves immature N-linked mannose-rich oligosaccharides attached in the ER but not the mature glycans attached in the trans-Golgi region. In contrast, PNGase F removes all oligosaccharides attached both in the ER and trans-Golgi regions (Maley et al., 1989). When expressed alone, γ2LHA subunits on Western blot ran as a single band that was sensitive to digestion by endoglycosidases Endo H and PNGase F, consistent with primarily immature glycosylation and suggesting that γ2LHA subunits were retained in the ER (Fig. 5D, WT subunit). When coexpressed with α1 and β2 subunits, γ2LHA subunits showed an extra band on Western blots that was insensitive to Endo H digestion, but was sensitive to PNGase F digestion (Fig. 5D, WT subunit). With coexpression of α1 and β2 subunits, γ2LHA subunits had mature glycosylation, suggesting processing in the Golgi apparatus and trafficking to the cell membrane.
With expression alone or with coexpression of α1 and β2 subunits, γ2-PTCHA subunits showed only one band on Western blot that was sensitive to both Endo H and PNGase F (Fig. 5D, mutant subunit), suggesting that γ2-PTCHA subunits were retained in the ER and not transported to the Golgi apparatus. The size of the digested γ2-PTCHA subunit protein band was consistent with the predicted size of the mature γ2-PTCHA subunit based on amino acid sequence. This phenomenon is consistent with the finding that γ2-PTCHA subunits were not secreted into the culture medium, which requires Golgi translocation. These data further suggested that the γ2-PTC subunit might not be trafficked to the cell membrane and might instead be retained in the ER under physiological conditions such as in patients or in Tg(hGABRG2IVS6+2T→G) mouse neurons.
The γ2-PTC subunits oligomerized with α1 and β2 subunits
GABAA receptor subunit oligomerization is determined by sequences at the extracellular N-terminal domain (Taylor et al., 1999), and the γ2-PTC subunit included >90% of the wild-type γ2 subunit N-terminal extracellular domain. A benzodiazepine-binding site is present at the αγ subunit interface. A radioligand binding study showed that the amount of radiolabeled benzodiazepine binding in cells expressing α1 and full-length γ2 subunits was comparable with the cells expressing α1 subunits and the N terminus of γ2 subunits, but that cells expressing α1, β2, and γ2 subunits had much higher benzodiazepine binding (Klausberger et al., 2001). There is a 15 aa sequence in the γ2 subunit N-terminal extracellular domain around residue R82 (residue numbered in the immature peptide) that was sufficient to pull down β2 subunits, but the presence of an R82Q mutation in this peptide abolished the interaction, suggesting that this site was involved in the oligomerization of β2 and γ2 subunits (Hales et al., 2005). The γ2-PTC subunit includes this 15 aa sequence, and so to explore whether γ2-PTC subunits can oligomerize with partnering subunits, wild-type γ2SHA, γ2LHA, and mutant γ2-PTCHA subunits were coexpressed with α1 and β2 subunits in HEK293T cells. HA-tagged proteins were pulled down using HA beads and blotted for α1 and β2 subunits or for the HA tag (Fig. 6A). The amount of pulled-down α1 or β2 subunits reflected binding between the γ2 subunits and α1 or β2 subunits, respectively. The eluted HA-tagged proteins showed a band pattern that was similar to that of the total HA-tagged proteins. The wild-type γ2SHA or γ2LHA subunits both pulled down substantial amounts of α1 and β2 subunits (Fig. 6A, lanes 2, 3). The γ2-PTCHA subunit pulled down α1 and β2 subunits (Fig. 6A, lane 4), but the amounts pulled down were less than those pulled down by wild-type γ2L or γ2S subunits (Fig. 6A, lane 4). This was an expected finding because the mutant γ2-PTCHA subunits contain the entire extracellular N-terminal domain of γ2 subunits including the 15 aa peptide sequence that has been shown to be sufficient to pull down β2 subunits. These data suggested that γ2-PTC subunits in physiological conditions would be nonfunctional and decrease GABAergic inhibition by decreasing surface γ2 subunit levels and having a dominant-negative action to reduce heteropentameric GABAA receptor assembly and trafficking because of its direct interaction with α1 and β2 subunits.
Figure 6.
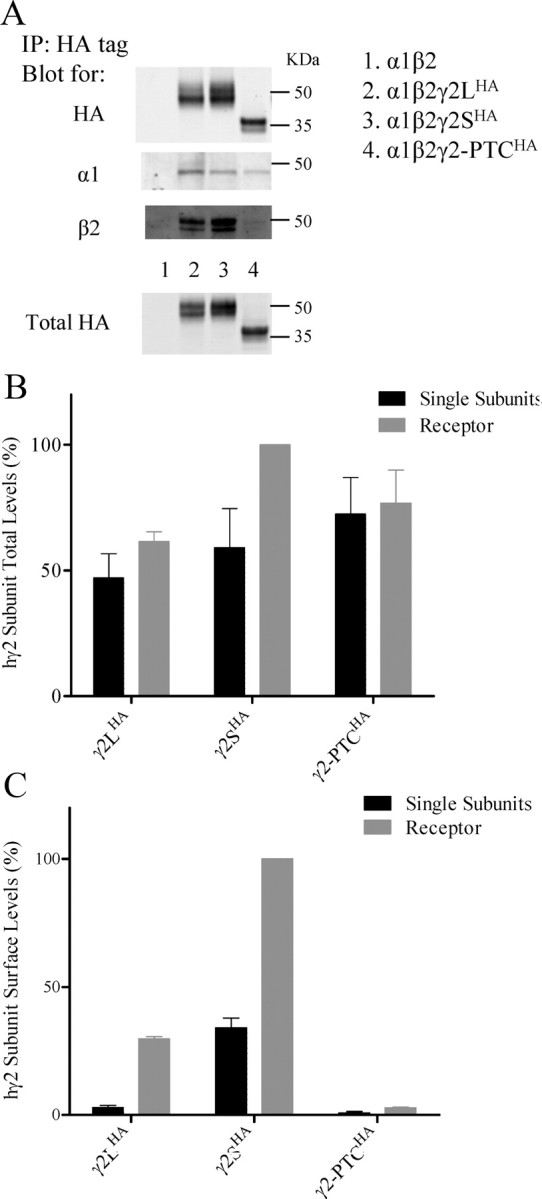
The γ2-PTC subunits oligomerized weakly with α1 and β2 subunits and had impaired membrane trafficking. A, HA-tagged wild-type and mutant γ2 subunits were coexpressed with α1 and β2 subunits in HEK293T cells. Total cell lysate from each condition were collected and incubated with HA beads. Pull down products were eluted with HA peptide and blotted with antibodies against α1 subunits, β2 subunits, and the HA epitope tag. Western blot on total cell lysate with HA antibodies are also shown. Total cell lysates were also blotted for α1 and β2 subunits in Western blot, but data are not shown. The experiment was repeated six times and a representative gel was shown. B, The normalized mean fluorescence intensity showing the total HA level from wild-type and mutant γ2HA subunits expressed as either single subunit (black) or with α1β2 subunit coexpression (gray). The total HA level with α1, β2, and γ2SHA subunit coexpression was used as 100%. The total HA level of pcDNA mock-transfected cells was taken as a 0% baseline. Data were analyzed with a two-way ANOVA with Bonferroni's multiple-comparison test. C, The surface HA level from wild-type and mutant γ2HA subunits expressed as either single subunit (black) or with α1 and β2 subunits coexpression (gray). The surface HA level with coexpression of α1, β2, and γ2SHA subunits was used as 100%. The surface HA level of pcDNA mock-transfected cells is 0% baseline. Data were analyzed with a two-way ANOVA with Bonferroni's multiple-comparison test. Error bars indicate SEM.
The γ2-PTC subunit was a stable intracellular protein
The hGABRG2HA BAC had a CNS expression pattern that was similar to that of the endogenous mouse γ2 subunit gene (Fig. 3B), and mutant γ2-PTC subunits were identified in Tg(hGABRG2IVS6+2T→G) mouse brain. Since mutant γ2-PTC subunits had immature glycosylation and impaired oligomerization, it is likely that they had impaired assembly into receptors and impaired membrane trafficking. Furthermore, it may be recognized as a misfolded or misassembled protein that was subject to ER-associated degradation by the proteasome. To evaluate this, we quantified mutant subunit expression and membrane trafficking using high-throughput flow cytometry. We expressed wild-type or mutant γ2HA subunits in HEK293T cells either as single subunits or coexpressed with α1 and β2 subunits and evaluated total and surface levels of each subunit in >50,000 cells. All subunits were transcribed from the same pcDNA3.1 vector. The total HA level obtained with cotransfection of α1, β2, and γ2SHA subunits was used as a control (100%) for other γ2 subunit levels, and total levels obtained from pcDNA mock-transfected cells were used as a baseline control (0%). Total levels of the γ2-PTCHA subunit did not differ from those of wild-type γ2LHA or γ2SHA subunits with single subunit expression (p = 0.02, 0.06, respectively; n = 4), or when coexpressed with α1 and β2 subunits (Fig. 6B). Thus, the mutant γ2-PTCHA subunit was not degraded and was as stable in these cells as wild-type γ2S subunits.
The γ2-PTC subunit had impaired membrane trafficking
To assess surface trafficking of the mutant γ2-PTCΗΑ subunit, we used the quantitative technique of flow cytometry without cell permeabilization. We cotransfected cells using the same subunit combinations used to assess total cell levels of α1β2γ2SHA subunits by measuring surface HA levels for each subunit (Fig. 6C). The surface HA level obtained with α1β2γ2SHA subunit coexpression was used as a 100% normalization control for γ2HA subunit surface level, and surface HA level obtained with pcDNA mock-transfected cells was used as baseline (0%). The single wild-type γ2LHA subunit had a low surface level (2.93 ± 0.76%; n = 4), which was increased substantially (29.71 ± 0.88%; p < 0.01; n = 4) by coexpression with α1 and β2 subunits. The wild-type γ2SHA single-subunit surface level was much higher than γ2LHA subunit surface level, probably because the γ2SHA single subunits have higher trafficking efficiency and lower PKC-dependent endocytosis (Connolly et al., 1999a,b). Its single-subunit surface level (34.08 ± 3.80%; n = 4) was substantially higher than the γ2LHA subunits single-subunit surface level. Its surface level with α1 and β2 subunit coexpression was 100%, also substantially higher than with α1β2γ2LHA coexpression (n = 4). Compared with γ2LHA and γ2SHA subunits, the surface levels of the γ2-PTCHA subunit were substantially smaller with both expression conditions. The γ2-PTCHA single-subunit surface level was low (0.76 ± 0.56%; n = 4) and did not increase significantly with α1 and β2 subunit coexpression (2.76 ± 0.30%; n = 4; not significant). The surface levels of γ2-PTCΗΑ subunits with or without α1 and β2 subunit coexpression were not significantly greater than the mock control level (p value: single subunit, >0.05; with α1 and β2 subunit coexpression: >0.05; n = 4). These results suggest that, even though the mutant γ2-PTCΗΑ subunit oligomerized with α1 and β2 subunits, it was not trafficked to the cell surface.
The γ2-PTC subunits were retained in the ER
We have demonstrated that γ2-PTC subunits are stable in cells and minimally trafficked to the cell surface when coexpressed with α1 and β2 subunits. Given this impaired trafficking, it is likely that the mutant γ2-PTC subunits with or without oligomerization with α1 and β2 subunits are retained in the ER with little localization in the trans-Golgi or surface membrane. Because the mutant hGABRG2(IVS6+2T→G) BAC in the Tg(hGABRG2IVS6+2T→G) mouse does not have an HA tag, and the antibodies against endogenous γ2 subunits had a high nonspecific signal, we could not stain the Tg(hGABRG2IVS6+2T→G) mouse brain to detect where the γ2-PTC subunit was expressed. Therefore, we coexpressed wild-type and mutant γ2-PTCHA subunits with α1 and β2 subunits in HEK293T cells, stained the permeabilized cells with fluorescence-conjugated antibodies against the α1 subunit or the HA tag, and obtained confocal microscope images to visualize the cellular localization of the subunits (Fig. 7A). The ER was visualized using antibodies against the ER marker BIP. In addition, membrane expression was confirmed further by confocal microscope images taken from unpermeabilized HEK293T cells cotransfected with α1 and β2 subunits and wild-type or mutant γ2HA subunits (data not shown). BIP staining was not detected in any of these samples, showing that paraformaldehyde fixation did not permeabilize the membrane (data not shown). With coexpression of α1 and β2 subunits without γ2 subunits, the α1 subunit signal overlapped the ER signal, but also showed a ring structure that surrounded the ER signal (Fig. 7A, first row) and outlined the cell membrane (data not shown), consistent with low levels of α1β2γ2-PTC and higher levels of α1β2 receptor expression on the cell membrane. With coexpression of α1, β2, and γ2HA subunits, wild-type γ2LHA or γ2SHA subunits were both visualized in regions that overlapped α1 subunits (Fig. 7A, second and third row; surface staining not shown), consistent with coassembly with α1 and β2 subunits into receptors that were trafficked to the cell surface. The γ2-PTCHA subunit signal overlapped that of the ER signal (Fig. 7A, fourth row) and was absent from the surface membrane (data not shown). The wild-type α1, γ2L, and γ2S subunits often showed HA signals in the region that was recognized by Golgi-specific antibodies (data not shown), but the γ2-PTCHA subunit was not. Thus, the γ2-PTCHA subunit was retained primarily in the ER, consistent with its background levels on the surface membrane and its absence in the culture medium.
Figure 7.
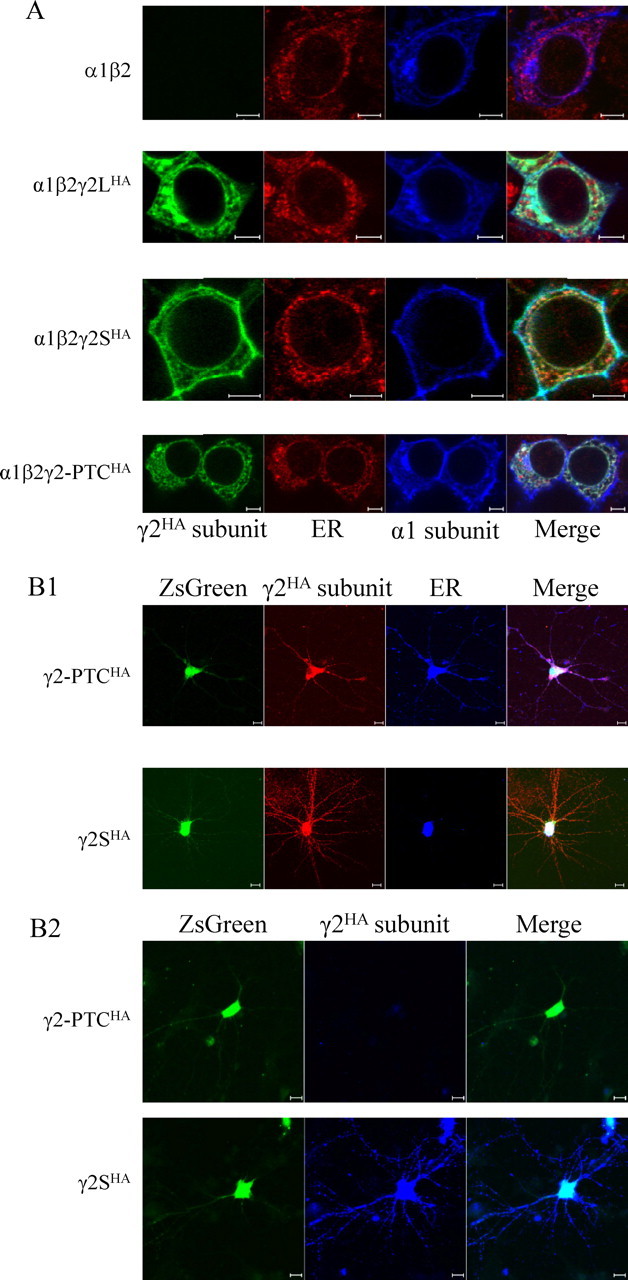
When expressed with α1 and β2 subunits, mutant γ2-PTC subunits were trapped in the ER. A, Confocal images were obtained of the distribution of GABAA receptor subunits in permeabilized HEK293T cells. In HEK293T cells, α1 and β2 subunits were coexpressed with a blank pcDNA vector, and wild-type and mutant γ2HA subunits were coexpressed with α1 and β2 subunits at 1 μg of each cDNA. The transfected cells were permeabilized and stained with Alexa fluorophore-conjugated antibodies against HA tag (Alexa 488 conjugated; green) or α1 subunits (Alexa 647 conjugated; blue). The ER was visualized with primary rabbit antibodies against the ER marker BIP, and Alexa 568-conjugated secondary antibodies against rabbit IgG (red). Scale bars, 5 μm. B, Confocal images were obtained of transfected cultured cortical neurons expressing γ2HA subunits in pLVX-IRES-ZsGreen1 vectors. B1, Cultured neurons were permeabilized and immunostained with mouse monoclonal antibody against the HA epitope tag and rabbit polyclonal antibodies against ER marker BIP, followed by Alexa 568-conjugated donkey anti-mouse IgG antibodies (red) and Alexa 647-conjugated donkey anti-rabbit IgG antibodies (blue). The ZsGreen, shown in green, labeled transfected neurons. Scale bars, 10 μm. B2, Cultured neurons were immunostained without permeabilization, showing surface expression of HA epitope-tagged γ2 subunits. Neurons were stained with mouse monoclonal antibody against the HA epitope tag and Alexa 647-conjugated donkey anti-mouse IgG (blue). The ZsGreen, shown in green, labeled transfected neurons. Scale bars, 10 μm.
We then cloned the γ2SHA and γ2-PTCHA subunit cDNAs into pLVX-γ2HA-IRES-ZsGreen1 vectors expressing γ2HA subunits and green fluorescent protein ZsGreen from the same mRNA but separated by internal ribosome entry site sequence (IRES) and expressed them in cultured cortical neurons (Fig. 7B). Cytoplasmic ZsGreen protein showed green fluorescence in both soma and processes in transfected neurons. The γ2HA subunit fluorescence signal was detected in all ZsGreen-positive neurons, but also in a few ZsGreen-negative neurons, suggesting that the cDNA downstream of IRES has a lower probability for translation to proteins (data not shown). The γ2-PTCHA subunit fluorescence signal in permeabilized neurons was diffusely distributed over the neuron soma and less intensely over the process, and it colocalized well with ER marker, BIP (Fig. 7B1). On the contrary, the γ2SHA subunit signal was present in widespread, large clusters on neuronal somata and dendritic arbors, and spread outside of the ER marker signal (Fig. 7B1). The clustered expression pattern of γ2SHA subunit immunoreactivity resembled that of endogenous γ2 subunits in cultured neurons (Mangan et al., 2005). The surface staining on cortical neurons further confirmed that the γ2-PTCHA subunit was absent from cell membranes (Fig. 7B2). While the γ2SHA subunit showed strong γ2HA clusters on unpermeabilized transfected neurons, both on cell somata and processes, the γ2-PTCHA subunit had only background level of the HA epitope tag signal (Fig. 7B2), similar to the HA epitope tag signal from pLVX-IRES-ZsGreen1 empty vector mock-transfected neurons (data not shown). These data confirmed that the γ2-PTCHA subunit was retained in the ER when expressed in neurons and was not expressed on synaptic membranes.
The GABA-evoked current from α1β2γ2-PTC subunit coexpression was similar to α1β2 receptor current
The data above suggest that the majority of receptors on the surface of cells with coexpression of α1, β2, and γ2-PTC subunits would likely be α1β2 receptors. To explore this, we coexpressed α1 and β2 subunits and α1 and β2 subunits with γ2-PTC or γ2S subunits in HEK293T cells and recorded GABA-evoked current evoked by a saturating concentration (1 mm) of GABA (Fig. 8A). With coexpression of α1 and β2 subunits, GABA-evoked currents had a small peak amplitude of ∼400 pA and very fast desensitization (Fig. 8A). With coexpression of α1, β2, and γ2S subunits, GABA-evoked currents were much larger and desensitization was slower. With coexpression of α1, β2, and γ2-PTC subunits, GABA-evoked currents had fast desensitization, and a small peak amplitude that was more similar to α1β2 receptor currents than α1β2γ2S receptor currents.
Figure 8.
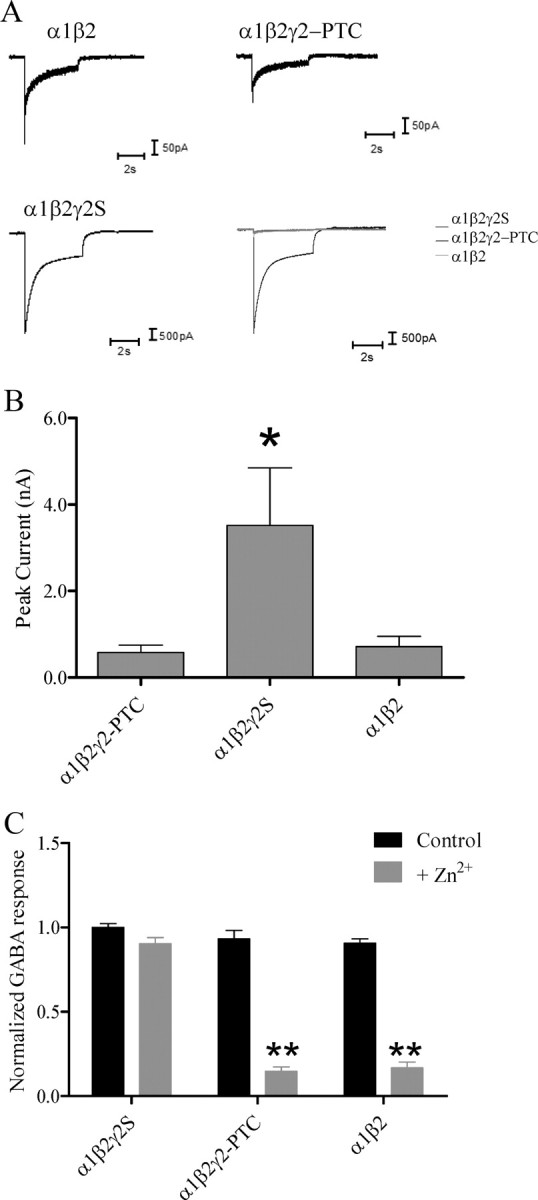
GABA-evoked currents recorded from cells coexpressing α1, β2, and γ2-PTC subunits were similar to those obtained with expression of α1and β2 subunits. A, GABAergic currents were recorded from coexpressed α1β2, α1β2γ2-PTC, and α1β2γ2S subunits. The merged picture showed the relative peak amplitude of currents recorded from α1β2γ2-PTC subunits was much smaller than that from α1β2γ2S subunits, but close to those obtained from α1β2 subunits. B, Peak current amplitudes from wild-type and mutant receptors were plotted. *p < 0.05, one-way ANOVA with Bonferroni's multiple-comparison test. C, The currents recorded from coexpressed α1β2γ2-PTC subunits had a Zn2+ sensitivity that was similar to that of coexpressed α1β2 subunits. Cells expressing α1β2γ2S, α1β2γ2-PTC, or α1β2 subunits were exposed to two 1 mm GABA applications 4 s apart or one 1 mm GABA application followed by 10 μm Zn2+ washed 4 s and 10 μm Zn2+ with 1 mm GABA application. The peak currents ratio of each cell was plotted. **p < 0.01, compared with the control conditions (two-way ANOVA with Bonferroni's multiple-comparison test). Error bars indicate SEM.
We then recorded from cells coexpressing α1, β2, and γ2S or γ2-PTC subunits (n = 8 cells) and measured peak current amplitudes (Fig. 8B). With coexpression of α1, β2, and γ2-PTC subunits, the average peak current amplitude was 575.5 pA. With coexpression of α1, β2, and γ2S subunits, the average peak current amplitude was 3516.0 pA, and with coexpression of α1 and β2 subunits, the peak current amplitude was 710.4 pA. The peak current amplitudes of these two wild-type receptors fall in the normal range of reported values. The α1β2γ2-PTC receptor peak current amplitude was significantly decreased from α1β2γ2S receptors but was similar to that of α1β2 receptors.
The divalent cation Zn2+ is an endogenous neuromodulator (Kay and Tóth, 2008). Its ability to inhibit GABAA receptor currents depends on receptor subunit composition (Nagaya and Macdonald, 2001). The α1β2 receptors are highly sensitive to Zn2+, with IC50 values of ∼0.1–1 μm, while the α1β2γ2 receptors are very insensitive to Zn2+, with IC50 values of ∼200–600 μm. We bathed cells with an external solution containing 10 μm Zn2+, applied 1 mm GABA alone or with 10 μm Zn2+, and then compared the peak currents with and without Zn2+ (Fig. 8C). We also applied GABA at the same time interval but without Zn2+, and compared the peak currents to quantify current rundown. With coexpression of α1 and β2, α1, β2, and γ2S, or α1, β2, and γ2-PTC subunits, currents showed minimum peak amplitude decreases with repetitive GABA applications. The α1β2 receptor peak current amplitude decreased 10%, the α1β2γ2S receptor peak current amplitude did not decrease, and the α1β2γ2-PTC receptor peak current amplitude decreased 17%. However, with Zn2+ application, α1β2 receptor peak current amplitude decreased 83.2%, α1β2γ2-PTC receptor peak current amplitude decreased 85.3%, and α1β2γ2S receptor peak current amplitude decreased 9.6%. Zn2+ inhibited all three receptor currents with different efficiencies. The α1β2γ2-PTC receptors had the same high sensitivity to Zn2+ as the α1β2 receptors consistent with formation primarily of surface α1β2 receptors with coexpression of α1, β2, and γ2-PTC subunits.
The γ2-PTC subunits induced an increase in the ER stress marker BIP
Although γ2-PTC subunit mRNA was decreased by NMD, and the translation product was expressed poorly on the cell membrane, patients carrying one mutant allele had seizures. The mutant γ2-PTC subunit was clearly expressed in the transgenic mouse brain and was stable in HEK293T cells and produced haploinsufficiency and a dominant-negative effect on receptor assembly. Since the γ2-PTC subunit was so stable in transgenic mouse brain, the possibility that the mutant subunit had additional functions that might contribute to epilepsy pathogenesis.
The γ2-PTC subunits were stable ER proteins with a sequence similar to a γ2 subunit truncated in the middle of the first transmembrane domain. While the γ2-PTC subunits were stable and not degraded, the subunit may not have the same conformation as wild-type γ2 subunits and could induce the unfolded protein response (UPR). The level of the ER chaperone BIP is an indicator of UPR-induced ER stress. We expressed 3 μg of wild-type or mutant γ2-PTCHA subunits in HEK293T cells and then evaluated cellular BIP levels by Western blot (Fig. 9A). The BIP band intensity for each condition was normalized to that of pcDNA mock-transfected cells (100%) (Fig. 9B). BIP levels in untreated and mock-transfected cells were not different, but treatment of untransfected cells with tunicamycin, an ER stress inducer, significantly increased BIP levels to 339.3 ± 69.3% (n = 5; p < 0.01). Wild-type γ2SHA subunits induced an increase in BIP levels to 143.9 ± 22.4% (n = 5), but it was not significantly different from the mock-transfected condition or untreated untransfected cells (not significant). The γ2-PTCHA subunits increased BIP levels to 244.8 ± 31.3% (n = 5), which was significantly more than that of the γ2SHA subunits (p < 0.05) or mock-transfected cells (p < 0.01). The γ2-PTCHA subunit induced BIP more efficiently than the wild-type subunit. The ER-retained γ2(Q390X) subunit has a strong dominant-negative effect on GABAA receptor assembly (Kang et al., 2009b). The mutant subunit bound to α1 and wild-type γ2HA subunits when coexpressed in the HEK293T cells, retained them in the ER, and decreased their surface expression. Expressing γ2S(Q390X)HA subunits in HEK293T cells increased the BIP level to 224.6 ± 27.2% (n = 5), which was also significantly higher than the γ2SHA subunits (p < 0.05), but had a trend to be lower than the γ2-PTC subunits (not significant). We evaluated the number of apoptotic cells using Annexin V and found that the expression of γ2-PTCHA subunits did not significantly increase cell apoptosis (data not shown). Thus, γ2-PTCHA subunits increased cell stress but did not induce apoptosis in these cells.
Figure 9.
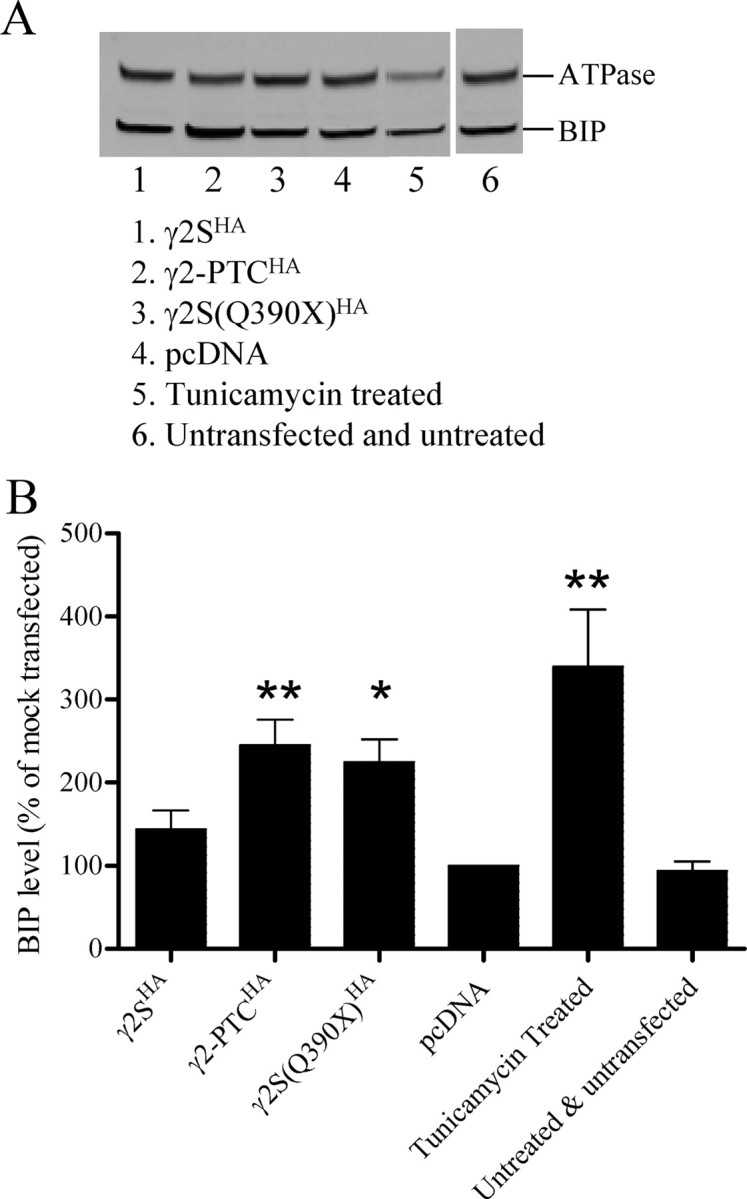
The γ2-PTC subunits induced an increase in the ER stress marker BIP. A, BIP protein levels in γ2HA subunit-transfected cells or tunicamycin-treated cells were evaluated. HEK293T cells were either transfected with 3 μg of γ2SHA, γ2-PTCHA, or γ2S(Q390X)HA subunit cDNA, or were treated with 1 μm tunicamycin for 3 h. Total proteins were collected and analyzed with Western blot detecting ATPase and BIP (n = 5). B, The band intensities of BIP and ATPase protein bands were quantified using Odyssey, version 3.0, software. The BIP intensity of each condition was normalized to ATPase band intensity. The normalized BIP band intensities were plotted. **p < 0.01 and *p < 0.05, compared with the normalized BIP level obtained from either pcDNA mock-transfected cells or untreated and untransfected cells (one-way ANOVA with Bonferroni's multiple-comparison test). Error bars indicate SEM.
Discussion
The intronic GABRG2 mutation, IVS6+2T→G, resulted in activation of a cryptic mRNA splice donor site
The GABRG2 intronic mutation, IVS6+2T→G, mutated the intron 6 splice donor site sequence from GT to GG, thus destroying the function of the site. It was suggested that the most likely pathway for splicing of this mutant mRNA was via exon 5 skipping (Kananura et al., 2002), but the actual splice pattern was not determined. Analysis of mammalian EST sequences revealed that 98.7% of introns contained canonical GU–AG junctions and that 0.56% contained noncanonical GC–AG junctions (Burset et al., 2001). The large mutant rabbit β-globin intron with an IVS+2T→G mutation was cleaved at the first step at the correct 5′ site with reduced efficiency, but the splicing intermediate was not cleaved at the 3′ site leading to accumulation of the lariat intermediate (Aebi et al., 1987). These findings suggested that intron 6 of the mutant GABRG2(IVS6+2T→G) gene was unlikely to be normally spliced.
DBASS5 is a database of aberrant 5′ splice sites in human disease genes (Buratti et al., 2007) and contains 40 mutations at the U of the 5′ GU sequence. In this database, the 5′ GU sequence was mutated most frequently to GG (35%), GC (35%), or GA (22.5%) sequences. Interestingly, 92.5% of the mutations activated a cryptic 5′ donor site within ∼100 bp of the wild-type donor site (data not shown). When expressed in HEK293T cells, the GABRG2 mutation, IVS6+2T→G, activated a cryptic 5′ donor site 53 bp downstream of the wild-type site, consistent with the function of these mutations in the DBASS5 database. While alternative intron splicing is regulated differently among cell types, the core intron splicing machinery is distributed ubiquitously in every cell. To confirm that the splicing pattern found HEK293T cells in vitro is also found in mouse brain in vivo, we expressed the human hGABRG2 BACs driven by its endogenous promoter in C57BL/6J mice. The Tg(hGABRG2HA) mice expressed the HA-tagged BAC clone RP11-1035I20, and the Tg(hGABRG2IVS6+2T→G) mice expressed the untagged BAC clone carrying the IVS6+2T→G mutation. There is a 20 kbp human chromosome 5 fragment upstream of the GABRG2 genomic sequence in this BAC clone, which is predicted to contain the endogenous human γ2 subunit promoter. This BAC clone was recognized by the mouse transcription and translation machineries, resulting in expression of wild-type and mutant human γ2 subunits in the transgenic mice brain. When the BACs were expressed in mouse brain, the mutant intron 6 splicing pattern was the same as the mutant BACs expressed in HEK293T cells.
Pre-mRNA intron splicing is regulated by functional interactions among transcription, splicing, and chromatin epigenetic modifications (Kornblihtt, 2005; Alexander and Beggs, 2010). The CMV-GABRG2 BACs were driven by a CMV promoter, which could recruit a different set of transcription factors and interact with the splicing machinery differently than with the endogenous GABRG2 promoter. However, the GABRG2 BAC and the GABRG2(IVS6+2T→G) BAC driven by the endogenous promoter and the CMV promoter-driven BACs had the same intron splicing pattern. Thus, the CMV promoter and the endogenous promoter had the same effect on GABRG2 gene intron splicing.
The intronic GABRG2 mutation, IVS6+2T→G, resulted in partial intron 6 retention and a frameshift resulting in a PTC in exon 7 that activated NMD
The mature mutant mRNA retained a 53 bp intron 6 fragment that resulted in an open reading frameshift in exon 7 and generated a PTC in exon 7. Thus, the mutant mRNA was NMD susceptible, and we confirmed this by demonstrating that the mutant mRNA was rescued partially by abolishing NMD function. NMD-susceptible mRNAs have lower translational efficiency, and protein translated from NMD-susceptible mRNA is often not stable, probably because such proteins are truncated (Kang et al., 2009a; Zhang et al., 2010). Our study suggested that the IVS6+2T→G mutation could significantly decrease mutant γ2 subunit mRNA levels due to NMD, suggesting that the disease may be, at least in part, caused by GABRG2 haploinsufficiency.
Transcription of the mutant mRNA resulted in production of a stable truncated protein, the γ2-PTC subunit
Although γ2-PTC subunit mRNAs were subject to degradation by NMD, they were not necessarily completely degraded since different cell types have different NMD efficiency (Linde et al., 2007). For example, we demonstrated that 39.1% of α1(S326fs328X) subunit mRNA survived NMD in HEK293T cells, 17% survived in HeLa cells, and 24% survived in cortical neuronal cell culture (Kang et al., 2009a). In cell types with less NMD efficiency than HEK293T cells, the amount of mutant transcript could be >40%, and mRNA not degraded by NMD could be translated to a stable protein. The mutant GABRG2(IVS6+2T→G) mRNA translation product was shown to contain the N-terminal 217 aa of the wild-type γ2 subunit and a novel 29 aa peptide tail (the γ2-PTC subunit) composed of retained intron 6 sequence and frameshifted exon 7 sequence at the C terminus of the mutant protein. The γ2-PTC subunit is homologous to a γ2 subunit truncated in the middle of TM1. The sequence homology between the first 246 aa of wild-type γ2 subunit and γ2-PTC subunit is 88.2%. Surprisingly, the γ2-PTC subunit was a stable intracellular protein in HEK293T cells, transfected rat cortical neurons, and Tg(hGABRG2IVS6+2T→G) mouse brain, and while the γ2-PTC subunit total level was comparable with wild-type γ2S or γ2L subunits in HEK293T cells, its surface level was significantly lower than the wild-type subunits.
The γ2-PTC subunit has a structure similar to AChBPs but has different functions
The γ2-PTC subunit contains the N terminus of the γ2 subunit, and the sequence identity between γ2-PTC subunit and AChBPs was high (21–29%) (Brejc et al., 2001). Thus, the γ2-PTC subunit is homologous to AChBPs. AChBPs form homopentamers in glial cells (Brejc et al., 2001; Hansen et al., 2004; Celie et al., 2005), and the crystal structure of AChBP homopentamers resembles the N terminus of assembled cys loop receptors. When wild-type γ2 subunits were coexpressed with α1 and β2 subunits in HEK293T cells or adult rat brain, they oligomerized with α1 and β2 subunits to form α1β2γ2 heteropentamers, but not γ2 homopentamers or α1β2 heteropentamers (Angelotti and Macdonald, 1993; Klausberger et al., 2001), suggesting that the binding efficiency of γ2 subunits to α1 and β2 subunits is higher than it is between γ2 subunits. Given the sequence similarity to AChBPs, the γ2-PTC subunit might also be able to oligomerize with α1 and β2 subunits and might even be assembled into heteropentameric receptors. Our studies showed that γ2-PTC subunits did oligomerize with α1 and β2 subunits; however, they did not produce heteropentamers that were secreted or trafficked to the cell membrane. Instead, they were retained in the ER and had a dominant-negative effect on surface trafficking of α1β2γ2 receptors. Thus, although γ2-PTC subunits and AChBPs are highly homologous, these two proteins clearly have very different functions.
The GABRG2(IVS6+2T→G) mutation could induce epilepsy by both γ2 subunit haploinsufficiency and γ2-PTC subunit dominant-negative functions
We demonstrated in vitro in transfected HEK293T cells and in vivo in mice that the mutant mRNA was degraded partially by NMD, and that the mRNA that was not degraded was translated to the stable γ2-PTC subunit. The γ2-PTC subunit was primarily retained in the ER and only minimally expressed on cell membranes. GABAA receptors containing γ2 subunits are predominately synaptic receptors that mediate phasic synaptic neurotransmission, and the γ2 subunit is required for synaptic GABAA receptor clustering. The IVS6+2T→G mutation decreased the γ2 subunit mRNA level and generated a mutant protein that was poorly trafficked to the cell membrane. It should decrease the membrane level of γ2 subunit-containing GABAA receptors, decrease the amount of synaptic GABAA receptors, and impair inhibitory GABAA receptor currents. Thus, the GABRG2(IVS6+2T→G) mutation could produce epilepsy, at least in part, by γ2 subunit haploinsufficiency. The γ2-PTC subunit oligomerized with α1 and β2 subunits and had a dominant-negative effect on surface trafficking of α1β2γ2 receptors. The GABRG2(IVS6+2T→G) mutation could also produce with epilepsy due to the dominant-negative effects of the γ2-PTC subunit.
The GABRG2(IVS6+2T→G) mutation could induce epilepsy also by inducing chronic ER stress
The γ2-PTC subunit is retained in the ER and increased the ER stress marker BIP level significantly higher than wild-type γ2S subunits. Another GABRG2 epilepsy mutation, the autosomal dominant GABRG2(Q390X) mutation, is associated with Dravet syndrome (Harkin et al., 2002). The mutant γ2(Q390X) subunit is also retained in the ER and not expressed on the cell membrane (Kang et al., 2009b). The γ2(Q390X) subunit also increased BIP level in HEK293T cells, but to a level slightly less than that the increase produced by the γ2-PTC subunit. Increased BIP level during UPR induced ER-stress induces both apoptosis and protective responses such as reduced translation, enhanced ER protein-folding capacity, and clearance of misfolded ER proteins (Lin et al., 2008). These adaptation and apoptosis responses are designed to help adaptation to the stress or to remove affected cells, depending on the nature and severity of the stress (Rutkowski and Kaufman, 2007). The fact that γ2-PTC subunit-transfected cells did not induce apoptosis suggested that the γ2-PTC subunit induced mild, chronic stress in the cell, but the adaptive responses induced by γ2-PTC subunits would affect how cells respond to other stress factors. ER stress responses contribute to the pathogenesis of diseases including diabetes mellitus, cancer, and AIDS (Rutkowski and Kaufman, 2007). Neurodegenerative diseases such as Alzheimer's disease and Huntington's disease are often associated with ER stress responses induced by mutant proteins. Thus, ER stress responses may contribute to the pathogenic mechanism of both GABRG2(Q390X) and GABRG2(IVS6+2T→G) mutations.
Footnotes
This work was supported by NIH Grant R01 NS051590 (R.L.M.) and an Epilepsy Foundation predoctoral research training fellowship (M.T.). Confocal experiments were performed in part using the Vanderbilt University Medical Center Cell Imaging Shared Resource (supported by NIH Grants CA68485, DK20593, DK58404, HD15052, DK59637, and EY08126). Flow Cytometry experiments were performed in the Vanderbilt Medical Center Flow Cytometry Shared Resource, which is supported by the Vanderbilt Ingram Cancer Center (NIH Grant P30 CA68485) and the Vanderbilt Digestive Disease Research Center (NIH Grant DK058404). We thank Xuan Huang, Wangzhen Shen, Ningning Hu, and Kelienne Verdier for technical assistance, Drs. Feng Huajun and Andre Lagrange for instructions in electrophysiology, Dr. Douglas Mortlock for sharing BAC recombineering protocols, Cara Sutcliffe and Ping Mayo for instructions in TaqMan quantitative PCR, Sean Schaffer for help with confocal microscopy experiments, and Dr. Ron Emeson for helpful discussions and suggestions.
References
- Abraham DJ, Leo AJ. Extension of the fragment method to calculate amino acid zwitterion and side chain partition coefficients. Proteins. 1987;2:130–152. doi: 10.1002/prot.340020207. [DOI] [PubMed] [Google Scholar]
- Aebi M, Hornig H, Weissmann C. 5′ cleavage site in eukaryotic pre-mRNA splicing is determined by the overall 5′ splice region, not by the conserved 5′ GU. Cell. 1987;50:237–246. doi: 10.1016/0092-8674(87)90219-4. [DOI] [PubMed] [Google Scholar]
- Alexander R, Beggs JD. Cross talk in transcription, splicing and chromatin: who makes the first call? Biochem Soc Trans. 2010;38:1251–1256. doi: 10.1042/BST0381251. [DOI] [PubMed] [Google Scholar]
- Angelotti TP, Macdonald RL. Assembly of GABAA receptor subunits: α1β1 and α1β1γ2S subunits produce unique ion channels with dissimilar single-channel properties. J Neurosci. 1993;13:1429–1440. doi: 10.1523/JNEUROSCI.13-04-01429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker G, Goslin K. Cellular and molecular neuroscience. Ed 2. Cambridge, MA: MIT; 1998. Culturing nerve cells. [Google Scholar]
- Beghi M, Beghi E, Cornaggia CM, Gobbi G. Idiopathic generalized epilepsies of adolescence. Epilepsia. 2006;47(Suppl 2):107–110. doi: 10.1111/j.1528-1167.2006.00706.x. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Mulley JC, Scheffer IE, Petrou S. Human epilepsies: interaction of genetic and acquired factors. Trends Neurosci. 2006;29:391–397. doi: 10.1016/j.tins.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Song L, Zhang H, Macdonald RL. Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci. 2002;22:5321–5327. doi: 10.1523/JNEUROSCI.22-13-05321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Buratti E, Chivers M, Královicová J, Romano M, Baralle M, Krainer AR, Vorechovsky I. Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35:4250–4263. doi: 10.1093/nar/gkm402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burset M, Seledtsov IA, Solovyev VV. SpliceDB: database of canonical and non-canonical mammalian splice sites. Nucleic Acids Res. 2001;29:255–259. doi: 10.1093/nar/29.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel I, Tal S, Vig I, Ast G. Comparative analysis detects dependencies among the 5′ splice-site positions. RNA. 2004;10:828–840. doi: 10.1261/rna.5196404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celie PH, Klaassen RV, van Rossum-Fikkert SE, van Elk R, van Nierop P, Smit AB, Sixma TK. Crystal structure of acetylcholine-binding protein from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. J Biol Chem. 2005;280:26457–26466. doi: 10.1074/jbc.M414476200. [DOI] [PubMed] [Google Scholar]
- Chandler RL, Chandler KJ, McFarland KA, Mortlock DP. Bmp2 transcription in osteoblast progenitors is regulated by a distant 3′ enhancer located 156.3 kilobases from the promoter. Mol Cell Biol. 2007;27:2934–2951. doi: 10.1128/MCB.01609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra D, Korpi ER, Miralles CP, De Blas AL, Homanics GE. GABAA receptor gamma 2 subunit knockdown mice have enhanced anxiety-like behavior but unaltered hypnotic response to benzodiazepines. BMC Neurosci. 2005;6:30. doi: 10.1186/1471-2202-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ. Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J Biol Chem. 1996;271:89–96. doi: 10.1074/jbc.271.1.89. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Uren JM, Thomas P, Gorrie GH, Gibson A, Smart TG, Moss SJ. Subcellular localization and endocytosis of homomeric gamma2 subunit splice variants of gamma-aminobutyric acid type A receptors. Mol Cell Neurosci. 1999a;13:259–271. doi: 10.1006/mcne.1999.0746. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Kittler JT, Thomas P, Uren JM, Brandon NJ, Smart TG, Moss SJ. Cell surface stability of gamma-aminobutyric acid type A receptors. Dependence on protein kinase C activity and subunit composition. J Biol Chem. 1999b;274:36565–36572. doi: 10.1074/jbc.274.51.36565. [DOI] [PubMed] [Google Scholar]
- Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, Belzung C, Fritschy JM, Lüscher B, Mohler H. Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat Neurosci. 1999;2:833–839. doi: 10.1038/12207. [DOI] [PubMed] [Google Scholar]
- Dredge BK, Darnell RB. Nova regulates GABAA receptor gamma2 alternative splicing via a distal downstream UCAU-rich intronic splicing enhancer. Mol Cell Biol. 2003;23:4687–4700. doi: 10.1128/MCB.23.13.4687-4700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Lüscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Eugène E, Depienne C, Baulac S, Baulac M, Fritschy JM, Le Guern E, Miles R, Poncer JC. GABAA receptor γ2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J Neurosci. 2007;27:14108–14116. doi: 10.1523/JNEUROSCI.2618-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Paysan J, Enna A, Mohler H. Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J Neurosci. 1994;14:5302–5324. doi: 10.1523/JNEUROSCI.14-09-05302.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MJ, Shen W, Song L, Macdonald RL. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the alpha1 subunit. J Biol Chem. 2005;280:37995–38004. doi: 10.1074/jbc.M508305200. [DOI] [PubMed] [Google Scholar]
- Günther U, Benson J, Benke D, Fritschy JM, Reyes G, Knoflach F, Crestani F, Aguzzi A, Arigoni M, Lang Y. Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 1995;92:7749–7753. doi: 10.1073/pnas.92.17.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, Tang H, Bollan KA, Johnson SJ, King DP, McDonald NA, Cheng A, Connolly CN. The epilepsy mutation, gamma2(R43Q) disrupts a highly conserved inter-subunit contact site, perturbing the biogenesis of GABAA receptors. Mol Cell Neurosci. 2005;29:120–127. doi: 10.1016/j.mcn.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Talley TT, Radic Z, Taylor P. Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J Biol Chem. 2004;279:24197–24202. doi: 10.1074/jbc.M402452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH, Richards MC, Williams DA, Mulley JC, Berkovic SF, Scheffer IE, Petrou S. Truncation of the GABAA-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–536. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, Rebstock J, Heils A, Steinlein OK. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. Making sense of nonsense GABAA receptor mutations associated with genetic epilepsies. Trends Mol Med. 2009;15:430–438. doi: 10.1016/j.molmed.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABAA receptor GABRA1 PTC mutation, 975delC, S326fs328X. J Neurosci. 2009a;29:2833–2844. doi: 10.1523/JNEUROSCI.4512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J Neurosci. 2009b;29:2845–2856. doi: 10.1523/JNEUROSCI.4772-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR, Tóth K. Is zinc a neuromodulator? Sci Signal. 2008;1:re3. doi: 10.1126/stke.119re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Ehya N, Fuchs K, Fuchs T, Ebert V, Sarto I, Sieghart W. Detection and binding properties of GABAA receptor assembly intermediates. J Biol Chem. 2001;276:16024–16032. doi: 10.1074/jbc.M009508200. [DOI] [PubMed] [Google Scholar]
- Kornblihtt AR. Promoter usage and alternative splicing. Curr Opin Cell Biol. 2005;17:262–268. doi: 10.1016/j.ceb.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde L, Boelz S, Nissim-Rafinia M, Oren YS, Wilschanski M, Yaacov Y, Virgilis D, Neu-Yilik G, Kulozik AE, Kerem E, Kerem B. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117:683–692. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WY, Lagrange AH, Hernandez CC, Harrison R, Dell A, Haslam SM, Sheehan JH, Macdonald RL. Glycosylation of β2 subunits regulates GABAA receptor biogenesis and channel gating. J Biol Chem. 2010;285:31348–31361. doi: 10.1074/jbc.M110.151449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ, Feng HJ. GABAA receptor mutations associated with generalized epilepsies. Adv Pharmacol. 2006;54:147–169. doi: 10.1016/s1054-3589(06)54007-4. [DOI] [PubMed] [Google Scholar]
- Maley F, Trimble RB, Tarentino AL, Plummer TH., Jr Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem. 1989;180:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Sun C, Carpenter M, Goodkin HP, Sieghart W, Kapur J. Cultured hippocampal pyramidal neurons express two kinds of GABAA receptors. Mol Pharmacol. 2005;67:775–788. doi: 10.1124/mol.104.007385. [DOI] [PubMed] [Google Scholar]
- Nagaya N, Macdonald RL. Two gamma2L subunit domains confer low Zn2+ sensitivity to ternary GABAA receptors. J Physiol. 2001;532:17–30. doi: 10.1111/j.1469-7793.2001.0017g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Schneider Gasser EM, Straub CJ, Panzanelli P, Weinmann O, Sassoè-Pognetto M, Fritschy JM. Immunofluorescence in brain sections: simultaneous detection of presynaptic and postsynaptic proteins in identified neurons. Nat Protoc. 2006;1:1887–1897. doi: 10.1038/nprot.2006.265. [DOI] [PubMed] [Google Scholar]
- Sieghart W, Sperk G. Subunit composition, distribution and function of GABAA receptor subtypes. Curr Top Med Chem. 2002;2:795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- Sinkkonen ST, Lüscher B, Lüddens H, Korpi ER. Autoradiographic imaging of altered synaptic alphabetagamma2 and extrasynaptic alphabeta GABAA receptors in a genetic mouse model of anxiety. Neurochem Int. 2004;44:539–547. doi: 10.1016/j.neuint.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, Lodder H, van der Schors RC, van Elk R, Sorgedrager B, Brejc K, Sixma TK, Geraerts WP. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature. 2001;411:261–268. doi: 10.1038/35077000. [DOI] [PubMed] [Google Scholar]
- Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, Clarke AL, Dibbens L, Krestel H, Mulley JC, Jones MV, Seeburg PH, Sakmann B, Berkovic SF, Sprengel R, Petrou S. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A. 2007;104:17536–17541. doi: 10.1073/pnas.0708440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PM, Thomas P, Gorrie GH, Connolly CN, Smart TG, Moss SJ. Identification of amino acid residues within GABAA receptor β subunits that mediate both homomeric and heteromeric receptor expression. J Neurosci. 1999;19:6360–6371. doi: 10.1523/JNEUROSCI.19-15-06360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyndale RF, Hales TG, Olsen RW, Tobin AJ. Distinctive patterns of GABAA receptor subunit mRNAs in 13 cell lines. J Neurosci. 1994;14:5417–5428. doi: 10.1523/JNEUROSCI.14-09-05417.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade-Martins R, Smith ER, Tyminski E, Chiocca EA, Saeki Y. An infectious transfer and expression system for genomic DNA loci in human and mouse cells. Nat Biotechnol. 2001;19:1067–1070. doi: 10.1038/nbt1101-1067. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABAA receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PJ. GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discov Today. 2003;8:445–450. doi: 10.1016/s1359-6446(03)02703-x. [DOI] [PubMed] [Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ashiya M, Sherman TG, Grabowski PJ. Essential nucleotides direct neuron-specific splicing of gamma 2 pre-mRNA. RNA. 1996;2:682–698. [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Liu W, Grabowski PJ. Coordinate repression of a trio of neuron-specific splicing events by the splicing regulator PTB. RNA. 1999;5:117–130. doi: 10.1017/s1355838299981530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhou L, Hu L, Zhu Y, Xu H, Liu Y, Chen X, Yi X, Kong X, Hurst LD. Nonsense-mediated decay targets have multiple sequence-related features that can inhibit translation. Mol Syst Biol. 2010;6:442. doi: 10.1038/msb.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]