

Summary
Tumor necrosis factor receptor (TNFR) superfamily members mediate the cellular response to a wide variety of biological inputs. The responses range from cell death, survival, differentiation, proliferation, to the regulation of immunity. All these physiological responses are regulated by a limited number of highly pleiotropic kinases. The fact that the same signaling molecules are involved in transducing signals from TNFR superfamily members that regulate different and even opposing processes raises the question of how their specificity is determined. Regulatory strategies that can contribute to signaling specificity include scaffolding to control kinase specificity, combinatorial use of several signal transducers, and temporal control of signaling. In this review, we discuss these strategies in the context of TNFR superfamily member signaling.
Keywords: TNFR, MAPK, IKK, NFκB, scaffold, signaling specificity
Diverse biological responses to TNFR superfamily members
The tumor necrosis factor receptor (TNFR) superfamily consists of 29 receptors that mediate cellular responses to 19 ligands. While most ligands bind to a single receptor, some bind to numerous receptors. For example, BAFF can associate with three receptors, and TNF-related apoptosis-inducing ligand (TRAIL) can even bind to five receptors (1, 2). All TNFRs are characterized as type I transmembrane proteins, with an extracellular N-terminus and intracellular C-terminus necessary for signaling initiation (3). The TNFR superfamily can be categorized into three overlapping classes: activating receptors, death receptors, and decoy receptors. Activating receptors such as TNFR1 and CD40 mediate activation of nuclear factor-jB (NFκB) and mitogen-activated protein kinase (MAPK) pathways. Death domain containing receptors (such as TNFR1 and FAS) contain an 80 amino acid death domain in their cytoplasmic domain. Its deletion abolishes ligand-induced cell death. Through sequestration of the ligand, decoy receptors (e.g. DCR1, OPG) have been shown to inhibit cell signaling.
Most receptors are expressed on a wide variety of cell types. Receptor engagement by members of the TNF superfamily can trigger diverse cellular responses, such as apoptosis [for example TNF, lymphotoxin (LT), FAS ligand (FASL)], survival [receptor activator of NFκB ligand (RANKL) and B-cell activating factor belonging to the TNF family (BAFF)], differentiation (such as TNF, RANKL), or proliferation (such as TNF, CD40L, OX40L, BAFF). These cellular responses are mediated by the activation of transcription factors NFκB, which comprise NFκB1 (p50 and its precursor p105), NFκB2 (p52 and its precursor p100), c-Rel, RelA (p65), and RelB, and activator protein-1 (AP-1), composed of Fos (c-Fos, FosB, Fra-1, and Fra-2) and Jun (c-Jun, JunB, and JunD) family members, as well as closely related transcription factors CREB, ATF2, ATF3, and B-ATF), which are activated by MAPK [p38, c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK)] and inhibitor of NFκB kinase (IKK) signaling cascades (1, 2).
Signaling by TNF superfamily members is essential for a large variety of physiological processes including hematopoiesis, protection from bacterial infection, immune surveillance, and tumor regression. Ligands, including TNF, LTb, and RANKL, provide crucial signals for the morphogenesis of secondary lymphoid organs; TNF, FAS, and TRAIL contribute to the function of cytotoxic effector cells in the recognition and destruction of virus-infected cells. The expression of FASL on activated T cells induces their cell death, a mechanism to modulate the immune response. Importantly, misregulation of TNFR signaling has been associated with a diverse range of diseases including autoimmunity, liver disease, tumorigenesis, lymphproliferative diseases, diabetes, and even allergic asthma (1, 2, 4–6). The diverse pathological effects caused by TNFR misregulation reflect the large variety of biological processes they are involved in and highlight the importance of precise regulation of TNFR superfamily signaling. Interestingly, the MAPK and IKK cascades are the critical signal transducers for all TNFRSFs raising the question of how the functional specificity of these kinases is determined to ensure signaling specificity.
Pleiotropic signal transducers within the TNFR signaling network
The key signal transducers of the TNFR superfamily members are the kinases IKK2, IKK1, JNK, p38, and ERK. Below we have summarized some key facts about each kinase, indicating the wide variety of substrates and biological functions that have been ascribed to each (Fig. 1).
Fig. 1. Kinases involved in TNFRSF-member signaling are highly pleiotropic.

The pleiotropic kinases JNK, p38, and ERK1/2 (MAPKs), canonical IKK2, and non-canonical IKK1 are key regulators in signal transduction in response to a large variety of cellular signals and can trigger highly diverse biological responses.
IKK2
IKK2 is a key regulator of NFκB activation induced by inflammatory cytokines, pathogens, environmental and metabolic stress and some developmental signals. By phosphorylating the classical IκBs (IκBα, IκBβ, IκBε), it triggers their proteasomal degradation and release of NFκB to the nucleus to allow for transcriptional activation. The primary canonical NFκB effectors are RelA, cRel, and p50 in both homodimeric or heterodimeric forms. Induction of their activity by IKK2 leads, in most cell types, to a general cell activation response, that may involve the secretion of inflammatory molecules, increasing the resistance to cell death-inducing stimuli, and, in lymphocytes particularly, activation of a proliferative program (7, 8). Apart from its role in activating NFκB, IKK2 has many additional substrates that are not part of the NFκB signaling system. The IKK2-dependent phosphorylation of the tumor suppressor p53 at S362 and S366 is thought to be a mechanism for regulating its stability (9). Upon TNFα stimulation, IKK2 phosphorylates insulin receptor (IR) substrate 1 (IRS-1) at S307, resulting in the termination of metabolic insulin signaling (10). In addition, IKK2 phosphorylates the tumor suppressor tuberous sclerosis 1 (TSC1) at position S487 and S511, which leads to its suppression and coincides with the activation of the mTOR pathway, enhanced angiogenesis and tumor development (11). β-catenin, a key molecule in Wnt signaling, and FOXO3a, which acts downstream of growth factor signaling (PI3K/Akt), have also been reported to be specifically phosphorylated by IKK2 (12). Recently, IKK2 has also been found to play a critical role in starvation-induced autophagy, in an NFκB independent manner (13, 14). The substrates however remain to be identified.
IKK1
The IKK1 kinase functions as a homodimer and as a heterodimer with IKK2. As a homodimer it may be activated through the NFκB-inducing kinase (NIK) by developmental signals such as BAFF and LTβ (6, 8). IKK1 dependent phosphorylation of p100 results in p100 processing and induction of transcriptional activation. The main mediators of transcription are RelA, RelB, p50, and p52 controlling cell survival and developmental processes (6, 8). Similar to IKK2, IKK1 has also been reported to have additional NFκB independent substrates. Like IKK2, IKK1 also phosphorylates β-catenin (15). Interestingly, while IKK2-induced phosphorylation negatively regulates β-catenin activity, phosphorylation by IKK1 increases β-catenin-dependent transcription, indicating distinct biological roles for these kinases. Many other IKK1 substrates localize to the nucleus, including the coactivators SMRT, SRC3, and CBP, as well as the histone H3, and they are thought to regulate cell proliferation in an NFκB-independent manner (16–18).
JNK
JNK is a member of the MAPK family of signaling proteins (19, 20). It consists of 10 JNK isoforms, which are derived through alternative splicing of mRNA transcripts generated from the three genes JNK1, JNK2, and JNK3. It is activated in response to cellular stress, cytokines, pathogens, and mitogens. JNK activation leads to the phosphorylation of a large number of transcription factors, the most prominent of which is c-Jun, a component of AP-1, as well as of numerous cellular proteins most of which are associated with apoptosis (such as Bcl2 and p53). Gene disruption in mice revealed its essential role in TNFα-induced c-Jun phosphorylation and AP-1 transcription factor activity. JNK is activated through phosphorylation of the Thr-Pro-Tyr motif in its activation loop by MAPK kinases (MAPKKs) MKK4 and MKK7. Activation of these kinases in turn is initiated by a cascade of kinases linked through stimulus-dependent association with different scaffold proteins (see below). The complex regulation of JNK is indicative of its critical role in multiple physiological processes. Indeed, JNK has been implicated in the regulation of cell survival and apoptosis, inflammation, metabolism, and development. By phosphorylating several pro-apoptotic members of the Bcl2-related protein family (Bim, Bmf, Bad) and through JNK-dependent activation of AP-1, it can activate the mitochondrial apoptotic pathway, while its phosphorylation of several anti-apoptotic members (Bcl2, Bcl-xL, Mcl-1) promote cell survival. In addition, JNK has been implicated to play important roles in other types of death including necrosis and autophagic/lysosomal cell death. JNK is also a potent inducer of inflammatory gene expression primarily through the activation of the transcription factors AP-1 and ATF-2. Accordingly, many autoimmune diseases (rheumatoid arthritis, multiple sclerosis, psoriasis) correlate with hyperactivity of JNK. The activation of JNK causes insulin resistance at least in part through the phosphorylation of Insulin receptor substrate-1 (IRS-1) at the inhibitory site Ser-307, thus suppressing insulin receptor signaling, indicating its role in metabolism (19–21).
p38
p38 is another member of the MAPK family. It consists of four isoforms, p38α, p38β, p38γ, and p38δ, each encoded by a distinct gene. They have similar in vitro substrate specificity but differ in regards to how their activity is regulated or their responsiveness to ligands; the p38 pathway is activated by a large number of growth factors, GPCR agonists, environmental stresses, pathogens, and cytokines. Dependent on the isotype, they are activated by dual kinases (threonine and tyrosine) MKK3, and the JNK kinases MKK4 and MKK7. Once activated, p38 phosphorylates several cytoplasmic substrates (including MBP, HSP27, and MAPKAPK2) and upon nuclear translocation activates the transcription factor ATF-2 through direct phosphorylation (22–24) as well as CREB (25). p38 can also indirectly regulate transcription through AP-1. One of the main consequences of p38 activation is increased expression of cytokines and receptors involved in inflammation and immunity (19, 21, 26). However, it remains unclear whether the aforementioned transcription factors account for p38’s function in gene expression or another reported p38 function in stimulus-induced mRNA stabilization (27). In addition, p38 is involved in inflammation, cell death, cell cycle regulation, and cell differentiation. Inhibition of p38 using specific pharmacological inhibitors results in reduced proliferation and cell cycle arrest (19, 21, 26).
ERK
ERKs are encoded by two genes, ERK1 (MAPK3), and ERK2 (MAPK1), that encode two main proteins, p44 (ERK1), and p42 (ERK2). They are preferentially activated by mitogens but, similar to JNK and p38, are also activated in response to GPCR agonists, cytokines, and environmental stress. Activation occurs through the Raf/MEK/ERK signaling cascade through different isoforms of the small GTP-binding protein Ras. Activated Raf binds to and phosphorylates MEK1 and MEK2, which in turn phosphorylate ERK1/2. Markedly, ERK1/2 have been reported to phosphorylate more than 160 substrates, including numerous important transcriptional regulators such as NF-AT, Elk-1, c-Fos, c-Myc, and H3 to control the cell cycle progression and cell death. Apart from ERK’s critical role in cell proliferation, it is also critically involved in differentiation, migration, and cellular transformation (28, 29).
Activation of signal transducers downstream of specific TNFRSF members
The key kinases are connected to members of the TNFR superfamily via complex signaling mechanisms that involve a multitude of protein adapters and enzymatic functions. Their cooperative or sequential regulation control dose response and dynamic behavior of the key kinases. Therefore, knowledge about the signaling molecules involved in the activation of the key signaling kinases and their regulation is of major importance to gain insights into how signaling specificity may be achieved. Below we detail what is currently known about these signaling mechanisms for three TNFRSF members (Fig. 2).
Fig. 2. Activation of signal transducers downstream of TNFR1, CD40, and LTβR engagement.

(A) TNF triggers the assembly of a signaling complex involving the TRADD, RIPK1, and ubiquitin ligase complexes. TAK1 is recruited and activated by binding of its scaffolds TAB 2/3 to polyubiquitin chains. Subsequent binding of NEMO to ubiquitin chains allows for the activation of IKK2. MAP3Ks activate MAPKs through a cascade of phosphorylation events. (B) CD40 engagement triggers the assembly of a complex containing TRAF2, TRAF3, TRAF6, cIAPs, MEKK1, and the LUBAC complex to activate MAPKs, NEMO-IKK2, and non-canonical IKK1. (C) Binding of LTβ to the LTβR triggers degradation of TRAF2 and TRAF3 resulting in NIK stabilization, which activates non-canonical IKK1 (see text for details).
TNFR1
Activation of TNFR1 triggers a wide variety of biological responses from cell death to survival, effects on metabolism, differentiation, adhesion, and motility. TNFR1 is particularly important in inflammatory responses (2). TNFR1 knockout mice show impaired clearance of bacterial pathogens and resistance to LPS (30–34). Although lymph nodes develop normally, these mice show defects in germinal center formation and impaired development of Peyer’s patches, indicating a critical role of TNFR1 in acute immune responses (30, 31). TNFR1 mediates its physiological functions through activation of canonical IKK, JNK, MAPK, and ERK.
Upon binding of TNF to the TNF receptor, the first steps in signaling are the recruitment of the adapter molecules TRADD and RIPK1 and then of TRAF2/5 to TRADD (35). TRADD is essential for TNF-induced signaling in MEFs as in TRADD-deficient MEFs TNFR-induced IκBα phosphorylation and degradation are completely abolished (36–38). In contrast, in macrophages the TRADD requirement is less complete, suggesting that in these cells TRAF2 may be recruited in a TRADD-independent manner (38). The requirement for RIP1 varies between cell types; while largely dispensable in MEFs (39–41), it is absolutely required for TNF-induced NFκB activation in T and B cells, as human T cells lacking RIPK1 and pre-B-cell lines derived from RIPK1 knockout mice show defects in NFκB activity (42–44). Notably, its kinase activity is not required (39). TRAF2-deficient mice show only a slight reduction in TNFR-induced NFκB activation, while TRAF2/5 double deficiency results in complete impairment, suggesting redundant functions for these molecules (39). However, this topic remains controversial.
Subsequently, cIAP1/2 are recruited to the complex, which is not only important for NEMO-containing IKK activation (45–47) but also for inhibition of the non-canonical IKK1 activity, as these E3 ligases are also involved in constitutive degradation of NIK (48, 49) (see below). Within this complex, RIPK1, TRAFs, and NEMO undergo several forms of ‘non-destructive’ ubiquitination, primarily conjugation of K63 or head-to tail (linear) polyubiquitin chains. While K63 ubiquitination of RIPK1 and NEMO is catalyzed by the E3 ligases cIAP1/2 or TRAF2 together with Uev1A and Ubc13 (44, 45, 50), the LUBAC complex, consisting of HOIL-1, HOIP, and SHARPIN, exclusively modifies RIPK1 and NEMO with linear polyubiquitin chains (51–54). Conjugation of K63 and linear ubiquitin chains to RIP1 triggers the recruitment of IKK activating kinases. First, the IKK-kinase TAK1 (IKK-K) is recruited by specific binding of its scaffolds TAB2/3 to the ubiquitin chains (39, 50, 55). TAK1 itself is K63 ubiquitinated, which triggers its autophosphorylation and activation (55–57). In vitro unanchored K63 polyubiquitin chains have been shown to be sufficient for TAK1 activation (58), but whether conjugation of ubiquitin chains to components of the complex may contribute to dose-responsiveness or specificity in vivo remains to be investigated.
Binding of NEMO to these ubiquitin chains brings IKK in close proximity to TAK1, allowing for IKK activation by phosphorylation of the activation loop serines (50–52, 59). TAK1 appears to be the major kinase responsible for IKK activation, as TAK1-deficient cells are largely defective in TNF-induced NFκB activation (60, 61). However, MEKK3 has also been reported to act as an IKK kinase. Similar to TAK1, MEKK3 binds to RIPK1 and TRAF2 and can phosphorylate IKK2 (62). Consistently, MEKK3 deficiency impairs IKK and MAPK activation in fibroblasts (62). Whether MEKK3 can replace TAK1 or whether they act cooperatively is currently not clear. Activation of the NEMO-IKK complex results in phosphorylation and degradation of the canonical IjBs and NFκB activation.
In addition to the activation of IKK, TNFR1 also leads to the potent activation of JNK, p38, and ERK1/2. Activation of JNK depends on TRAF2 and on the formation of ‘non-destructive’ K63 ubiquitin chains (63–70). The involvement of linear chains is still controversial (53, 54, 71). Activation of p38 similarly depends on TRAF2 and RIPK1; however, the contribution of ubiquitin chains is unclear (72, 73). Several MAP3Ks have been implicated in the activation, including TAK1 (74), MEKK1 (74), TPL-2 (75), and ASK1 (74, 76, 77). Genetic deletions of MAP3Ks have not revealed an absolute requirement for any kinase, indicating some redundancy in TRAF2-dependent JNK and p38 activation. MAP3Ks then phosphorylate and activate MAP2Ks, which are somewhat pathway specific. While MKK3 and MKK6 specifically phosphorylate and activate p38, MKK7 activates JNK. MKK4 is less specific and can activate both p38 and JNK (reviewed in 78). The main activator of the TNF-induced ERK1/2 cascade is TPL-2 (79). Macrophages from Tpl2 knockout mice are defective in ERK1/2 activation induced by TNF, while activation of other MAPKs and NFκB remain unaffected (79). In contrast, TNF stimulation of Tpl2-deficient MEFs results in defective activation of JNK, p38, ERK1/2 as well as NFκB (75), indicating a cell type-specific role for TPL-2.
CD40
CD40 is expressed on DCs, B cells, and endothelial cells. In DCs, CD40 signaling promotes cytokine production, induction of costimulatory molecules, and facilitates cross presentation; in B cells, CD40 signaling can promote germinal center formation, isotype switching, somatic hypermutation, and formation of plasma cells and memory B cells. In addition, it has been demonstrated to be important for the survival of many cell types, including GC B cells, DCs, and endothelial cells. CD40-deficient mice show defective B-cell development, Ig class switching, and GC formation, ultimately causing immunodeficiency (80–83). In similarity to TNFR1, CD40’s physiological functions are also mediated by the signal transducers IKK1, IKK2, JNK, p38, and ERK.
In contrast to TNFR1 signaling, activation of CD40 signaling does not require the adapter TRADD but instead is initiated by binding of ubiquitin ligases TRAF2, TRAF3, and TRAF6 directly to the receptor (84, 85). In the case of CD40 activation, the TRAF molecules do not appear to have redundant functions. TRAF2 has been shown to be the primary mediator of JNK and p38 activation (67, 68, 86). Engagement of TRAF2 to CD40 results in the recruitment of MEKK1 (87), which drives the phosphorylation of JNK and p38. TRAF2-deficient fibroblasts are defective in CD40L-induced JNK and p38 activation, with only little defect in NFκB activation (67, 68, 86). Attenuation of MAPK activation was also observed in TRAF2-deficient B cells (88). In addition, TRAF2 deficiency caused constitutive p100 processing and elevated c-Rel activity (88). This reflects the fact that in contrast to TNFR1, CD40 engagement also triggers the activation of non-canonical IKK through NIK (89). In unstimulated cells, NIK expression is very low due to its constitutive degradation by cIAPs, TRAF2, and TRAF3 (48, 49). CD40 engagement triggers self-degradation of TRAF2 and cIAP-dependent degradation of TRAF3, resulting in stabilization and accumulation of NIK (90–93). Indeed, germ line inactivation of either TRAF2 or TRAF3 leads to NIK accumulation and constitutive p100 processing, indicating that NIK accumulation is sufficient for its activation of non-canonical NFκB pathway (94–96). It subsequently activates IKK1 through phosphorylation of activation loop serines. NIK and IKKa phosphorylate p100 resulting in its processing and induction of NFκB transcription (97–99).
Binding of TRAF6 to CD40 is important for activation of JNK and p38 activation as well as NFκB (100). Its deficiency results in reduction or abrogation in the activation of canonical NFκB, JNK, and p38 (101, 102). Similar to TNFR signaling, the cIAP1/2 and LUBAC ubiquitin ligase complexes have also been shown to be recruited to the CD40 receptor (103–105). Whereas TNFR1 signaling involves conjugation of K63 and linear Ub-chains to recruit RIPK1 and NEMO and mediate activation of TAK1 and downstream MAPK and canonical IKKs (44, 45, 50–54), CD40 signaling requires ubiquitination of TRAF2 and TRAF6 for these downstream signaling events. Interestingly, K63 ubiquitination appears to be critical for JNK and p38 activation and less important for CD40-induced NFκB activation, as B cells and macrophages from ubc13–/– mice, which are defective in catalyzing K63 chains, show strong defects in JNK and p38 activation, while NFκB activation is largely intact (105). In contrast, interfering with components of the LUBAC complex results in defective NFκB and JNK activation (52, 54), indicating differential requirements of Ub chains for MAPK and NFκB activation. A more detailed understanding of the nature of Ub-chains important for signaling, the proteins they are conjugated to (if any) and the kinetics of Ub-chain formation will be important for a better understanding of signaling specificity.
In similarity to TNFR1 signaling, both TAK1 and MEKK3 are implicated as the activating kinases for canonical IKK by CD40 ligand engagement (57, 106, 107). Several kinases have been suggested to mediate activation of MAPK pathways. As mentioned above, MEKK1 is recruited to the CD40 complex through its interaction with TRAF2 to induce JNK and p38 activation (87). Accordingly, in MEKK1-deficient B cells no JNK and p38 activation can be detected, while there are conflicting results on the effects on NFκB activation (108–111). Several other MAP3Ks have been suggested to be important for JNK and p38 activation, including TAK1, MEKK3, and TPL-2. Although TAK1 is critical for TNF, BCR, and TLR ligand induced activation of MAPKs, it appears to only play a minor role in CD40-induced signaling, as CD40 engagement in TAK1-deficient B cells shows only modest defects in JNK and p38 activation (61). In overexpression experiments MEKK3 has been demonstrated to be able to induce JNK and p38 activation (112), due to embryonic lethality the physiological relevance for CD40-induced MAPK activation however remains unclear (113). Interestingly, the MAP3K TPL-2 does not appear to be involved in JNK, p38, or NFκB activation in CD40 stimulated B cells (79) but instead plays a critical role in the activation of the ERK pathway (79). Tpl2-deficient mice show a partial activation defect of ERK in response to CD40 and TLR activation resulting in partial inhibition of IgE production (79, 114). TPL-2 has also been implicated to be involved in processing of NFκB1 p105 (115); however, processing appeared to be normal in Tpl2 knockout mice (79).
LTβR
LTβR signaling primarily controls the development of secondary lymphoid organs (lymph nodes and Peyer’s patches). LTβR-deficient mice lack all lymph nodes, Peyer’s patches, and display a disturbed splenic architecture (116–118). In adults, LTβR also controls the maturation and maintenance of the microarchitecture of lymphoid organs through expression of specific chemokines (such as CXCL13 and CCL19), which induce stromal cell differentiation (117, 119).
In contrast to CD40 signaling, which leads to the activation of MAPKs, the canonical NEMO-IKK2 complex and non-canonical-NIK-IKK1, LTβ, and BAFFR only induce NEMO-independent, non-canonical IKK1. In similarity to CD40, the cytoplasmic domain of LTβR associates with TRAF3 and TRAF2 (120, 121). Ligation of LTαβ or LIGHT to the LTβ receptor triggers the degradation of TRAF2 and TRAF3 resulting in accumulation of NIK and subsequent activation of IKK1 (see CD40 signaling). Through IKK1-induced degradation and processing of p100 RelA:p50 and RelB:p52 dimers are released into the nucleus to activate transcription (122–124). NFκB activation induced by developmental signals such as LTβ is considerably slower and weaker as compared to inflammatory signals. LTβR engagement also weakly induces JNK activation in a TRAF2-dependent manner (125, 126); the exact mechanism however remains elusive.
Mechanisms that ensure signaling specificity
As described above, TNFRSF signal transduction pathways involve a small number of signaling enzymes, yet their biological responses are highly diverse. Over the past few years, many studies using a variety of genetic tools have revealed that the functional requirements of specific signaling proteins in signal transduction are cell type specific. Cell type-specific genetic requirements may be the result of differential expression of parallel, potentially compensating pathway components. However, it may also be the result of cell-type specific expression of proteins that modulate the function or kinetics of the key signaling enzymes that thereby control signaling specificity.
Broadly, the specificity of signaling can be controlled at the level of substrate specificity of the key kinases, differential wiring of signaling inputs through scaffolding, the kinetics of the kinase activities, as well as through combinatorial and temporal control mechanisms. Below we discuss some of these mechanisms of regulation and how they may contribute to signaling specificity.
Regulation of the enzymatic substrate specificity of kinases
MAPKs and IKKs are highly pleiotropic kinases, involved in numerous distinct biological responses by phosphorylating a diverse array of substrates. MAPKs have been estimated to have as many as 200–300 substrates each (127–129). One mechanism by which specificity may be ensured in vivo may involve co-factors that alter the intrinsic enzymatic specificity. However, unlike other enzymes such as prokaryotic RNA polymerases whose DNA binding specificity is regulated by sigma factors, surprisingly little information has emerged about specificity factors for the pleiotropic kinases in the TNFR signaling network.
MAPKs are known to have substrate binding sites, usually referred to as docking domains (130–132). These domains are distinct from the serine/threonine phospho-acceptor sites and consist of positively and negatively charged residues. Docking interactions themselves can be regulated by post-translational modifications (130–132). Apart from regulating interactions with substrates, binding can trigger allosteric conformational changes that can in turn affect strength and duration of MAPK signaling (133). However, not all known substrates have docking sites, suggesting that additional mechanisms are likely to exist in order to achieve substrate selectivity to ensure efficient and specific signaling (127).
IKK1 and IKK2 are also known to be pleiotropic kinases that can phosphorylate a wide range of substrates influencing diverse cellular responses (134, 135). Interestingly, in in vitro assays IKKs do not show a high degree of substrate specificity, and in contrast to MAPKs, IKKs do not appear to have docking sites raising the question of how their activity is regulated to ensure signaling specificity. To understand the molecular basis for signaling specificity in cellular pathways, it will be of major importance to further elucidate mechanisms that control substrate specificity in vivo.
Signaling specificity via scaffolding
When analyzing signaling responses to diverse biological inputs, it becomes clear that many signal transducers are shared, and, as described above, their substrate specificities appear limited. Yet, the biological outputs are highly specific. Signaling specificity can be achieved by organizing discrete subsets of proteins in space and time. One way to achieve this is by sequestering functionally interacting proteins into specific subcellular compartments, such as organelles or the plasma membrane. Another prevalent strategy is the assembly of functionally interacting proteins into specific complexes through protein scaffolds. Scaffolds bind to two or more components of a cascade, bringing them in close proximity, thereby not only facilitating efficient propagation of the signal but also mediating its insulation from other signals. In addition to organizing signaling molecules into signalosomes, scaffolds can also have allosteric effects on the kinases thus regulating kinase activity itself.
The most prominent signaling cascades that are regulated by scaffolds are the MAPK pathways. As mentioned above, initiation of JNK and p38 pathways is triggered by many MAP3Ks including TAK1, ASK1, MEKKs, and TPL-2 (74–77), which, among others, can phosphorylate MKK4 and MKK7 to activate JNK, and MKK3, MKK4, and MKK6 to activate p38 (78). The usage of alternative kinases at each step of the cascade might allow for the precise stimulus-dependent control of MAPKs. Indeed, the stimulus-specific organization of different kinases into cascades by scaffolds can create functional signaling modules to control specificity of signal transduction (Fig. 3). For example, filamin has been identified as a scaffold that may coordinate the specific activation of JNK upon stimulation with TNF (136). The actin binding protein interacts with the TNFR1 adapter TRAF2, the MAP2K MKK4, and JNK, thereby coordinating a module that channels the upstream TNFR1 signal to JNK (137). Accordingly, filamin deficiency causes a selective loss of TNF-induced JNK activation (136). The dual specificity protein phosphatase SKRP1 has also been reported to be involved in TNF-induced JNK activation through a scaffolding function facilitating the interaction of the MAP3K ASK1, MAP2K MKK7, and JNK (138–141). Similar examples for a stimulus-specific organization of MAPK cascades by scaffolds can be found for the activation of p38. The osmo-sensing scaffold for MEKK3 (OSM) for example associates with Rac, MEKK3, and MKK3 to regulate p38 activation specifically in response to osmotic shock (142). Other p38-specific scaffolds include JIP4 (143) and JLP1, which is implicated in p38-dependent regulation of differentiation (144–146). Interestingly, the MAP3K MEKK1 not only acts as a kinase to activate JNK upon CD40 ligation, but itself possesses a scaffolding function by interacting with components of the JNK module (108, 147–151). It also constitutively interacts with Raf-1, MEK1, and ERK2 to facilitate stimulus-induced activation of ERK1/2 (147, 152, 153). TAK1, which is involved in transducing signals from diverse upstream receptors (e.g. TNFRs, TLRs, NLRs) to activate downstream JNK, p38, as well as NFκB pathways, associates with the scaffolds TAB1, TAB2, and TAB3. Apart from mediating TAK1 activation (see below), through binding to poly-ubiquitin chains, TAB2/3 also recruit TAK1 to NEMO-IKK2 to mediate NFκB activation.
Fig. 3. Scaffolding to achieve signaling specificity.

Scaffolds can organize pleiotropic kinases into stimulus-specific signaling modules to direct them to one specific pathway. (A) Filamin interacts with TRAF2, MEKK4, and JNK to allow for JNK activation upon TNFR1 engagement. ERK1/2 and p38 might similarly be organized into stimulus-specific modules through yet unidentified scaffolding proteins. (B) Binding of NEMO to IKK2 allows for its activation but is also required for proper signaling propagation to IκBα in response to inflammatory signals. (C) NIK is not only the activating kinase of IKK1 but also channels IKK1 activity to p100.
A well-known scaffold necessary for the activation of the canonical IKKs is NEMO (154–156). Binding of NEMO to stimulus-induced polyubiquitin chains brings IKKs in close proximity to each other (allowing for trans-autophosphorylation) and to the IKK-kinase TAK1, which then activates IKK2 through phosphorylation of its activation loop serines (39, 50–52, 55, 59). Thus, NEMO links upstream inflammatory signals to IKK2 through binding to ubiquitin chains. How is active IKK2 able to phosphorylate IjBs so efficiently and rapidly while phosphorylation of alternative substrates is much weaker and occurs with much slower kinetics? Our unpublished data suggest that IKK2-dependent phosphorylation of IjB is also mediated by NEMO (BS and AH, unpublished data). Binding of NEMO to IjBs specifically channels IKK2 kinase activity towards inflammatory responses. Thus, scaffolding of NEMO ensures downstream signaling specificity by directing the pleiotropic IKK2 kinase to the canonical NFκB pathway. The complex between NEMO-IKK2 and IκBα appears to be constitutive, which might not only ensure specific phosphorylation of IjBs but also enables the rapid response, necessary for efficient inflammatory signal transduction. In analogy to stimulus-specific MAPK kinase modules, NEMO assembles an inflammation-specific IKK module by linking upstream IKK activators with the downstream effector IκBα. The specificity of IKK1 for p100 also seems to be regulated through scaffolding. NIK forms a complex with IKK1 and p100, thereby not only activating IKK1 but also directing IKK1s kinase activity towards p100. Thus, active IKK1 will only induce p100 processing when NIK is stabilized by specific developmental signals (e.g. LTβ, CD40) (99).
In summary, pathway-specific scaffolds can direct a pleiotropic kinase to one specific pathway and at the same time prevent the activation of pathways irrelevant to that particular stimulus. Thus, scaffolds ensure stimulus-specific functions of kinases. The identification of scaffolds that are specific for each TNFR superfamily member will be critical to gain a better understanding about the regulation of signaling specificity.
Combinatorial coding
Signaling specificity may also be mediated through the combinatorial use of several pleiotropic signal transducers (Fig. 4). In this scenario, stimulation of a single receptor triggers the formation of a signalosome that allows for the activation of a subset of signal transducers. Downstream effectors (e.g. the promoters of potential target genes) may integrate the combinations of signal transducers (e.g. transcription factors) to determine the stimulus-specific activity level (e.g. of target genes).
Fig. 4. Combinatorial control of kinases facilitates stimulus specificity.
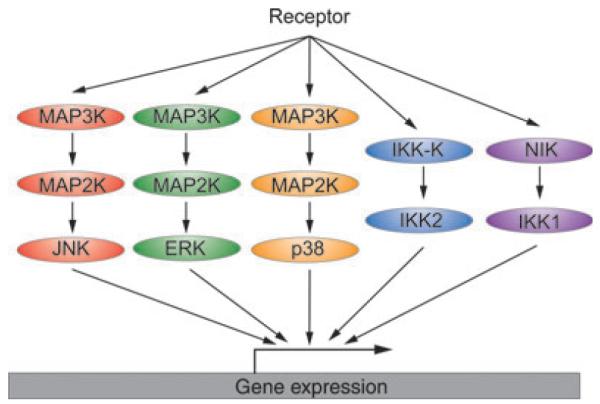
Engagement of TNFRSFs activates pathways leading to the activation of ERK, JNK, p38, IKK2, and IKK1. Phosphorylation of downstream effectors induces a transcriptional program. Some genes require a synergistic function of several transcription factors and may thus be transcribed in a stimulus-specific manner. Specificity can also be achieved post-transcriptionally through p38-dependent regulation of mRNA stability.
Because they lack intrinsic enzymatic activities, TNFRs associate with TRAF proteins to initiate intracellular signaling. TRAF protein recruitment forms the basis for the encoding of a combinatorial code by which a receptor-associated complex leads to the activation of several pleiotropic kinases. The family of TRAF proteins consists of 6 members, TRAF1-6, that have both overlapping as well as distinct roles. The TRAF proteins with which the receptor associates at least in part determines the pathways that are activated. TRAFs are not only critically involved in signaling of TNFR superfamily members but also play important roles in TLR/IL-1R signaling, where TRAF3 is indispensable for IRF3 activation upon TLR3 engagement (157), or TRAF6 is necessary for activation of NFκB by LPS (101, 158). TRAFs have a RING-finger domain (RING) with an associated E3 ligase activity, a Zinc-finger (ZF) motif, and a highly conserved C-terminal domain that mediates homo- and heterodimerization of TRAFs as well as association with cell surface receptors (85, 159).
The association of TRAF2 to CD40 is important for activation of MAPKs and to a lesser extent for the activation of canonical IKKs (67, 68, 86). Accordingly, TRAF2 deficiency results in defective JNK activation and shifts the balance of signaling towards non-canonical IKK, despite a defect in TRAF3 degradation (86, 88). This observation highlights the importance of TRAF2 in negatively regulating the activation of non-canonical IKKs. In B cells, TRAF3 deficiency similarly results in enhanced activation of NIK-IKK but also in enhanced canonical IKK and JNK activity (48, 49, 159, 160). Thus, although both TRAF2 and TRAF3 are important negative regulators of non-canonical IKK, they have different functions in regulating JNK activity. TRAF6 is dispensable for TNFR1 and LTβ signaling but is essential for CD40-induced JNK, p38, ERK1/2, and canonical IKK activation (84, 85).
For activation of downstream kinases, TRAFs do not just act as adapters or scaffolds, but their E3 ubiquitin ligase activity is required. In conjunction with Uev1A and Ubc13, TRAFs mediate the conjugation of K63-linked ubiquitin chains onto other TRAFs, RIPK1, and NEMO, which has been shown to be critical for activation of NEMO-IKK2 and JNK by TNF as well as CD40L (44, 50, 70, 103–105). In fact, non-destructive ubiquitin chains (such as K63 and linear chains) might add an additional level of regulation to achieve signaling specificity. The type of ubiquitin chain as well as the molecules they are conjugated onto are likely to be stimulus specific. Interestingly, TNF and IL1β signaling appear to have differential requirements for ubiquitin chain types (161). While for IL-1β-induced IKK activation the Uev1A/Ubc13/TRAF2 E3 ligase complex that is restricted to K63 chain linkage is necessary for IKK activation, TNF-induced signaling does not require catalysis of K63 chains (161). Recently, mass spec analysis revealed that RIPK1 and NEMO are heavily ubiquitinated with K63, linear and K11-linked chains upon stimulation with TNF, indicating the high level of diversity in ubiquitin modifications that occur during signaling (54). More work is required to get a better understanding of the stimulus specific role of ubiquitin chains. The use of ubiquitin-chains as scaffolds allows for highly flexible and dynamic assembly of diverse signaling complexes but also for the recruitment of specific activators and inactivators. Binding of the scaffolds TAB 2/3 to polyubiquitin chains for example recruits the MAP3K TAK1 to the TNFR1 complex to allow for IKK2 activation, which in turn also depends on the ability of its scaffold NEMO to bind to ubiquitin chains (39, 50–52, 55, 59). In addition to coordinating a signaling cascade, one can imagine that ubiquitin chains may branch one upstream signal to multiple downstream kinases through association of distinct ubiquitin binding proteins to different types of polyubiquitin chains, thereby playing a key role in encoding a combinatorial signaling code.
The decoding of combinatorially coded signals has been studied primarily at the level of gene expression. Gene promoters or enhancers often contain binding sites for several signal-responsive transcription factors that are the down-stream effectors of pleiotropic kinases. Synergistic function by several transcription factors has been documented in synthetic experimental systems (162). The multivalent interactions by the ubiquitous co-activators CBP/p300 suggest a possible decoding mechanism in vivo. For some specific genes, a combinatorial requirement has been established and examined at a mechanistic level. The classic example is the control of the IFNβ enhancer, which requires the activity of AP-1, IFN response factor 3 (IRF3), and NFκB. A nucleosome located over the transcription start site was identified as the block for transcription initiation, and only the assembly of an ‘enhanceosome’ (163) allowed for the recruitment of chromatin remodeling factors that in turn allow for pre-initiation complex assembly (164). Interestingly, although AP-1 and NFκB are separated by four IRFs on the enhancer, protein-protein interactions between them were found to be critical (165). These are facilitated by HMG proteins capable of inducing DNA bends (166). Similarly, expression of the chemokine MCP1/CCL2 requires communication between a proximally bound SP-1 and a distally bound NFκB that suggests chromosome looping (167). A different mechanism of combinatorial control has been proposed for the TNF gene. Expression of TNF requires NFκB binding to its promoter but also ERK and p38 signals to control mRNA nuclear-cytoplasmic transport, mRNA stability, and translation (114, 168, 169). These examples indicate different mechanisms in the gene regulatory network may be targeted by the combinatorial control of TNFRSF-induced intracellular signals.
Temporal coding
Biochemical cell population timecourse studies and single cell studies with in vivo reporters have revealed that the activity profile of TNFRS-induced transcription factors is dynamic and that the observed temporal profiles are stimulus-specific (170, 171). These observations led to the proposal that stimulus-specific temporal control of pleiotropic transducers may allow for stimulus-specific signaling (7). The temporal code may be encoded via receptor-associated mechanisms, transmitted via stimulus-specific temporal control of a pleiotropic transducer, and decoded by mechanisms associated with the effector, for example the gene regulatory network controlling the expression of a target gene.
Evidence for the temporal coding model for generating stimulus-specific signaling can be found in JNK, p38, and ERK signaling (170, 172). A prominent example demonstrating the importance of temporal control in MAPK signaling is the neuronal cell line PC12, in which ERK signaling can induce proliferation (in response to EGF), as well as differentiation (in response to NGF) depending on the duration of the signal (173). While EGF induced MAPK signaling is transient, MAPK activity induced by NGF is sustained. Extensive evidence for the importance of temporal coding also comes from canonical NFκB signaling. Quantitative measurements of the canonical IKK activity revealed that its induced activity profile is stimulus-specific (171). Whereas TNF activates a transient IKK activity, LPS leads to prolonged IKK activity. In the former case, the negative feedback regulator A20 limits late IKK activity, whereas in the latter, cytokine feedback via TNF ensures an elevated late phase. Within the TNFRSF network, temporal control of effectors is mediated by the kinetics of key reaction mechanisms, such as receptor internalization, recycling, and replenishment, ubiquitin chain formation rates and their degradation through deubiquitin enzymes (DUSPs) such as A20, nuclear translocation rates, and negative feedback mechanism impacting IKK or NFκB(Fig. 5).
Fig. 5. Temporal control can facilitate stimulus specificity.
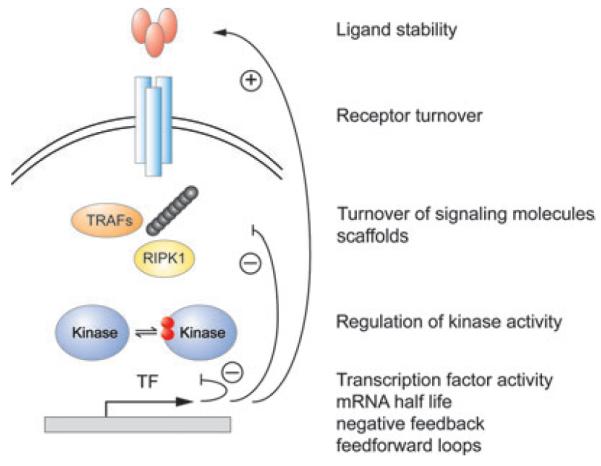
The temporal profile of key signal transducers, whether pleiotropic kinases or transcription factors, may determine signaling specificity. Temporal profiles are encoded and transduced through kinetic mechanisms that control receptor internalization and recycling, half-life control of signaling mediators, including ubiquitin chain second messengers or scaffolds, as well as negative and positive feedback. Signaling dynamics may be decoded to determine the activity of effectors, such as downstream genes, by mRNA half-life control, cooperativity with other transcription factors or chromatin regulatory mechanisms.
How may target genes distinguish between different temporal profiles of NFκB activity? We may imagine a variety of mechanisms by which the temporal code may be decoded by gene regulatory networks (GRNs). At its simplest, short versus long mRNA half-life may sensitize a gene to transient versus prolonged NFκB activity. Coupled to non-linear or thresholded dose response curves of promoters, the GRN may decode complex temporal profiles of NFκB in specific ways. In more sophisticated GRNs, NFκB may need to coincident with or phase shifted relative to other transcription factors to allow for gene activation. On others yet, NFκB may be required to be coincident with a transcription factor induced by the early NFκB activity, forming a feed forward control loop.
Conclusion
The signaling network innervated by members of the TNFR superfamily consists of a number of kinases with highly pleiotropic functions. Generally, these kinases show too little intrinsic specificity in vitro to account for their apparent signaling specificity in cells, but several models have emerged that begin to explain the mechanisms by which signaling specificity is achieved. The most prominent of these may be the utilization of scaffolding proteins, which may recruit kinases to upstream activators or to downstream substrates. Indeed when such scaffolds are multivalent, the same protein may determine both upstream activators and downstream targets, thus placing the kinase within a cascade and effectively insulating it from other signaling pathways. Within the MAPK field, prominent examples of such scaffolds are the yeast Ste5/Pbs2 scaffold modules, and in mammals, the MAPK kinase cascades that are organized by various stimulus specific scaffolds. In NFκB signaling, NEMO appears to play a similar role (Schrö-felbauer et al. unpublished data). However, we expect that ongoing efforts to understand specificity of signaling within the TNFRSF network will lead to the identification of other such scaffolding proteins and the characterization of their roles in signaling insulation and as mediators of signaling crosstalk.
Acknowledgements
This work was supported by RO1 grant CA141722. BS is a Leukemia and Lymphoma Society fellow.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Aggarwal BB. Signaling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 2.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 3.Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci. 2002;27:19–26. doi: 10.1016/s0968-0004(01)01995-8. [DOI] [PubMed] [Google Scholar]
- 4.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 5.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 6.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;9:491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 7.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 8.Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NF-kappaB system for both canonical and non-canonical signaling. Cell Res. 2011;21:86–102. doi: 10.1038/cr.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia Y, Padre RC, De Mendoza TH, Bottero V, Tergaonkar VB, Verma IM. Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci USA. 2009;106:2629–2634. doi: 10.1073/pnas.0812256106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao Z, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277:48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- 11.Lee DF, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 12.Lamberti C, et al. Regulation of beta-catenin function by the IkappaB kinases. J Biol Chem. 2001;276:42276–42286. doi: 10.1074/jbc.M104227200. [DOI] [PubMed] [Google Scholar]
- 13.Comb WC, Cogswell P, Sitcheran R, Baldwin AS. IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene. 2011;30:1727–1732. doi: 10.1038/onc.2010.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Criollo A, et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010;29:619–631. doi: 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carayol N, Wang CY. IKKalpha stabilizes cytosolic beta-catenin by inhibiting both canonical and non-canonical degradation pathways. Cell Signal. 2006;18:1941–1946. doi: 10.1016/j.cellsig.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Hoberg JE, Yeung F, Mayo MW. SMRT derepression by the IkappaB kinase alpha: a prerequisite to NF-kappaB transcription and survival. Mol Cell. 2004;16:245–255. doi: 10.1016/j.molcel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Park KJ, Krishnan V, O’Malley BW, Yamamoto Y, Gaynor RB. Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell. 2005;18:71–82. doi: 10.1016/j.molcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26:75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang G, Shi LZ, Chi H. Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine. 2009;48:161–169. doi: 10.1016/j.cyto.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 21.Rincon M, Davis RJ. Regulation of the immune response by stress-activated protein kinases. Immunol Rev. 2009;228:212–224. doi: 10.1111/j.1600-065X.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- 22.Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 23.Livingstone C, Patel G, Jones N. ATF-2 contains a phosphorylation-dependent transcriptional activation domain. EMBO J. 1995;14:1785–1797. doi: 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995;14:1798–1811. doi: 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 26.Ashwell JD. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol. 2006;6:532–540. doi: 10.1038/nri1865. [DOI] [PubMed] [Google Scholar]
- 27.Winzen R, et al. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activatedprotein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death – apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 30.Pfeffer K, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 31.Rothe J, et al. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 32.Peschon JJ, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- 33.O’Brien DP, Briles DE, Szalai AJ, Tu AH, Sanz I, Nahm MH. Tumor necrosis factor alpha receptor I is important for survival from Streptococcus pneumoniae infections. Infect Immun. 1999;67:595–601. doi: 10.1128/iai.67.2.595-601.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deckert-Schluter M, Bluethmann H, Rang A, Hof H, Schluter D. Crucial role of TNF receptor type 1 (p55), but not of TNF receptor type 2 (p75), in murine toxoplasmosis. J Immunol. 1998;160:3427–3436. [PubMed] [Google Scholar]
- 35.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 36.Chen NJ, et al. Beyond tumor necrosis factor receptor: TRADD signaling in Toll-like receptors. Proc Natl Acad Sci USA. 2008;105:12429–12434. doi: 10.1073/pnas.0806585105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ermolaeva MA, et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol. 2008;9:1037–1046. doi: 10.1038/ni.1638. [DOI] [PubMed] [Google Scholar]
- 38.Pobezinskaya YL, et al. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat Immunol. 2008;9:1047–1054. doi: 10.1038/ni.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279:33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- 40.Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12:419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 41.Wong WW, Gentle IE, Nachbur U, Anderton H, Vaux DL, Silke J. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ. 2010;17:482–487. doi: 10.1038/cdd.2009.178. [DOI] [PubMed] [Google Scholar]
- 42.Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 43.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006;281:13636–13643. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 45.Bertrand MJ, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 46.Mahoney DJ, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varfolomeev E, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vallabhapurapu S, et al. Non-redundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zarnegar BJ, et al. Non-canonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adapters cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires sitespecific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 51.Tokunaga F, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 52.Haas TL, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 53.Ikeda F, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature. 2011;471:637–641. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerlach B, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 55.Kanayama A, et al. TAB 2 and TAB 3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 56.Scholz R, Sidler CL, Thali RF, Winssinger N, Cheung PC, Neumann D. Autoactivation of transforming growth factor beta-activated kinase 1 is a sequential bimolecular process. J Biol Chem. 2010;285:25753–25766. doi: 10.1074/jbc.M109.093468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, Matsumoto K. Role of the TAB 2-related protein TAB 3 in IL-1 and TNF signaling. EMBO J. 2003;22:6277–6288. doi: 10.1093/emboj/cdg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xia ZP, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 60.Fan Y, et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alphaand interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J Biol Chem. 2010;285:5347–5360. doi: 10.1074/jbc.M109.076976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sato S, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 62.Yang J, et al. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001;2:620–624. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- 63.Reinhard C, Shamoon B, Shyamala V, Williams LT. Tumor necrosis factor alpha-induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO J. 1997;16:1080–1092. doi: 10.1093/emboj/16.5.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Natoli G, et al. Activation of SAPK/JNK by TNF receptor 1 through a non-cytotoxic TRAF2-dependent pathway. Science. 1997;275:200–203. doi: 10.1126/science.275.5297.200. [DOI] [PubMed] [Google Scholar]
- 65.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 66.Song HY, Regnier CH, Kirschning CJ, Goeddel DV, Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-kappaB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc Natl Acad Sci USA. 1997;94:9792–9796. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee SY, Reichlin A, Santana A, Sokol KA, Nussenzweig MC, Choi Y. TRAF2 is essential for JNK but not NF-kappaB activationand regulates lymphocyte proliferation and survival. Immunity. 1997;7:703–713. doi: 10.1016/s1074-7613(00)80390-8. [DOI] [PubMed] [Google Scholar]
- 68.Yeh WC, et al. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 69.Habelhah H, Takahashi S, Cho SG, Kadoya T, Watanabe T, Ronai Z. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-kappaB. EMBO J. 2004;23:322–332. doi: 10.1038/sj.emboj.7600044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fukushima T, et al. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc Natl Acad Sci USA. 2007;104:6371–6376. doi: 10.1073/pnas.0700548104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tokunaga F, et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature. 2011;471:633–636. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- 72.Yuasa T, Ohno S, Kehrl JH, Kyriakis JM. Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. Germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase 1 and SAPK while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of MKK6 and p38. J Biol Chem. 1998;273:22681–22692. doi: 10.1074/jbc.273.35.22681. [DOI] [PubMed] [Google Scholar]
- 73.Lee TH, et al. The death domain kinase RIP1 is essential for tumor necrosis factor alpha signaling to p38 mitogen-activated protein kinase. Mol Cell Biol. 2003;23:8377–8385. doi: 10.1128/MCB.23.22.8377-8385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takaesu G, et al. TAB 2, a novel adapter protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 75.Das S, et al. Tpl2/cot signals activate ERK, JNK, and NF-kappaB in a cell-type and stimulus-specific manner. J Biol Chem. 2005;280:23748–23757. doi: 10.1074/jbc.M412837200. [DOI] [PubMed] [Google Scholar]
- 76.Ichijo H, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 77.Nishitoh H, et al. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 78.Wang X, Destrument A, Tournier C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta. 2007;1773:1349–1357. doi: 10.1016/j.bbamcr.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 79.Eliopoulos AG, Wang CC, Dumitru CD, Tsichlis PN. Tpl2 transduces CD40 and TNF signals that activate ERK and regulates IgE induction by CD40. EMBO J. 2003;22:3855–3864. doi: 10.1093/emboj/cdg386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 81.Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. 2004;22:307–328. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- 82.Danese S, Sans M, Fiocchi C. The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut. 2004;53:1035–1043. doi: 10.1136/gut.2003.026278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ha H, Han D, Choi Y. TRAF-mediated TNFR-family signaling. Curr Protoc Immunol. 2009;87:11.9D.1–11.9D.19. doi: 10.1002/0471142735.im1109ds87. [DOI] [PubMed] [Google Scholar]
- 85.Bishop GA, Moore CR, Xie P, Stunz LL, Kraus ZJ. TRAF proteins in CD40 signaling. Adv Exp Med Biol. 2007;597:131–151. doi: 10.1007/978-0-387-70630-6_11. [DOI] [PubMed] [Google Scholar]
- 86.Hostager BS, Haxhinasto SA, Rowland SL, Bishop GA. Tumor necrosis factor receptorassociated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem. 2003;278:45382–45390. doi: 10.1074/jbc.M306708200. [DOI] [PubMed] [Google Scholar]
- 87.Gallagher E, et al. Kinase MEKK1 is required for CD40-dependent activation of the kinases Jnk and p38, germinal center formation, B cell proliferation and antibody production. Nat Immunol. 2007;8:57–63. doi: 10.1038/ni1421. [DOI] [PubMed] [Google Scholar]
- 88.Grech AP, Amesbury M, Chan T, Gardam S, Basten A, Brink R. TRAF2 differentially regulates the canonical and non-canonical pathways of NF-kappaB activation in mature B cells. Immunity. 2004;21:629–642. doi: 10.1016/j.immuni.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 89.Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009;228:225–240. doi: 10.1111/j.1600-065X.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- 90.Brown KD, Hostager BS, Bishop GA. Regulation of TRAF2 signaling by self-induced degradation. J Biol Chem. 2002;277:19433–19438. doi: 10.1074/jbc.M111522200. [DOI] [PubMed] [Google Scholar]
- 91.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 92.Moore CR, Bishop GA. Differential regulation of CD40-mediated TNF receptor-associated factor degradation in B lymphocytes. J Immunol. 2005;175:3780–3789. doi: 10.4049/jimmunol.175.6.3780. [DOI] [PubMed] [Google Scholar]
- 93.Varfolomeev E, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 94.He JQ, et al. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med. 2006;203:2413–2418. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253–267. doi: 10.1016/j.immuni.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391–401. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 97.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 98.Senftleben U, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 99.Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKK-alpha) to p100 and IKKalpha-mediated phosphorylation. J Biol Chem. 2004;279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 100.Rowland SL, Tremblay MM, Ellison JM, Stunz LL, Bishop GA, Hostager BS. A novel mechanism for TNFR-associated factor 6-dependent CD40 signaling. J Immunol. 2007;179:4645–4653. doi: 10.4049/jimmunol.179.7.4645. [DOI] [PubMed] [Google Scholar]
- 101.Lomaga MA, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsuzawa A, et al. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science. 2008;321:663–668. doi: 10.1126/science.1157340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deng L, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 104.Hofmann RM, Pickart CM. In vitro assembly and recognition of Lys-63 polyubiquitin chains. J Biol Chem. 2001;276:27936–27943. doi: 10.1074/jbc.M103378200. [DOI] [PubMed] [Google Scholar]
- 105.Yamamoto M, et al. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat Immunol. 2006;7:962–970. doi: 10.1038/ni1367. [DOI] [PubMed] [Google Scholar]
- 106.Blonska M, You Y, Geleziunas R, Lin X. Restoration of NF-kappaB activation by tumor necrosis factor alpha receptor complex-targeted MEKK3 in receptor-interacting protein-deficient cells. Mol Cell Biol. 2004;24:10757–10765. doi: 10.1128/MCB.24.24.10757-10765.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 108.Yujiri T, et al. MEK kinase 1 gene disruption alters cell migration and c-Jun NH2-terminal kinase regulation but does not cause a measurable defect in NF-kappa B activation. Proc Natl Acad Sci USA. 2000;97:7272–7277. doi: 10.1073/pnas.130176697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xia Y, et al. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc Natl Acad Sci USA. 2000;97:5243–5248. doi: 10.1073/pnas.97.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee FS, Peters RT, Dang LC, Maniatis T. MEKK1 activates both IkappaB kinase alpha and IkappaB kinase beta. Proc Natl Acad Sci USA. 1998;95:9319–9324. doi: 10.1073/pnas.95.16.9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee FS, Hagler J, Chen ZJ, Maniatis T. Activation of the IkappaB alpha kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- 112.Blank JL, Gerwins P, Elliott EM, Sather S, Johnson GL. Molecular cloning of mitogen-activated protein/ERK kinase kinases (MEKK) 2 and 3. Regulation of sequential phosphorylation pathways involving mitogen-activated protein kinase and c-Jun kinase. J Biol Chem. 1996;271:5361–5368. doi: 10.1074/jbc.271.10.5361. [DOI] [PubMed] [Google Scholar]
- 113.Yang J, et al. Mekk3 is essential for early embryonic cardiovascular development. Nat Genet. 2000;24:309–313. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 114.Dumitru CD, et al. TNF-alpha induction by LPS is regulated post-transcriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103:1071–1083. doi: 10.1016/s0092-8674(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 115.Belich MP, Salmeron A, Johnston LH, Ley SC. TPL-2 kinase regulates the proteolysis of the NF-kappaB-inhibitory protein NF-kappaB1 p105. Nature. 1999;397:363–368. doi: 10.1038/16946. [DOI] [PubMed] [Google Scholar]
- 116.Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7:344–353. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 117.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 118.Gommerman JL, Browning JL. Lymphotoxin/light, lymphoid microenvironments and autoimmune disease. Nat Rev Immunol. 2003;3:642–655. doi: 10.1038/nri1151. [DOI] [PubMed] [Google Scholar]
- 119.McCarthy DD, Summers-Deluca L, Vu F, Chiu S, Gao Y, Gommerman JL. The lymphotoxin pathway: beyond lymph node development. Immunol Res. 2006;35:41–54. doi: 10.1385/IR:35:1:41. [DOI] [PubMed] [Google Scholar]
- 120.Force WR, Glass AA, Benedict CA, Cheung TC, Lama J, Ware CF. Discrete signaling regions in the lymphotoxin-beta receptor for tumor necrosis factor receptor-associated factor binding, subcellular localization, and activation of cell death and NF-kappaB pathways. J Biol Chem. 2000;275:11121–11129. doi: 10.1074/jbc.275.15.11121. [DOI] [PubMed] [Google Scholar]
- 121.Li C, et al. Structurally distinct recognition motifs in lymphotoxin-beta receptor and CD40 for tumor necrosis factor receptor-associated factor (TRAF)-mediated signaling. J Biol Chem. 2003;278:50523–50529. doi: 10.1074/jbc.M309381200. [DOI] [PubMed] [Google Scholar]
- 122.Basak S, et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mordmuller B, Krappmann D, Esen M, Wegener E, Scheidereit C. Lymphotoxin and lipopolysaccharide induce NF-kappaB-p52 generation by a co-translational mechanism. EMBO Rep. 2003;4:82–87. doi: 10.1038/sj.embor.embor710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: critical roles for p100. J Biol Chem. 2003;278:23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- 125.Kim YS, Nedospasov SA, Liu ZG. TRAF2 plays a key, non-redundant role in LIGHT-lymphotoxin beta receptor signaling. Mol Cell Biol. 2005;25:2130–2137. doi: 10.1128/MCB.25.6.2130-2137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chang YH, Hsieh SL, Chen MC, Lin WW. Lymphotoxin beta receptor induces interleukin 8 gene expression via NF-kappaB and AP-1 activation. Exp Cell Res. 2002;278:166–174. doi: 10.1006/excr.2002.5573. [DOI] [PubMed] [Google Scholar]
- 127.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 128.Chuderland D, Seger R. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol Biotechnol. 2005;29:57–74. doi: 10.1385/MB:29:1:57. [DOI] [PubMed] [Google Scholar]
- 129.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 130.Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000;2:110–116. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 131.Enslen H, Davis RJ. Regulation of MAP kinases by docking domains. Biol Cell. 2001;93:5–14. doi: 10.1016/s0248-4900(01)01156-x. [DOI] [PubMed] [Google Scholar]
- 132.Bardwell L, Shah K. Analysis of mitogen-activated protein kinase activation and interactions with regulators and substrates. Methods. 2006;40:213–223. doi: 10.1016/j.ymeth.2006.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Good M, Tang G, Singleton J, Remenyi A, Lim WA. The Ste5 scaffold directs mating signaling by catalytically unlocking the Fus3 MAP kinase for activation. Cell. 2009;136:1085–1097. doi: 10.1016/j.cell.2009.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 135.Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009;19:404–413. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 136.Marti A, et al. Actin-binding protein-280 binds the stress-activated protein kinase (SAPK) activator SEK-1 and is required for tumor necrosis factor-alpha activation of SAPK in melanoma cells. J Biol Chem. 1997;272:2620–2628. doi: 10.1074/jbc.272.5.2620. [DOI] [PubMed] [Google Scholar]
- 137.Leonardi A, Ellinger-Ziegelbauer H, Franzoso G, Brown K, Siebenlist U. Physical and functional interaction of filamin (actin-binding protein-280) and tumor necrosis factor receptor-associated factor 2. J Biol Chem. 2000;275:271–278. doi: 10.1074/jbc.275.1.271. [DOI] [PubMed] [Google Scholar]
- 138.Chen AJ, et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J Biol Chem. 2002;277:36592–36601. doi: 10.1074/jbc.M200453200. [DOI] [PubMed] [Google Scholar]
- 139.Zama T, Aoki R, Kamimoto T, Inoue K, Ikeda Y, Hagiwara M. A novel dual specificity phosphatase SKRP1 interacts with the MAPK kinase MKK7 and inactivates the JNK MAPK pathway. Implication for the precise regulation of the particular MAPK pathway. J Biol Chem. 2002;277:23909–23918. doi: 10.1074/jbc.M200837200. [DOI] [PubMed] [Google Scholar]
- 140.Zama T, Aoki R, Kamimoto T, Inoue K, Ikeda Y, Hagiwara M. Scaffold role of a mitogen-activated protein kinase phosphatase, SKRP1, for the JNK signaling pathway. J Biol Chem. 2002;277:23919–23926. doi: 10.1074/jbc.M200838200. [DOI] [PubMed] [Google Scholar]
- 141.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 142.Uhlik MT, et al. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol. 2003;5:1104–1110. doi: 10.1038/ncb1071. [DOI] [PubMed] [Google Scholar]
- 143.Kelkar N, Standen CL, Davis RJ. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol Cell Biol. 2005;25:2733–2743. doi: 10.1128/MCB.25.7.2733-2743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Takaesu G, et al. Activation of p38alpha/beta MAPK in myogenesis via binding of the scaffold protein JLP to the cell surface protein Cdo. J Cell Biol. 2006;175:383–388. doi: 10.1083/jcb.200608031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kang JS, et al. A Cdo-Bnip-2-Cdc42 signaling pathway regulates p38alpha/beta MAPK activity and myogenic differentiation. J Cell Biol. 2008;182:497–507. doi: 10.1083/jcb.200801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oh JE, et al. Cdo promotes neuronal differentiation via activation of the p38 mitogen-activated protein kinase pathway. FASEB J. 2009;23:2088–2099. doi: 10.1096/fj.08-119255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Minden A, et al. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266:1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 148.Su YC, Han J, Xu S, Cobb M, Skolnik EY. NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 1997;16:1279–1290. doi: 10.1093/emboj/16.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Xia Y, Wu Z, Su B, Murray B, Karin M. JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev. 1998;12:3369–3381. doi: 10.1101/gad.12.21.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xu S, Cobb MH. MEKK1 binds directly to the c-Jun N-terminal kinases/stress-activated protein kinases. J Biol Chem. 1997;272:32056–32060. doi: 10.1074/jbc.272.51.32056. [DOI] [PubMed] [Google Scholar]
- 151.Yan M, et al. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- 152.Xu S, Robbins DJ, Christerson LB, English JM, Vanderbilt CA, Cobb MH. Cloning of rat MEK kinase 1 cDNA reveals an endogenous membrane-associated 195-kDa protein with a large regulatory domain. Proc Natl Acad Sci USA. 1996;93:5291–5295. doi: 10.1073/pnas.93.11.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yujiri T, Sather S, Fanger GR, Johnson GL. Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science. 1998;282:1911–1914. doi: 10.1126/science.282.5395.1911. [DOI] [PubMed] [Google Scholar]
- 154.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 155.Yamaoka S, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 156.Mercurio F, et al. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–1538. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Oganesyan G, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 158.Naito A, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353–362. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- 159.He JQ, Oganesyan G, Saha SK, Zarnegar B, Cheng G. TRAF3 and its biological function. Adv Exp Med Biol. 2007;597:48–59. doi: 10.1007/978-0-387-70630-6_4. [DOI] [PubMed] [Google Scholar]
- 160.Xie P, Hostager BS, Bishop GA. Requirement for TRAF3 in signaling by LMP1 but not CD40 in B lymphocytes. J Exp Med. 2004;199:661–671. doi: 10.1084/jem.20031255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Xu M, Skaug B, Zeng W, Chen ZJ. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol Cell. 2009;36:302–314. doi: 10.1016/j.molcel.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Carey M, Lin YS, Green MR, Ptashne M. A mechanism for synergistic activation of a mammalian gene by GAL4 derivatives. Nature. 1990;345:361–364. doi: 10.1038/345361a0. [DOI] [PubMed] [Google Scholar]
- 163.Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 164.Lomvardas S, Thanos D. Modifying gene expression programs by altering core promoter chromatin architecture. Cell. 2002;110:261–271. doi: 10.1016/s0092-8674(02)00822-x. [DOI] [PubMed] [Google Scholar]
- 165.Kim TK, Maniatis T. The mechanism of transcriptional synergy of an in vitro assembled interferon-beta enhanceosome. Mol Cell. 1997;1:119–129. doi: 10.1016/s1097-2765(00)80013-1. [DOI] [PubMed] [Google Scholar]
- 166.Thanos D, Maniatis T. The high mobility group protein HMG I(Y) is required for NF-kappa B-dependent virus induction of the human IFN-beta gene. Cell. 1992;71:777–789. doi: 10.1016/0092-8674(92)90554-p. [DOI] [PubMed] [Google Scholar]
- 167.Ping D, Boekhoudt GH, Rogers EM, Boss JM. Nuclear factor-kappa B p65 mediates the assembly and activation of the TNF-responsive element of the murine monocyte chemoattractant-1 gene. J Immunol. 1999;162:727–734. [PubMed] [Google Scholar]
- 168.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 169.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 170.Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:701–710. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 171.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 172.Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 173.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]