

Abstract
Oxidative stress arises when there is a marked imbalance between the production and removal of reactive oxygen species (ROS) in favor of the prooxidant balance, leading to potential oxidative damage. ROSs were considered traditionally to be only a toxic byproduct of aerobic metabolism. However, recently, it has become apparent that ROS might control many different physiological processes such as induction of stress response, pathogen defense, and systemic signaling. Thus, the imbalance of the increased antioxidant potential, the so-called antioxidative stress, should be as dangerous as well. Here, we synthesize increasing evidence on “antioxidative stress-induced” beneficial versus harmful roles on health, disease, and aging processes. Oxidative stress is not necessarily an un-wanted situation, since its consequences may be beneficial for many physiological reactions in cells. On the other hand, there are potentially harmful effects of “antioxidative stress,” especially in the cases of overconsumption of synthetic antioxidants. Antioxidants can neutralize ROS and decrease oxidative stress; however, this is not always beneficial in regard to disease formation or progression (of, e.g., cancer) or for delaying aging.
1. Introduction
The process of aging or senescence is complex; it may derive from a variety of different mechanisms and is caused by a variety of different factors. In recent years, oxidative stress has been implicated in a wide variety of degenerative processes, diseases, and syndromes, including the mutagenesis, cell transformation, and cancer; atherosclerosis/arteriosclerosis, heart attacks, strokes, and ischemia/reperfusion injury; chronic inflammatory diseases, such as rheumatoid arthritis, lupus erythematosus, and psoriatic arthritis; acute inflammatory problems, such as wound healing; photooxidative stresses to the eye, such as cataract; central nervous system disorders, such as certain forms of familial amyotrophic lateral sclerosis, certain glutathione peroxidase-linked adolescent seizures, Parkinson's disease and Alzheimer's dementia; a wide variety of age-related disorders, perhaps even including factors underlying the aging process itself [1]. There are many theories trying to explain the aging process, each from a different angle. The most recent studies support the idea that oxidative stress is a significant marker of senescence; this was established in different species [2]. Harman first proposed the free radical theory of aging in the 1950s and extended this idea to implicate mitochondrial production of reactive oxygen species in the 1970s [3]. According to the free radical theory of aging [4–7], enhanced and unopposed metabolism-driven oxidative stress plays a major role in diverse chronic age-related disorders. The free-radical theory of aging states that organisms age because their cells accumulate free radical damage over time. Halliwell and Gutteridge later suggested to rename this free radical theory of aging as the “oxidative damage theory of aging” [8], since aging and diseases are caused not only by free radicals but also by other reactive oxygen and nitrogen species. Theory links oxygen consumption, metabolism, ATP, and ROS formation and holds that increases in ROS accompany aging and lead to functional alterations, pathological conditions, and even death [9]. Furthermore, impairment of mitochondrial activity is assumed to be one of the main causes of the aging process [10–12]. Mitochondria are the main site of intracellular oxygen consumption and the main source of ROS formation [10, 13, 14]. Mitochondrial ROSs originate from the electron transport chain and the nitric oxide synthase reactions. Nonmitochondrial sources of ROS include environmental pollutants, pollutants in food, radiation, or they are the by-products of other metabolic processes within organisms. Majority of free radicals are generated inside the cell rather than coming from the environment [10, 15, 16]. The mitochondrial damage theory has been recently reviewed by Wilken [17]. Age-related functional deficits have been observed in some, but not all, studies of aging mitochondria, adding support to the mitochondrial damage theory. The age-related increases in the levels of both oxidative damage and mutational load of mtDNA predicted by the mitochondrial theory of aging have been confirmed in multiple species and organ systems [18]. However, whether this damage affects mitochondrial function or significantly modulates, the physiology of aging has remained controversial [19, 20]. On the other hand, the “vicious cycle” theory, which states that free radical damage to mitochondrial DNA leads to mitochondria that produce more superoxide, has been questioned by some scientists since the most damaged mitochondria are degraded by autophagy (mitophagy), whereas some defective mitochondria (which produce less ATP as well as less superoxide) remain to reproduce themselves [21]. Several lines of direct and indirect evidence generated over the past two decades have demonstrated a positive relationship between the increased oxidative stress in vivo and biological aging. In reality, the oxidative damage potential is greater than antioxidant defense, and thus there is a constant free radicals formation in low amounts, which escapes the cell defenses. Estimates of how much oxygen is turned into free radicals vary; however, typically cited values are around 1.5–5% of the total consumed oxygen [7, 22]. These estimates have been questioned by Hansford et al. [23] and Staniek and Nohl [24], who suggested that H2O2 production rates were lower than 1% of consumed O2. Yet, even if we accept a conservative value of 0.15%, it still represents a substantial amount of free radicals formation [25]. Thus, high levels of reactive oxygen species (ROS) compared to antioxidant defenses are considered to play a major role in diverse chronic age-related diseases and aging. Reduction of oxidative stress was reported to be associated with prolongation of life expectancy, and ROS-lowering interventions were proposed as antiaging strategies [26–40].
Since a biological antioxidant has been defined as any substance that, when present at low concentrations compared to those of an oxidizable substrate, significantly delays or prevents oxidation of that substrate [8], the questions arise whether reduction of oxidative stress in cell environment with antioxidant treatment would be beneficial; how it would influence the health outcome and what adverse effects could this trigger. At this point, it should be stressed that the antioxidants cannot distinguish among the radicals that play a beneficial physiological role and those that cause oxidative damage to biomolecules.
Oxidative stress was first defined by Sies as “a disturbance in the pro-oxidant/antioxidant balance in favor of the former, leading to potential damage.” The term “antioxidative stress” was used for the first time by Dundar and Aslan [41] for the negative effects of antioxidants. Here, we present and discuss the evidence on how “antioxidative” and oxidative stresses or antioxidative imbalance can be damaging for the organism on the examples of cancerogenesis and aging process.
2. Antioxidants and Human Trials
The results of many clinical trials in which individuals received one or more synthetic antioxidants failed to obtain beneficial results. Recent evidence suggests that antioxidant supplements (although highly recommended by the pharmaceutical industry and taken by many individuals) do not offer sufficient protection against oxidative stress, oxidative damage or increase the lifespan. Some recent studies showed that antioxidant therapy has no effect and can even increase mortality [42–53]. Schulz and coworkers showed that nutritive antioxidants completely abolish the extension of lifespan by inhibiting an adoptive reaction to ROS called mitohormesis [54]. It was demonstrated by in vivo and in vitro studies that vitamin C, vitamin E, SOD, glutathione and beta carotene have a potential to cause also “antioxidative stress” in addition to prooxidative stress under certain conditions [55–59]. There are evidently homeostatic mechanisms in cells that govern the amount of allowable antioxidant activity. This indicates that other substances in fruits and vegetables, or a complex mix of substances (e.g., inhibitors of P450 from grapefruit, garlic; inhibitors of cell proliferation, e.g., resveratrol, green tea polyphenols, curcumin; antagonists of estrogen, e.g., some flavonoids; inhibitors of metastases, like some flavonoids; inhibitors of angiogenesis, like genistein, epigallocatechin gallate), contribute to better cardiovascular health and decreased cancer incidence observed in individuals who consume more of fruit and vegetable [60, 61].
Dosing the cells with exogenous antioxidants might decrease the rate of synthesis or uptake of endogenous antioxidants, so that the total “cell antioxidant potential” remains unaltered. Cutler described “the oxidative stress compensation model” which explains why dietary supplements of antioxidants have minimum effect on longevity [62, 63]. Most humans are able to maintain their set point of oxidative stress and so no matter how much additional antioxidant supplement they consumed in their diet further decrease in oxidative stress does not occur. Antioxidant supplements thus do not appear to significantly decrease oxidative stress or/and increase life expectancy in humans [64]. Besides, to exert beneficial physiological effects, antioxidants should be well absorbed in the body and reach the site of ROS formation in cells, where they should be present and in appropriate amounts for a sufficient time.
3. ROS as Signaling Molecules, Regulators of Transcription Factor Activities, and Other Determinants of Gene Expression
Human cells also generate some hydrogen peroxide and other ROS molecules deliberately to use them as chemical signals to regulate everything from glucose metabolism to cellular growth and proliferation [65]. The most commonly synthesized ROSs are superoxide radical and NO, which are produced by NADPH oxidases and NO synthases, respectively, in different places of the organism [66]. These enzymes are highly active in most of the reproductive tissues, indicating that some ROSs are required for reproduction.
ROSs induce various biological processes that include a transient elevation of intracellular Ca2+ concentration, phosphorylation of specific proteins, activation of specific transcription factors, modulation of eicosanoid metabolism, and stimulation of cell growth [67]. Nitric oxide was identified as a signaling molecule as early as 1987 [68] and is now a well-known regulator of some transcription factor activities and other determinants of gene expression. Hydrogen peroxide and superoxide have similar intracellular effects [69]. ROS can affect directly conformation and/or activities of all sulfhydryl-containing molecules, such as proteins or glutathione (GSH), by oxidation of their thiol moiety. Among many other enzymes and membrane receptors, this type of redox regulation affects many proteins important in signal transduction and carcinogenesis, such as protein kinase C, Ca2+-ATPase, collagenase, and tyrosine kinase [70]. For several transcription factors, ROSs are physiological mediators of transcription control [71]. The well-known examples of redox-sensitive transcription factors are nuclear factor-κB (NF-κB) and activator Protein-1 (AP-1). Thus, increased oxidative stress is not beneficial for the cell; however, an increase in cellular antioxidants might impact redox regulation and signal transduction. The complete elimination of free radicals would thus disrupt, rather than extend, the normal functioning of the body.
In addition to ROS, regulators of transcription factor activities and other determinants of gene expression also act as signaling molecules. Even the products oxidized by ROS (e.g., lipids, proteins, sugars, and nucleic acids) may act in the similar way. For example, lipid peroxidation products may modulate signal transduction pathways and mediate biological processes through receptors or receptor-independent pathways. At the same time, it has been shown that the lipid peroxidation products induce adaptive response and increase tolerance against forthcoming oxidative stress by upregulating defense capacity [72].
4. “Antioxidative Stress” and Cancer
Increasing cellular viability by antioxidants prior or after the toxic compound-induced toxicity (e.g., Cr(VI), UV-radiation, ionizing radiation) might not always be beneficial. Carcinogen-induced growth arrest and apoptosis are at the molecular decision point between carcinogen toxicity and carcinogen carcinogenesis [73]. When normally growing cells are in contact with carcinogens, they may respond by undergoing growth arrest, apoptosis, or necrosis. A population of genetically modified cells may also emerge, which exhibits either intrinsic or induced resistance to apoptosis [73]. Control of intracellular concentration of ROS is critical for the survival of cancer cells. It was demonstrated recently in human lung cancer cells that acute increases in intracellular concentrations of ROS caused the inhibition of glycolysis through glycolytic enzyme pyruvate kinase M2 thus diverted the glucose flux into the pentose phosphate pathway [74]. This generated sufficient reducing potential (antioxidant response) for detoxification of ROS. This promoted the metabolic changes required for proliferation and enabled the cancer cells to withstand oxidative stress. Therefore, precancer cells may lead to neoplasia as a result of their altered growth/death ratio, due to disrupted cell cycle control, genomic instability, or altered metabolism. This, however, raises the question whether decreasing carcinogen toxicity by antioxidants might actually increase the incidence of cancer by allowing the inappropriate survival of altered cells. This hypothesis was confirmed by human intervention studies in which smoking male volunteers were exposed for 5–8 years to daily supplements of vitamin E and beta carotene. The overall mortality of male smokers increased in those taking supplements of beta carotene and was most probably due to its effect on cell proliferation [8, 75]. Supplementation by alpha-tocopherol or beta-carotene did not prevent lung cancer in older men who smoke [75]. Beta-carotene supplementation at pharmacologic levels may modestly increase lung cancer incidence in cigarette smokers, and this effect may be associated with heavier smoking and higher alcohol intake. Randomized human trials have demonstrated the antineoplastic and neoplastic effects of antioxidants, with neoplastic effects in patients from higher risk groups due to smoking and alcohol consumption, or patients undergoing chemo- or radiation therapy. The influence of “antioxidative” stress and cancer was confirmed also by the study of Schafer and coworkers [76], who revealed an unanticipated mechanism for survival of epithelial cells detached from the extracellular matrix. Normally such cells lose the ability to take up glucose, which results in ATP deficiency. Such cells do not survive normally in these conditions but can be rescued through two different pathways. The expression of cancer-promoting oncogene ErbB2 in detached endothelial cells restores the cell's ability to take up glucose and reduces the levels of ROS through the antioxidant-generating pentose phosphate pathway. Alternatively, the ATP deficiency could be rescued by antioxidant treatment, which stimulates fatty acid oxidation otherwise inhibited by detachment-induced ROS. The latter cell rescue occurs without the rescue of glucose uptake. Therefore, the addition of antioxidants promotes the survival of preinitiated tumor cells even in unnatural matrix environments by altering metabolic regulation, which results in enhanced malignancy [76].
Antioxidants may thus have dichotomous activities with respect to tumorigenesis, namely, suppressing tumorigenesis by preventing oxidative damage to DNA [77, 78] and promoting tumorigenesis by allowing survival of cells that are metabolically impaired (e.g., in altered matrix environments).
Also, iron metabolism, iron balance, and iron stores in relations with antioxidants (Fenton's chemistry) are important in ROS-induced damage (see Section 4.1). Given that the cells require iron, restricting its supply could also limit the growth of cells, including tumor cells [79–90]. Conversely, the iron carrier neutrophil gelatinase-associated lipocalin (NGAL, Siderocalin) is overexpressed in tumours [91–93]. The role of iron-promoting oncogenesis is documented well [94–110].
4.1. Is Iron Overload the Reason Why Human Trials with Synthetic Antioxidative Supplements Failed to Obtain Beneficial Results?
Potential strategies to decrease oxidative stress may involve not only overall reduction of oxidative stress but also the use of iron and other metal chelators hampering Fenton-type chemistry [111]. Harmful reactions, in which partially reduced oxygen species such as superoxide (O2 ∙−) and hydrogen peroxide (H2O2) may be formed, are, in part, mediated by redox-active metals, the most physiologically relevant of which are iron and copper. These transition metal ions can play an important role in ROS production in the cell. Reduced forms of redox active metal ions participate in the Fenton reaction where the hydroxyl radical (HO∙) is generated from hydrogen peroxide.
Fenton Reaction —
(1) For example,
(2)
Furthermore, the Haber-Weiss reaction, which involves the oxidized forms of redox active metal ions and superoxide anion, generates the reduced form of metal ion, which can be coupled to Fenton chemistry to generate hydroxyl radical (see also below). Superoxide and H2O2 themselves are not excessively reactive; thus, they are not especially damaging at physiological concentrations. However, their reactions with poorly liganded iron species can lead to the production of hydroxyl radical, which is extremely damaging, and a major cause of chronic inflammation [112].
Haber-Weiss Reaction —
(3) For example,
(4) AA ascorbate
AA∙ ascorbate∙.
In the net reaction molecule of hydrogen peroxide is converted into hydroxyl radical and water in the presence of iron as a catalyst. Because the iron concentration in biological systems is often very low, the very important factor for Fenton chemistry activity in biological systems is the presence of functional metal redox-cycling mechanism [113]. Superoxide anion radical (O2 ∙−) or certain antioxidants (e.g., ascorbate) play the role of such reducing agent in biological systems. Antioxidants reduce Fe(III) to Fe(II) which reenters the Fenton reaction. Thus, although ascorbate (or some other antioxidant) is “reducing” and an “antioxidant” agent, its reaction with O2, especially when catalyzed by Fe(II), produces superoxide and thence HO• radicals that have prooxidant properties. It is clear that any increase in the levels of superoxide anion, hydrogen peroxide or the redox active metal ions are likely to lead to the formation of high levels of hydroxyl radical by the chemical mechanisms listed above.
Some compounds contribute to antioxidant defense by chelating transition metals and preventing them from catalyzing the production of free radicals in the cell. Metal-chelating antioxidants such as transferrin, albumin, and ceruloplasmin avoid radical production by inhibiting the Fenton reaction catalyzed by copper and iron. Particularly important is the ability to sequester iron, which is the function of iron-binding proteins such as transferrin and ferritin [114]. The cellular pools of low-molecular-weight iron are not characterized well [115, 116]. If these are in contact with ascorbate (or other antioxidants), prooxidant effects may occur. As already mentioned, ascorbic acid reduces Fe(III) to Fe(II) which reduces oxygen to hydroxyl radical [117]. According to Herbert [118], every advertisement and label for iron supplements and/or vitamin C should warn the consumers: “Do not take this product until your blood iron status has been determined.” Six percent of Americans have the negative iron balance, and such multivitamin/mineral product may help them. Twelve percent of Americans have positive iron balance, and such product may hurt them. If the inappropriate iron balance is a confounding factor in trials with antioxidants, then the ranges of negative effects from these studies should overlap with those of iron balance. Indeed, there was approximately 5–15% rise when different antioxidants were assessed for increased mortality. For example, when the different antioxidants were assessed separately in the study of Bjelakovic et al. [49], there was a significantly increased mortality by vitamin A (RR 1.16, 95% CI 1.10 to 1.24), beta-carotene (RR 1.07, 95% CI 1.02 to 1.11), and vitamin E (RR 1.04, 95% CI 1.01 to 1.07); however, there was no significant detrimental effect of vitamin C (RR 1.06, 95% CI 0.94 to 1.20). The effects of a combination of beta carotene and of retinol (vitamin A) on the incidence of lung cancer were compared by Omenn et al. [42]. The relative risk of death from any cause was 1.17 (95 percent confidence interval, 1.03 to 1.33) in the treated group: 1.46 (95 percent confidence interval, 1.07 to 2.00) of death from lung cancer and 1.26 (95 percent confidence interval, 0.99 to 1.61) of death from cardiovascular diseases. As there is no direct evidence to connect the inappropriate iron balance with antioxidants treatment, the only conclusion could be that iron status should be considered in further clinical trials with antioxidants.
Dietary antioxidants can therefore act in complex and synergistic ways depending on iron status and promote the production of OH∙ radicals in the presence of inappropriately or inadequately liganded Fe(II) [119–122]. In this regard, the use of elemental iron plus ascorbate in food supplements [123] does not seem appropriate. The problem of iron overload increases with aging since iron stores tend to increase with age [124–128], partly due to dietary reasons [129–131]. Several commonly used dietary vitamin and mineral supplements may be associated with increased total mortality risk in elderly women; this association is strongest when supplemented iron [47]. On the other hand, anemia was reported in older population [132, 133]. Killilea et al. [134] reported additionally that the iron content of cells increases also as the cells age normally. Some excessive iron can be removed from the body by regular blood transfusions, like in the case of haemochromatosis. Additionally, certain polyphenols inhibit the absorption of iron. Flavonoids act as antioxidants, through, both, free radical scavenging and metal chelation [135]. Many other iron chelators have been reviewed elsewhere (e.g., [85, 86, 112]). The reduction of redox stress thus requires appropriate amounts of both antioxidants and effective iron chelators.
Additionally, calorie restriction (CR) is thought to be the only experimental manipulation that considerably increases both mean and maximum lifespan in a phylogenetically wide variety of animals [136]. CR provokes a mild stress response, causing enhanced cell defences and metabolic changes, coordinated by the endocrine system. CR may improve iron status, since age-related iron accumulation was found to be markedly suppressed by dietary restriction (DR) in all tissues of 344 male Fischer rats [137]. This improved iron status could in part promote longevity [138]. While we have comparatively little influence on the production of superoxide and peroxide in mitochondria, we can try and improve the speciation of iron ions by pharmacological or dietary means [112].
5. ROS and Adaptive Responses
The key to the future success of regulating oxidative stress induced damage should be the suppression of oxidative damage without disruption of the well-integrated antioxidant defense network. Approach to neutralize free radicals with antioxidants should be changed into prevention of free radical formation in the first place. Evidence that such approaches are possible was summarized in the recent article by Poljsak [2].
The term “hormesis” describes beneficial actions resulting from the response of an organism to a low-intensity stressor. The basic concept behind the idea of hormesis is to provoke the intrinsic capability of a body rather than to supply exogenous natural or synthetic antioxidants to try to compensate for age-related decline of physiological activities in the overall maintenance mechanisms of life [139].
Hormetic pathways activated by phytochemicals may involve kinases and transcription factors that induce the expression of genes that encode antioxidant enzymes, protein chaperones, phase-2 enzymes, neurotrophic factors, and other cytoprotective proteins. Specific examples of such pathways include the sirtuin/FOXO pathways, the NF-κB pathway, and the Nrf-2/ARE pathway [140]. Examples of moderate (usually intermittent) stress inducers include ischemic preconditioning, exercise, and dietary energy restriction [141]. The work of Lee and Yu [142], Koizumi et al. [143], and Chen and Lowry [144] strongly suggest that food restriction (energetic stress) enhances the overall antioxidant capacity to maintain the optimal status of intracellular environments through the concerted interactions of cellular components to regulate ROS and membrane stability against peroxidative stress [137]. However, recent studies do not provide compelling evidence that the CR-associated longevity is mediated by antioxidants [145]. Attention is thus directed to other targets of nutrient signaling that result in the increased production of cytoprotective and restorative proteins in cells.
Finkel and Holbrook [146] stated that the best strategy to enhance endogenous antioxidant levels may actually be oxidative stress itself, based on the classical physiological concept of hormesis. Studies have reported the antiaging and life-prolonging effects of a wide variety of the so-called stressors, such as prooxidants, aldehydes, H2O2 [147], CR [148–151], shear stress [152, 153], irradiation [154], radiation stress [155], UV-radiation [156], physical exercise [157], heat shock, hyperbaric conditions [158], and hypergravity. ROS production is increased by several environmental factors of stress, such as exposition to high levels of light, drought, heavy metals, salt concentrations, temperature extremes, air pollution, UV radiation, herbicides, and pathogen attacks. Whether ROS will act as damaging, protective, or signaling factors depends on the delicate equilibrium between ROS production and scavenging at the proper site and time [159]. Heat and cold can also work as hormesis agents [160–164]. Thermal stress induces a heat-shock response involving increased expression of heat-shock proteins (HSP). Overexpression of certain HSPs in the mitochondria can significantly extend the longevity of normal-lived animals [165, 166]. The so-called adaptive response processes may explain how ROS formation could culminate in promotion of health and life span. For example, moderate physical activity enhanced mitochondrial activity and subsequently increased ROSs formation that ultimately induces an adaptive response, which culminates in metabolic health and extended longevity. On the other hand, health-promoting effects were demonstrated to be reduced, if the subjects exposed to physical activity were treated also with antioxidant supplements [167, 168].
According to Ristow and Schmeisser [145], endogenously produced ROS presumably not only induce ROSs defense enzymes, but also increase activities of phase II response enzymes that protect from damage beyond ROS. Increased oxidative stress over extended period of time is harmful as well as is the extended period of antioxidative stress. Increased ROSs induce endogenous defence mechanisms culminating in increased stress resistance. Interestingly, low doses of ROS with intermittent duration seem to exert the hormesis effects, whereas higher doses of ROS are unquestionably detrimental [145]. The question arises weather Le Chatelier's principle can be used also in situation of antioxidative stress to predict the effect of a change in conditions on a chemical equilibrium. The principle states
“If a system in equilibrium is subjected to a stress the equilibrium will shift in the direction which tends to relieve that stress.”
6. The Importance of Determination of the Oxidative/Antioxidative Status In Vivo
To establish the mechanism of toxicity as ROS mediated, there are direct and indirect methods. Direct methods relate to ROS measurement such as superoxide, H2O2, OH∙. These species are very reactive, and their quantitation can be difficult. The only technique that can detect free radicals directly is the spectroscopic technique of electron spin resonance (ESR), sometimes called electron paramagnetic resonance (EPR). The indirect methods are used in order to overcome these problems. Indirect methods usually measure changes in endogenous antioxidant defense systems or measure the products of damage by ROS among the cellular components [169]. Therefore, changes in endogenous antioxidant defense systems and damages on cellular components, caused by ROS, are measured by indirect methods. The principle behind fingerprinting methods is to measure products of damage by ROS, that is, to measure not the species themselves but the damage that they cause. Measuring the damage caused by ROS instead of direct measuring of ROS seems logical, since it is the damage caused by ROS that is important rather than the total amount of ROS generated. A good marker of oxidative damage must be increased in the presence of oxidative stress, and it must remain unchanged in its absence [170]. The marker must measure a product that is endogenously present, not produced during the isolation procedure. Multiple methods of measurement are available today, each has their own benefits and limitations. Ascertaining the true importance of ROS has been hard, as these evanescent species are difficult to measure in vivo. It is generally accepted that two or more assays should be used whenever possible to enhance their validity, since different parameters are measured by different assays. There are inherent limitations in all methods, and no method can be said to measure accurately the amounts of ROS by itself [171]. For example, in order to determine oxidative stress, both, the ROS potential as well as the antioxidative defense potential should be measured. Besides, it is not possible for a reactive free radical produced in an extravascular space of living tissue, with a lifetime of microseconds, to diffuse into the blood to be detected at the distant site. Numerous in vitro methods are described for the antioxidation potential determination that are easy to perform and largely used in screening. However, the results of such tests are relevant only partially for humans as certain active compounds (e.g., those with large molecular masses) are poorly absorbed from the gastrointestinal tract and/or may undergo metabolic degradation. Therefore, new experimental models are required to provide information if protective effects take place in humans under realistic conditions.
There are many challenges regarding the methodology for oxidative state detection on cellular level, for example, which method(s) to use, which antioxidant should be used for a standard, on the most appropriate positive control (e.g., an oxidising agent or catalysts of Fenton reaction), and so forth. There are no specific recommendations on methods which evaluate best the oxidative stress in biological systems although there are several test systems with different end-points [169]. Additionally, there are no reference values of what is the optimal antioxidative potential in urine, blood, or even intracellularly. It is not known how much antioxidants and in which combinations they are needed for a beneficial antioxidant effect in vivo. Besides, typical oxidative stress status of an individual is not established yet because it is difficult to measure [172]. Determination of antioxidative potential per se is thus not enough, since it is difficult to establish how the individual antioxidants work: by preventing the formation of ROS, by scavenging free radicals, by inducing the signaling pathways, or by repairing the oxidative damage. The improvement in methodology will likely overcome at least some of these drawbacks.
7. Antioxidant Supplements and the Consequences of Oxidative Imbalance
Mitochondria are the main site of intracellular oxygen consumption and the main source of ROS formation [10–14]. Cellular and tissue defenses against ROS include the enzymes superoxide dismutase (Mn-SOD, Cu/Zn-SOD, extracellular (EC)-SOD), catalase, glutathione peroxidase, peroxiredoxins, and the nonenzymatic antioxidants, like glutathione (GSH), thioredoxin, ascorbate, α-tocopherol, and uric acid [173]. In reality, the oxidative damage potential is grater than antioxidant defence, and thus there is a constant small amount of toxic free radical formation, which escapes the cellular defenses. These may lead to a conclusion that endogenous levels of antioxidants are not high enough to fight the stress insults and that the addition of exogenous antioxidants could enhance the cellular antioxidant capacity.
On the other hand, low levels of ROS function as signalling molecules to induce adaptive responses. Over-consumption of exogenous antioxidants could thus lead to the state of “antioxidative” stress, where antioxidants might attenuate or block the adaptive stress responses. Additionally, reactive oxygen species in moderate concentrations are essential mediators of defence against microorganisms and unwanted cells. Thus, if administration of antioxidant supplements decreases the levels of free radicals, this may interfere with essential defence mechanisms for removal of damaged cells, including those that are precancerous and cancerous [174]. Therefore, antioxidant supplements may actually cause some harm [44, 175–177]. Our diets typically contain safe levels of vitamins; however, supplementing them with high levels of antioxidant supplements can potentially upset an important physiological balance [44, 175–177]. A systematic review and meta-analysis done by Bjelakovic et al. [176] concluded that the long-term treatment with beta carotene, vitamin A, and vitamin E may increase mortality. It is not known whether the increased mortality is due to iron overload, adaptive stress response prevention, repression of immune system, or any other mechanism. Presented findings provide indirect evidences for the hypothesis that “anti-oxidative” stress influences ROS production and subsequently inappropriate induction of ROS defenses. On the other hand, also excessive oxidative stress for an extended period of time increases oxidative damage with negative influences on health and longevity.
Oxidative or “antioxidative” stresses occur early in the process that may lead to pathology; therefore, they are not associated with any specific clinical symptoms or signs at an early stage. Often they are not recognized until the development of irreversible damage to the patient that manifest itself as a symptom of a chronic disease. It is therefore important to recognise oxidative imbalance at an early stage in order to prevent the long-term oxidative and antioxidative stresses (Figure 1). These point to the need for determination of individual's oxidative status before starting or ending the supplement therapy with antioxidants. This may prevent or reverse the development of diseases which arise because of chronic oxidative imbalance.
Figure 1.
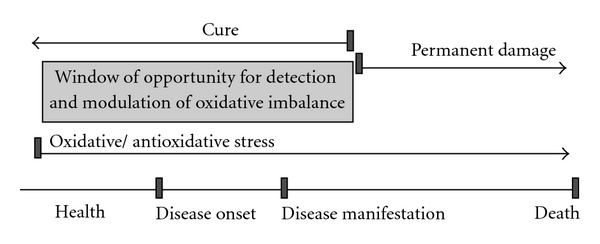
Early detection of “antioxidative” or oxidative stress enables successful prevention and treatment. The window of opportunity for detection and modulation of oxidative imbalance should be explored in order to timely recognise and ameliorate the oxidative imbalance to prevent or postpone its consequences, namely, the free radical-related disease development and premature aging.
The question arises weather determination of individual's oxidative balance/imbalance status would lead to a better preventive or therapeutic interventions that may slow down the aging process. If so, who would benefit from it most? We believe that individuals with oxidative stress because of their diets, lifestyle, metabolic errors, exposure to environmental pollution or iron overload would benefit from regular monitoring of their oxidative status. The individuals who overdose antioxidant supplements and enter the status of “antioxidative” stress may benefit even more.
8. Conclusion
The current level of awareness of safety and quality of supplements with antioxidative properties is inadequate, as there are still many gaps in our knowledge regarding the mechanisms of bioavailability, biotransformation, and action of these supplements. There are several studies in which the addition of antioxidants affected and even increased the mortality.
Exogenous antioxidants tend to depress the endogenous antioxidant levels. Changing the level of one antioxidant causes a compensatory change in the levels of others, while the overall antioxidant capacity remains unaffected. Dosing cells with exogenous antioxidants might decrease the rate of synthesis or uptake of endogenous antioxidants, so that the total “cell antioxidant potential” remains unaltered. Thus, the key to future success of dietary antioxidant supplementation should be in the suppression of oxidative damage without the disruption of the well-integrated antioxidant defense network.
References
- 1.Davies KJ. Oxidative stress: the paradox of aerobic life. Biochemical Society Symposium. 1995;61:1–31. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 2.Poljsak B. Strategies for reducing or preventing the generation of oxidative stress. Oxidative Medicine and Cellular Longevity. 2011;2011:15 pages. doi: 10.1155/2011/194586. Article ID 194586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harman D. The biologic clock: the mitochondria? Journal of the American Geriatrics Society. 1972;20(4):145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 4.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 5.Harman D. Aging: overview. Annals of the New York Academy of Sciences. 2001;928:1–21. doi: 10.1111/j.1749-6632.2001.tb05631.x. [DOI] [PubMed] [Google Scholar]
- 6.Harman D. Free radical theory of aging: effect of free radical reaction inhibitors on the mortality rate of male laf mice. Journals of Gerontology. 1968;23(4):476–482. doi: 10.1093/geronj/23.4.476. [DOI] [PubMed] [Google Scholar]
- 7.Beckman KB, Ames BN. The free radical theory of aging matures. Physiological Reviews. 1998;78(2):547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 8.Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine. 3rd edition. Oxford, UK: Clarendon Press; 1999. [Google Scholar]
- 9.Hagen TM. Oxidative stress, redox imbalance, and the aging process. Antioxidants and Redox Signaling. 2003;5(5):503–506. doi: 10.1089/152308603770310149. [DOI] [PubMed] [Google Scholar]
- 10.Ames BN, Liu J. Delaying the mitochondrial decay of aging. Annals of the New York Academy of Sciences. 2004;1019:406–411. doi: 10.1196/annals.1297.073. [DOI] [PubMed] [Google Scholar]
- 11.Trifunovic A, Larsson NG. Mitochondrial dysfunction as a cause of ageing. Journal of Internal Medicine. 2008;263(2):167–178. doi: 10.1111/j.1365-2796.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- 12.Bratic I, Trifunovic A. Mitochondrial energy metabolism and ageing. Biochimica et Biophysica Acta. 2010;1797(6-7):961–967. doi: 10.1016/j.bbabio.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Campo R, López-Torres M, Cadenas S, Rojas C, Barja G. The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. Journal of Comparative Physiology B. 1998;168(3):149–158. doi: 10.1007/s003600050131. [DOI] [PubMed] [Google Scholar]
- 14.Barja G. Mitochondrial free radical production and aging in mammals and birds. Annals of the New York Academy of Sciences. 1998;854:224–238. doi: 10.1111/j.1749-6632.1998.tb09905.x. [DOI] [PubMed] [Google Scholar]
- 15.Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear dna is extensive. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(17):6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sastre J, Pallardó FV, García de la Asunción J, Viña J. Mitochondria, oxidative stress and aging. Free Radical Research. 2000;32(3):189–198. doi: 10.1080/10715760000300201. [DOI] [PubMed] [Google Scholar]
- 17.Wilken R. Healthy aging: skeletal muscla. In: Stanner S, Thompson R, Buttriss J, editors. Healthy Aging—The Role of Nutrition and Lifestyle. Wiley-Blackwell; 2009. [Google Scholar]
- 18.Golden T, Morten K, Johnson F, Samper E, Melov S. Mitochondria: a critical role in aging. In: Masoro EJ, Austad S, editors. Handbook of the Biology of Aging. 6th edition. Elsevier; 2006. [Google Scholar]
- 19.Jacobs HT. The mitochondrial theory of aging: dead or alive? Aging Cell. 2003;2(1):11–17. doi: 10.1046/j.1474-9728.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 20.Pak JW, Herbst A, Bua E, Gokey N, McKenzie D, Aiken JM. Rebuttal to jacobs: the mitochondrial theory of aging: alive and well. Aging Cell. 2003;2(1):9–10. doi: 10.1046/j.1474-9728.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- 21.de Grey ADNJ. Reactive oxygen species production in the mitochondrial matrix: implications for the mechanism of mitochondrial mutation accumulation. Rejuvenation Research. 2005;8(1):13–17. doi: 10.1089/rej.2005.8.13. [DOI] [PubMed] [Google Scholar]
- 22.Casteilla L, Rigoulet M, Penicaud L. Mitochondrial ros metabolism: modulation by uncoupling proteins. Iubmb Life. 2002;52(3–5):181–188. doi: 10.1080/15216540152845984. [DOI] [PubMed] [Google Scholar]
- 23.Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. Journal of Bioenergetics and Biomembranes. 1997;29(1):89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 24.Staniek K, Nohl H. H2O2 detection from intact mitochondria as a measure for one-electron reduction of dioxygen requires a non-invasive assay system. Biochimica Et Biophysica Acta. 1999;1413(2):70–80. doi: 10.1016/s0005-2728(99)00083-3. [DOI] [PubMed] [Google Scholar]
- 25.Speakman JR, Selman C, McLaren JS, Harper EJ. Living fast, dying when? the link between aging and energetics. Journal of Nutrition. 2002;132(supplement 6):1583S–1597S. doi: 10.1093/jn/132.6.1583S. [DOI] [PubMed] [Google Scholar]
- 26.Harrington LA, Harley CB. Effect of vitamin e on lifespan and reproduction in Caenorhabditis elegans . Mechanisms of Ageing and Development. 1988;43(1):71–78. doi: 10.1016/0047-6374(88)90098-x. [DOI] [PubMed] [Google Scholar]
- 27.Phillips JP, Campbell SD, Michaud D, Charbonneau M, Hilliker AJ. Null mutation of copper/zinc superoxide dismutase in Drosophila confers hypersensitivity to paraquat and reduced longevity. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(8):2761–2765. doi: 10.1073/pnas.86.8.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster . Science. 1994;263(5150):1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 29.Parkes TL, Elia AJ, Dickinson D, Hilliker AJ, Phillips JP, Boulianne GL. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nature Genetics. 1998;19(2):171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 30.Melov S, Ravenscroft J, Malik S, et al. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289(5484):1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- 31.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(23):12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bakaev VV, Lyudmila MB. Effect of ascorbic acid on longevity in the nematode Caenorhabditis elegans . Biogerontology. 2002;3(supplement 1):12–16. [Google Scholar]
- 33.Ruan H, Tang XD, Chen ML, et al. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):2748–2753. doi: 10.1073/pnas.032671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishii N, Senoo-Matsuda N, Miyake K, et al. Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative stress. Mechanisms of Ageing and Development. 2004;125(1):41–46. doi: 10.1016/j.mad.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Huang TT, Naeemuddin M, Elchuri S, et al. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Human Molecular Genetics. 2006;15(7):1187–1194. doi: 10.1093/hmg/ddl034. [DOI] [PubMed] [Google Scholar]
- 36.Zou S, Sinclair J, Wilson MA, et al. Comparative approaches to facilitate the discovery of prolongevity interventions: effects of tocopherols on lifespan of three invertebrate species. Mechanisms of Ageing and Development. 2007;128(2):222–226. doi: 10.1016/j.mad.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Takahashi M, Shimizu T, et al. Effects of a potent antioxidant, platinum nanoparticle, on the lifespan of Caenorhabditis elegans . Mechanisms of Ageing and Development. 2008;129(6):322–331. doi: 10.1016/j.mad.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Quick KL, Ali SS, Arch R, Xiong C, Wozniak D, Dugan LL. A carboxyfullerene sod mimetic improves cognition and extends the lifespan of mice. Neurobiology of Aging. 2008;29(1):117–128. doi: 10.1016/j.neurobiolaging.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Dai DF, Santana LF, Vermulst M, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119(21):2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibamura A, Ikeda T, Nishikawa Y. A method for oral administration of hydrophilic substances to Caenorhabditis elegans: effects of oral supplementation with antioxidants on the nematode lifespan. Mechanisms of Ageing and Development. 2009;130(9):652–655. doi: 10.1016/j.mad.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 41.Dundar Y, Aslan R. Antioxidative stress. Eastern Journal of Medicine. 2000;5(2):45–47. [Google Scholar]
- 42.Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of β carotene and vitamin A on lung cancer and cardiovascular disease. The New England Journal of Medicine. 1996;334(18):1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 43.Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: a systematic review and meta-analysis. The Lancet. 2004;364(9441):1219–1228. doi: 10.1016/S0140-6736(04)17138-9. [DOI] [PubMed] [Google Scholar]
- 44.Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Annals of Internal Medicine. 2005;142(1):37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 45.Collins R, Armitage J, Parish S, Sleight P, Peto R. MRC/BHF heart protection study of antioxidant vitamin supplementation in 20, 536 high-risk individuals: a randomised placebo-controlled trial. The Lancet. 2002;360(9326):23–33. doi: 10.1016/S0140-6736(02)09328-5. [DOI] [PubMed] [Google Scholar]
- 46.Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E and β carotine for age-related cataract and vision loss: AREDS report no. 9. Archives of Ophthalmology. 2001;119(10):1439–1452. doi: 10.1001/archopht.119.10.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mursu J, Robien K, Harnack LJ, Park K, Jacobs DR., Jr. Dietary supplements and mortality rate in older women: the Iowa women’s health study. Archives of Internal Medicine. 2011;171(18):1625–1633. doi: 10.1001/archinternmed.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klein EA, Thompson IM, Jr., Tangen CM, et al. Vitamin E and the risk of prostate cancer: the selenium and vitamin E cancer prevention trial (SELECT) Journal of the American Medical Association. 2011;306(14):1549–1556. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database of Systematic Reviews. 2008;(2) doi: 10.1002/14651858.CD007176. Article ID CD007176. [DOI] [PubMed] [Google Scholar]
- 50.Hercberg S, Ezzedine K, Guinot C, et al. Antioxidant supplementation increases the risk of skin cancers in women but not in men. Journal of Nutrition. 2007;137(9):2098–2105. doi: 10.1093/jn/137.9.2098. [DOI] [PubMed] [Google Scholar]
- 51.Bardia A, Tleyjeh IM, Cerhan JR, et al. Efficacy of antioxidant supplementation in reducing primary cancer incidence and mortality: systematic review and meta-analysis. Mayo Clinic Proceedings. 2008;83(1):23–34. doi: 10.4065/83.1.23. [DOI] [PubMed] [Google Scholar]
- 52.Lawenda BD, Kelly KM, Ladas EJ, Sagar SM, Vickers A, Blumberg JB. Should supplemental antioxidant administration be avoided during chemotherapy and radiation therapy? Journal of the National Cancer Institute. 2008;100(11):773–783. doi: 10.1093/jnci/djn148. [DOI] [PubMed] [Google Scholar]
- 53.Myung SK, Kim Y, Ju W, Choi HJ, Bae WK. Effects of antioxidant supplements on cancer prevention: meta-analysis of randomized controlled trials. Annals of Oncology. 2010;21(1):166–179. doi: 10.1093/annonc/mdp286. [DOI] [PubMed] [Google Scholar]
- 54.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metabolism. 2007;6(4):280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 55.Ionescu JG, Poljsak B. Metal ions mediated pro-oxidative reactions with vitamin C: possible implications for treatment of different malignancies. International Journal of Cancer Prevention. 2010;3(3):149–174. [Google Scholar]
- 56.Poljsak B, Milisav I, Lampe T, Ostan I. Reproductive benefit of oxidative damage: an oxidative stress “malevolence”? Oxidative Medicine and Cellular Longevity. 2011;2011:9 pages. doi: 10.1155/2011/760978. Article ID 760978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fettman MJ, Valerius KD, Ogilvie GK, et al. Effects of dietary cysteine on blood sulfur amino acid, glutathione, and malondialdehyde concentrations in cats. American Journal of Veterinary Research. 1999;60(3):328–333. [PubMed] [Google Scholar]
- 58.Bast A, Haenen GRMM, Doelman CJA. Oxidants and antioxidants: state of the art. American Journal of Medicine. 1991;91(3C):2S–13S. doi: 10.1016/0002-9343(91)90278-6. [DOI] [PubMed] [Google Scholar]
- 59.Halliwell B, Murcia MA, Chirico S, Aruoma OI. Free radicals and antioxidants in food and in vivo: what they do and how they work. Critical Reviews in Food Science and Nutrition. 1995;35(1-2):7–20. doi: 10.1080/10408399509527682. [DOI] [PubMed] [Google Scholar]
- 60.Cherubini A, Vigna GB, Zuliani G, Ruggiero C, Senin U, Fellin R. Role of antioxidants in atherosclerosis: epidemiological and clinical update. Current Pharmaceutical Design. 2005;11(16):2017–2032. doi: 10.2174/1381612054065783. [DOI] [PubMed] [Google Scholar]
- 61.Lotito SB, Frei B. Consumption of flavonoid-rich foods and increased plasma antioxidant capacity in humans: cause, consequence, or epiphenomenon? Free Radical Biology and Medicine. 2006;41(12):1727–1746. doi: 10.1016/j.freeradbiomed.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 62.Cutler RG, Mattson MP. Measuring oxidative stress and interpreting its relevance in humans. In: Cutler RG, Rodriguez H, editors. Oxidative Stress and Aging. Hackensack, NJ, USA: World Scientific; 2003. [Google Scholar]
- 63.Cutler RG. Genetic stability, dysdifferentiation, and longevity determinant genes. In: Cutler RG, Rodriguez H, editors. Critical Reviews of Oxidative Stress and Damage. Hackensack, NJ, USA: World Scientific; 2003. [Google Scholar]
- 64.Green LJ. The Dermatologist’s Guide to Looking Younger. Freedom, Calif, USA: Crossing Press; 1999. [Google Scholar]
- 65.Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Experimental and Molecular Medicine. 1999;31(2):53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- 66.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 67.Kaul N, Forman HJ. Reactive oxygen species in physiology and toxicology: from lipid peroxidation to transcriptional activation. In: Rhodes Cr., editor. Toxicology of the Human Environment: The Critical Role of Free Radicals. New York, NY, USA: Taylor and Francis; 2000. pp. 310–335. [Google Scholar]
- 68.Palmer RMJ, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 69.Kamata H, Hirata H. Redox regulation of cellular signalling. Cellular Signalling. 1999;11(1):1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 70.Dalton TP, Shertzer HG, Puga A. Regulation of gene expression by reactive oxygen. Annual Review of Pharmacology and Toxicology. 1999;39:67–101. doi: 10.1146/annurev.pharmtox.39.1.67. [DOI] [PubMed] [Google Scholar]
- 71.Nordberg J, Arner ESJ. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biology and Medicine. 2001;31(11):1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 72.Chen ZH, Niki E. Two faces of lipid peroxidation products: the “Yin and Yang” principles of oxidative stress. Journal of Experimental and Integrative Medicine. 2011;1(4):215–219. [Google Scholar]
- 73.Carlisle DL, Pritchard DE, Singh J, et al. Apoptosis and p53 induction in human lung fibroblasts exposed to chromium (VI): effect of ascorbate and tocopherol. Toxicological Sciences. 2000;55(1):60–68. doi: 10.1093/toxsci/55.1.60. [DOI] [PubMed] [Google Scholar]
- 74.Anastasiou D, Poulogiannis G, Asara JM, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334(6060):1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Albanes D, Heinonen OP, Taylor PR, et al. α-Tocopherol and β-carotene supplements and lung cancer incidence in the α-tocopherol, β-carotene cancer prevention study: effects of base- line characteristics and study compliance. Journal of the National Cancer Institute. 1996;88(21):1560–1570. doi: 10.1093/jnci/88.21.1560. [DOI] [PubMed] [Google Scholar]
- 76.Schafer ZT, Grassian AR, Song L, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461(7260):109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gao P, Zhang H, Dinavahi R, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12(3):230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Narayanan BA. Chemopreventive agents alters global gene expression pattern: predicting their mode of action and targets. Current Cancer Drug Targets. 2006;6(8):711–727. doi: 10.2174/156800906779010218. [DOI] [PubMed] [Google Scholar]
- 79.Rakba N, Aouad F, Henry C, et al. Iron mobilization and cellular protection by a new synthetic chelator O-Trensox. Biochemical Pharmacology. 1998;55(11):1797–1806. doi: 10.1016/s0006-2952(98)00009-4. [DOI] [PubMed] [Google Scholar]
- 80.Le NTV, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochimica Et Biophysica Acta. 2002;1603(1):31–46. doi: 10.1016/s0304-419x(02)00068-9. [DOI] [PubMed] [Google Scholar]
- 81.Buss JL, Torti FM, Torti SV. The role of iron chelation in cancer therapy. Current Medicinal Chemistry. 2003;10(12):1021–1034. doi: 10.2174/0929867033457638. [DOI] [PubMed] [Google Scholar]
- 82.Lovejoy DB, Richardson DR. Iron chelators as anti-neoplastic agents: current developments and promise of the PIH class of chelators. Current Medicinal Chemistry. 2003;10(12):1035–1049. doi: 10.2174/0929867033457557. [DOI] [PubMed] [Google Scholar]
- 83.Buss JL, Greene BT, Turner J, Torti FM, Torti SV. Iron chelators in cancer chemotherapy. Current Topics in Medicinal Chemistry. 2004;4(15):1623–1635. doi: 10.2174/1568026043387269. [DOI] [PubMed] [Google Scholar]
- 84.Hanai JI, Mammoto T, Seth P, et al. Lipocalin 2 diminishes invasiveness and metastasis of Ras-transformed cells. The Journal of Biological Chemistry. 2005;280(14):13641–13647. doi: 10.1074/jbc.M413047200. [DOI] [PubMed] [Google Scholar]
- 85.Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacological Reviews. 2005;57(4):547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 86.Pahl PMB, Horwitz LD. Cell permeable iron chelators as potential cancer chemotherapeutic agents. Cancer Investigation. 2005;23(8):683–691. doi: 10.1080/07357900500359976. [DOI] [PubMed] [Google Scholar]
- 87.Nie G, Chen G, Sheftel AD, Pantopoulos K, Ponka P. In vivo tumor growth is inhibited by cytosolic iron deprivation caused by the expression of mitochondrial ferritin. Blood. 2006;108(7):2428–2434. doi: 10.1182/blood-2006-04-018341. [DOI] [PubMed] [Google Scholar]
- 88.Edgren G, Nyren O, Melbye M. Cancer as a ferrotoxic disease: are we getting hard stainless evidence? Journal of the National Cancer Institute. 2008;100(14):976–977. doi: 10.1093/jnci/djn225. [DOI] [PubMed] [Google Scholar]
- 89.Huang X. Does iron have a role in breast cancer? The Lancet Oncology. 2008;9(8):803–807. doi: 10.1016/S1470-2045(08)70200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zacharski LR, Chow BK, Howes PS, et al. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: results from a randomized trial. Journal of the National Cancer Institute. 2008;100(14):996–1002. doi: 10.1093/jnci/djn209. [DOI] [PubMed] [Google Scholar]
- 91.Stoesz SP, Friedl A, Haag JD, Lindstrom MJ, Clark GM, Gould MN. Heterogeneous expression of the lipocalin NGAL in primary breast cancers. International Journal of Cancer. 1998;79(6):565–572. doi: 10.1002/(sici)1097-0215(19981218)79:6<565::aid-ijc3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 92.Tong Z, Wu X, Ovcharenko D, Zhu J, Chen CS, Kehrer JP. Neutrophil gelatinase-associated lipocalin as a survival factor. Biochemical Journal. 2005;391(2):441–448. doi: 10.1042/BJ20051020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lim R, Ahmed N, Borregaard N, et al. Neutrophil gelatinase-associated lipocalin (NGAL) an early-screening biomarker for ovarian cancer: NGAL is associated with epidermal growth factor-induced epithelio-mesenchymal transition. International Journal of Cancer. 2007;120(11):2426–2434. doi: 10.1002/ijc.22352. [DOI] [PubMed] [Google Scholar]
- 94.Nelson RL, Davis FG, Sutter E, Sobin LH, Kikendall JW, Bowen P. Body iron stores and risk of colonic neoplasia. Journal of the National Cancer Institute. 1994;86(6):455–460. doi: 10.1093/jnci/86.6.455. [DOI] [PubMed] [Google Scholar]
- 95.Okada S. Iron-induced tissue damage and cancer: the role of reactive oxygen species-free radicals. Pathology International. 1996;46(5):311–332. doi: 10.1111/j.1440-1827.1996.tb03617.x. [DOI] [PubMed] [Google Scholar]
- 96.Toyokuni S. Iron-induced carcinogenesis: the role of redox regulation. Free Radical Biology and Medicine. 1996;20(4):553–566. doi: 10.1016/0891-5849(95)02111-6. [DOI] [PubMed] [Google Scholar]
- 97.Weinberg ED. The role of iron in cancer. European Journal of Cancer Prevention. 1996;5(1):19–36. [PubMed] [Google Scholar]
- 98.Weinberg ED. Iron loading and disease surveillance. Emerging Infectious Diseases. 1999;5(3):346–352. doi: 10.3201/eid0503.990305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Núñez M, Tapia V, Toyokuni S, Okada S. Iron-induced oxidative damage in colon carcinoma (Caco-2) cells. Free Radical Research. 2001;34(1):57–68. doi: 10.1080/10715760100300061. [DOI] [PubMed] [Google Scholar]
- 100.Glei M, Latunde-Dada GO, Klinder A, et al. Iron-overload induces oxidative DNA damage in the human colon carcinoma cell line HT29 clone 19A. Mutation Research. 2002;519(1-2):151–161. doi: 10.1016/s1383-5718(02)00135-3. [DOI] [PubMed] [Google Scholar]
- 101.Toyokuni S. Iron and carcinogenesis: from Fenton reaction to target genes. Redox Report. 2002;7(4):189–197. doi: 10.1179/135100002125000596. [DOI] [PubMed] [Google Scholar]
- 102.Deugnier Y. Iron and liver cancer. Alcohol. 2003;30(2):145–150. doi: 10.1016/s0741-8329(03)00129-0. [DOI] [PubMed] [Google Scholar]
- 103.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annual Review of Pharmacology and Toxicology. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 104.Storz P. Reactive oxygen species in tumor progression. Frontiers in Bioscience. 2005;10(2):1881–1896. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- 105.Lee SK, Lee JJ, Lee HJ, et al. Iron chelator-induced growth arrest and cytochrome c dependent apoptosis in immortalized and malignant oral keratinocytes. Journal of Oral Pathology and Medicine. 2006;35(4):218–226. doi: 10.1111/j.1600-0714.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 106.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 107.Wu J, Eckard J, Chen H, Costa M, Frenkel K, Huang X. Altered iron homeostasis involvement in arsenite-mediated cell transformation. Free Radical Biology and Medicine. 2006;40(3):444–452. doi: 10.1016/j.freeradbiomed.2005.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kallianpur AR, Lee SA, Gao YT, et al. Dietary animal-derived iron and fat intake and breast cancer risk in the Shanghai Breast Cancer Study. Breast Cancer Research and Treatment. 2008;107(1):123–132. doi: 10.1007/s10549-007-9538-3. [DOI] [PubMed] [Google Scholar]
- 109.Prá D, Franke SIR, Giulian R, et al. Genotoxicity and mutagenicity of iron and copper in mice. Biometals. 2008;21(3):289–297. doi: 10.1007/s10534-007-9118-3. [DOI] [PubMed] [Google Scholar]
- 110.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chemistry and Biology. 2008;15(3):234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Terman A, Brunk UT. Oxidative stress, accumulation of biological “garbage”, and aging. Antioxidants and Redox Signaling. 2006;8(1-2):197–204. doi: 10.1089/ars.2006.8.197. [DOI] [PubMed] [Google Scholar]
- 112.Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Medical Genomics. 2009;2, article 2 doi: 10.1186/1755-8794-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deguillaume L, Leriche M, Chaumerliac N. Impact of radical versus non-radical pathway in the Fenton chemistry on the iron redox cycle in clouds. Chemosphere. 2005;60(5):718–724. doi: 10.1016/j.chemosphere.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 114.Imlay JA. Pathways of oxidative damage. Annual Review of Microbiology. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 115.Jacobs A. Low molecular weight intracellular iron transport compounds. Blood. 1977;50(3):433–439. [PubMed] [Google Scholar]
- 116.Voogd A, Sluiter W, van Eijk HG, Koster JF. Low molecular weight iron and the oxygen paradox in isolated rat hearts. Journal of Clinical Investigation. 1992;90(5):2050–2055. doi: 10.1172/JCI116086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford, UK: Oxford University Press; 2005. [Google Scholar]
- 118.Herbert V. The antioxidant supplement myth. American Journal of Clinical Nutrition. 1994;60(2):157–158. doi: 10.1093/ajcn/60.2.157. [DOI] [PubMed] [Google Scholar]
- 119.Miller DM, Buettner GR, Aust SD. Transition metals as catalysts of “autoxidation” reactions. Free Radical Biology and Medicine. 1990;8(1):95–108. doi: 10.1016/0891-5849(90)90148-c. [DOI] [PubMed] [Google Scholar]
- 120.Lachili B, Hininger I, Faure H, et al. Increased lipid peroxidation in pregnant women after iron and vitamin C supplementation. Biological Trace Element Research. 2001;83(2):103–110. doi: 10.1385/BTER:83:2:103. [DOI] [PubMed] [Google Scholar]
- 121.Reif DW. Ferritin as a source of iron for oxidative damage. Free Radical Biology and Medicine. 1992;12(5):417–427. doi: 10.1016/0891-5849(92)90091-t. [DOI] [PubMed] [Google Scholar]
- 122.FáBIáN I, Csordás V. Metal ion catalyzed autoxidation reactions: kinetics and mechanisms. Advances in Inorganic Chemistry. 2003;54:395–461. [Google Scholar]
- 123.Hoppe M, Hulthen L, Hallberg L. The relative bioavailability in humans of elemental iron powders for use in food fortification. European Journal of Nutrition. 2006;45(1):37–44. doi: 10.1007/s00394-005-0560-0. [DOI] [PubMed] [Google Scholar]
- 124.Zacharski LR, Ornstein DL, Woloshin S, Schwartz LM. Association of age, sex, and race with body iron stores in adults: analysis of of NHANES III data. American Heart Journal. 2000;140(1):98–104. doi: 10.1067/mhj.2000.106646. [DOI] [PubMed] [Google Scholar]
- 125.Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O, Ledue TB, Craig WY. Reference distributions for serum iron and transferrin saturation: a comparison of a large cohort to the world’s literature. Journal of Clinical Laboratory Analysis. 2002;16(5):246–252. doi: 10.1002/jcla.10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O, Ledue TB, Craig WY. Reference distributions for serum iron and transferrin saturation: a practical, simple, and clinically relevant approach in a large cohort. Journal of Clinical Laboratory Analysis. 2002;16(5):237–245. doi: 10.1002/jcla.10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu JM, Hankinson SE, Stampfer MJ, Rifai N, Willett WC, Ma J. Body iron stores and their determinants in healthy postmenopausal US women. American Journal of Clinical Nutrition. 2003;78(6):1160–1167. doi: 10.1093/ajcn/78.6.1160. [DOI] [PubMed] [Google Scholar]
- 128.Doulias PT, Vlachou C, Boudouri C, Kanavaros P, Siamopoulos KC, Galaris D. Flow cytometric estimation of “labile iron pool” in human white blood cells reveals a positive association with ageing. Free Radical Research. 2008;42(3):253–259. doi: 10.1080/10715760801911649. [DOI] [PubMed] [Google Scholar]
- 129.Fleming DJ, Tucker KL, Jacques PF, Dallal GE, Wilson PWF, Wood RJ. Dietary factors associated with the risk of high iron stores in the elderly Framingham Heart Study cohort. American Journal of Clinical Nutrition. 2002;76(6):1375–1384. doi: 10.1093/ajcn/76.6.1375. [DOI] [PubMed] [Google Scholar]
- 130.Beard J. Dietary iron intakes and elevated iron stores in the elderly: is it time to abandon the set-point hypothesis of regulation of iron absorption? American Journal of Clinical Nutrition. 2002;76(6):1189–1190. doi: 10.1093/ajcn/76.6.1189. [DOI] [PubMed] [Google Scholar]
- 131.Cade JE, Moreton JA, O’Hara B, et al. Diet and genetic factors associated with iron status in middle-aged women. American Journal of Clinical Nutrition. 2005;82(4):813–820. doi: 10.1093/ajcn/82.4.813. [DOI] [PubMed] [Google Scholar]
- 132.Guralnik JM, Eisenstaedt RS, Ferrucci L, Klein HG, Woodman RC. Prevalence of anemia in persons 65 years and older in the united states: evidence for a high rate of unexplained anemia. Blood. 2004;104(8):2263–2268. doi: 10.1182/blood-2004-05-1812. [DOI] [PubMed] [Google Scholar]
- 133.Price EA. Aging and erythropoiesis: current state of knowledge. Blood Cells, Molecules, and Diseases. 2008;41(2):158–165. doi: 10.1016/j.bcmd.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 134.Killilea DW, Atamna H, Liao C, Ames BN. Iron accumulation during cellular senescence in human fibroblasts in vitro. Antioxidants and Redox Signaling. 2003;5(5):507–516. doi: 10.1089/152308603770310158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Glauce Socorro de Barros V, Luzia Kalyne A, Leal M, Fontenele JB. Role of plant extracts and polyphenolic compounds in oxidative stress-related diseases. In: Kozyrev D, Slutsky V, editors. Handbook of Free Radicals: Formation, Types and Effects. New York, NY, USA: Nova Science Publishers, Inc.; 2010. [Google Scholar]
- 136.Gredilla R, Barja G. Mitochondrial oxidative stress and caloric restriction. In: Mattson MP, editor. Metabolism and Lifespan Determination. Vol. 14. Advances in Cell Aging and Gerontology; 2003. pp. 105–122. [Google Scholar]
- 137.Cook CI, Yu BP. Iron accumulation in aging: modulation by dietary restriction. Mechanisms of Ageing and Development. 1998;102(1):1–13. doi: 10.1016/s0047-6374(98)00005-0. [DOI] [PubMed] [Google Scholar]
- 138.Reverter-Branchat G, Cabiscol E, Tamarit J, Ros J. Oxidative damage to specific proteins in replicative and chronological-aged Saccharomyces cerevisiae—common targets and prevention by calorie restriction. The Journal of Biological Chemistry. 2004;279(30):31983–31989. doi: 10.1074/jbc.M404849200. [DOI] [PubMed] [Google Scholar]
- 139.Goto S, Radak Z, Takahasi R. Biological implications of protein oxidation. In: Cutler R, Rodriguez H, editors. Critical Review of Oxidative Stress and Aging. Hackensack, NJ, USA: World Scientific; 2003. [Google Scholar]
- 140.Son TG, Camandola S, Mattson MP. Hormetic dietary phytochemicals. Neuromolecular Medicine. 2008;10(4):236–246. doi: 10.1007/s12017-008-8037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mattson MP. Hormesis defined. Ageing Research Reviews. 2008;7(1):1–7. doi: 10.1016/j.arr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee DW, Yu BP. Food restriction as an effective modulator of free radical metabolism in rats. Korean Biochemical Journal. 1991;24:148–154. [Google Scholar]
- 143.Koizumi A, Weindruch R, Walford RL. Influences of dietary restriction and age on liver enzyme activities and lipid peroxidation in mice. Journal of Nutrition. 1987;117(2):361–367. doi: 10.1093/jn/117.2.361. [DOI] [PubMed] [Google Scholar]
- 144.Chen LH, Lowry SR. Cellular antioxidant defense system. Progress in Clinical and Biological Research. 1989;287:247–256. [PubMed] [Google Scholar]
- 145.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radical Biology and Medicine. 2011;51(2):327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 146.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 147.Chen ZH, Yoshida Y, Saito Y, Niki E. Adaptation to hydrogen peroxide enhances PC12 cell tolerance against oxidative damage. Neuroscience Letters. 2005;383(3):256–259. doi: 10.1016/j.neulet.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 148.Masoro EJ. Hormesis and the antiaging action of dietary restriction. Experimental Gerontology. 1998;33(1-2):61–66. doi: 10.1016/s0531-5565(97)00071-5. [DOI] [PubMed] [Google Scholar]
- 149.Rattan SI, Demirovic D. Hormesis as a mechanism for the anti-aging effects of calorie restriction. In: Everitt AV, Rattan SIS, Couteur DG, Cabo RD, editors. Calorie Restriction, Aging and Longevity. Dordrecht, The Netherlands: Springer; 2010. pp. 233–245. [Google Scholar]
- 150.Yu BP, Chung HY. Stress resistance by caloric restriction for longevity. Annals of the New York Academy of Sciences. 2001;928:39–47. doi: 10.1111/j.1749-6632.2001.tb05633.x. [DOI] [PubMed] [Google Scholar]
- 151.Masoro E. The role of hormesis in life extension by dietary restriction. Interdisciplinary Topics in Gerontology. 2007;35:1–17. doi: 10.1159/000096552. [DOI] [PubMed] [Google Scholar]
- 152.Warabi E, Wada Y, Kajiwara H, et al. Effect on endothelial cell gene expression of shear stress, oxygen concentration, and low-density lipoprotein as studied by a novel flow cell culture system. Free Radical Biology and Medicine. 2004;37(5):682–694. doi: 10.1016/j.freeradbiomed.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 153.Warabi E, Takabe W, Minami T, et al. Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: role of reactive oxygen/nitrogen species. Free Radical Biology and Medicine. 2007;42(2):260–269. doi: 10.1016/j.freeradbiomed.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 154.Plews M, Simon SLR, Boreham DR, et al. A radiation-induced adaptive response prolongs the survival of prion-infected mice. Free Radical Biology and Medicine. 2010;49(9):1417–1421. doi: 10.1016/j.freeradbiomed.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 155.Yanase S, Hartman PS, Ito A, Ishii N. Oxidative stress pretreatment increases the X-radiation resistance of the nematode Caenorhabditis elegans . Mutation Research. 1999;426(1):31–39. doi: 10.1016/s0027-5107(99)00079-2. [DOI] [PubMed] [Google Scholar]
- 156.Poljsak B. Skin aging, Free Radicals and Antioxidants. New York, NY, USA: Nova Science Publisher; 2012. [Google Scholar]
- 157.Packer L, Cadenas E, Davies KJA. Free radicals and exercise: an introduction. Free Radical Biology and Medicine. 2008;44(2):123–125. doi: 10.1016/j.freeradbiomed.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 158.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. general properties and effect of hyperbaric oxygen. Biochemical Journal. 1973;134(3):707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gratão PL, Polle A, Lea PJ, Azevedo RA. Making the life of heavy metal-stressed plants a little easier. Functional Plant Biology. 2005;32(6):481–494. doi: 10.1071/FP05016. [DOI] [PubMed] [Google Scholar]
- 160.Rattan SIS, Gonzalez-Dosal R, Nielsen ER, Kraft DC, Weibel J, Kahns S. Slowing down aging from within: mechanistic aspects of anti-aging hormetic effects of mild heat stress on human cells. Acta Biochimica Polonica. 2004;51(2):481–492. [PubMed] [Google Scholar]
- 161.Rattan SIS. Hormesis in aging. Ageing Research Reviews. 2008;7(1):63–78. doi: 10.1016/j.arr.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 162.Conti B, Sanchez-Alavez M, Winsky-Sommerer R, et al. Transgenic mice with a reduced core body temperature have an increased life span. Science. 2006;314(5800):825–828. doi: 10.1126/science.1132191. [DOI] [PubMed] [Google Scholar]
- 163.Hosono R, Mitsui Y, Sato Y. Life span of the wild and mutant nematode Caenorhabditis elegans: effects of sex, sterilization, and temperature. Experimental Gerontology. 1982;17(2):163–172. doi: 10.1016/0531-5565(82)90052-3. [DOI] [PubMed] [Google Scholar]
- 164.Ali SS, Marcondes MCG, Bajova H, Dugan LL, Conti B. Metabolic depression and increased ROS production by isolated mitochondria at moderately lower temperatures. The Journal of Biological Chemistry. 2010;285(42):32522–32528. doi: 10.1074/jbc.M110.155432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kurapati R, Passananti HB, Rose MR, Tower J. Increased hsp22 RNA levels in Drosophila lines genetically selected for increased longevity. Journals of Gerontology Series A. 2000;55(11):B552–B559. doi: 10.1093/gerona/55.11.b552. [DOI] [PubMed] [Google Scholar]
- 166.Morrow G, Battistini S, Zhang P, Tanguay RM. Decreased lifespan in the absence of expression of the mitochondrial small heat shock protein hsp22 in Drosophila. Journal of Biological Chemistry. 2004;279(42):43382–43385. doi: 10.1074/jbc.C400357200. [DOI] [PubMed] [Google Scholar]
- 167.Ristow M, Zarse K, Oberbach A, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(21):8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gomez-Cabrera MC, Domenech E, Romagnoli M, et al. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. American Journal of Clinical Nutrition. 2008;87(1):142–149. doi: 10.1093/ajcn/87.1.142. [DOI] [PubMed] [Google Scholar]
- 169.Poljsak B, Jamik P. Methodology for oxidative state detection in biological systems. In: Kozyrev D, Slutsky V, editors. Handbook of Free Radicals: Formation, Types and Effects. New York, NY, USA: Nova Science Publishers; 2010. (Cell biology research progress series). [Google Scholar]
- 170.Miwa S, Muller FL, Beckman KB. The basics of oxidative biochemistry. In: Miwa S, Muller FL, Beckman KB, editors. Oxidative Stress in Aging. From Model Systems to Human Diseases. Totowa, NJ, USA: Humana Press; 2008. [Google Scholar]
- 171.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. New York, NY, USA: Oxford University Press; 1985. The chemistry of oxygen radicals and other oxygen-derived species; pp. 20–64. [Google Scholar]
- 172.Argüelles S, Gómez A, Machado A, Ayala A. A preliminary analysis of within-subject variation in human serum oxidative stress parameters as a function of time. Rejuvenation Research. 2007;10(4):621–636. doi: 10.1089/rej.2006.0528. [DOI] [PubMed] [Google Scholar]
- 173.Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radical Biology and Medicine. 2001;30(5):463–488. doi: 10.1016/s0891-5849(00)00373-7. [DOI] [PubMed] [Google Scholar]
- 174.Salganik RI. The benefits and hazards of antioxidants: controlling apoptosis and other protective mechanisms in cancer patients and the human population. Journal of the American College of Nutrition. 2001;20(5):464S–472S. doi: 10.1080/07315724.2001.10719185. [DOI] [PubMed] [Google Scholar]
- 175.Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. The Lancet. 2003;361(9374):2017–2023. doi: 10.1016/S0140-6736(03)13637-9. [DOI] [PubMed] [Google Scholar]
- 176.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Journal of the American Medical Association. 2007;297(8):842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 177.Caraballoso M, Sacristan M, Serra C, Bonfill X. Drugs for preventing lung cancer in healthy people. Cochrane Database of Systematic Reviews. 2003;(2) doi: 10.1002/14651858.CD002141. Article ID CD002141. [DOI] [PubMed] [Google Scholar]