

Abstract
Impaired T-cell responses in chronic hepatitis C virus (HCV) patients have been reported to be associated with the establishment of HCV persistent infection. However, the mechanism for HCV-mediated T-cell dysfunction is yet to be defined. Myeloid-derived suppressor cells (MDSCs) play a pivotal role in suppressing T-cell responses. In this study we examined the accumulation of MDSCs in human peripheral blood mononuclear cells (PBMCs) following HCV infection. We found that CD33+ mononuclear cells cocultured with HCV-infected hepatocytes, or with HCV core protein, suppress autologous T-cell responses. HCV core-treated CD33+ cells exhibit a CD14+CD11b+/lowHLADR−/low phenotype with up-regulated expression of p47phox, a component of the NOX2 complex critical for reactive oxygen species (ROS) production. In contrast, immunosuppressive factors, arginase-1 and inducible nitric oxide synthase (iNOS), were not up-regulated. Importantly, treatment with an inactivator of ROS reversed the T-cell suppressive function of HCV-induced MDSCs. Lastly, PBMCs of chronic HCV patients mirror CD33+ cells following treatment with HCV core where CD33+ cells are CD14+CD11b+HLADR−/low, and up-regulate the expression of p47phox.
Conclusion
These results suggest that HCV promotes the accumulation of CD33+ MDSC, resulting in ROS-mediated suppression of T-cell responsiveness. Thus, the accumulation of MDSCs during HCV infection may facilitate and maintain HCV persistent infection.
Hepatitis C virus (HCV) infection in humans is almost invariably associated with viral persistence leading to chronic hepatitis, which in turn predisposes the infected individual to hepatocellular carcinoma and the necessity of a liver transplant.1 CD8+ T cells play a pivotal role in controlling HCV infection; however, severe CD4+ and CD8+ T-cell dysfunction has been observed in chronic HCV patients.2 This suggests that HCV may employ mechanisms to evade or possibly suppress the host T-cell response. Innate immune cells play a pivotal role in controlling viral infection during the early phase of infection and in shaping adaptive immunity. Because monocytes/macrophages (M/Mϕ) and dendritic cells (DCs) are the major innate immune cell types at the site of viral infection, their interaction with effector T cells is crucial for determining the course of the immune response. However, during chronic viral infection M/Mϕ and DCs exhibit aberrant antigen-presenting cell (APC) activation and function, including abnormally low production of inflammatory cytokines (i.e., interferon-alpha [IFN-α], interleukin [IL]-12).3 Thus, it is possible that HCV actively suppresses the immune response by altering the differentiation of innate immune cells, resulting in an impairment of a subsequent robust antiviral adaptive response.
HCV infection and replication mainly occurs in hepatocytes.4 Due to fenestrations in liver endothelial cells, innate immune cells recruited to the liver following HCV infection directly interact with HCV-infected hepatocytes. Intriguingly, HCV core protein (21 kDa) is secreted from HCV-infected hepatocytes and is present extracellularly in the plasma of chronically infected patients.5 Extracellular core exerts an immunomodulatory role in human M/Mϕ and DCs resulting in inhibition of Toll-like receptor (TLR)-induced proinflammatory cytokine production including IFN-α and IL-12.6,7 Furthermore, HCV core activates signal transducer and activator of transcription 3 (STAT3), a transcription factor that is critical for the development of regulatory APCs, through the up-regulation of IL-6.8 These studies suggest that HCV core alters APC activation and differentiation. Thus, T-cell responses against HCV are likely impaired through viral factor-mediated alteration of myeloid cells, allowing the establishment of persistent infection in the liver.
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous subset of regulatory APCs that are responsible for the inhibition of T-cell responses. MDSCs have been well described in multiple severe human diseases such as cancer, autoimmune disease, and bacterial infections.9 In the mouse, the MDSC populations have been divided into two groups; polymorphonuclear MDSCs (PMN-MDSC) described as CD11b+Gr-1highLy6G+Ly6Clow/int cells and mononuclear MDSCs (Mo-MDSC) described as CD11b+Gr-1intLy6G−Ly6Chigh cells.10,11 However, the phenotypic markers of MDSCs are less clear in humans. Although MDSCs have been described as CD33+CD11b+HLADRlow/− in some cancer models, the expression level of CD14 is variable in different experimental systems.9,12 Although the precise molecular mechanism for the differentiation of MDSCs is yet to be defined, the expansion and accumulation of these cells are mediated by tumor-derived factors including M-CSF, granulocyte-macrophage colony-stimulating factor (GM-CSF), transforming growth factor beta (TGF-β), vascular endothelial cell growth (VEGF), and IL-6.13 The vast majority of these factors activate STAT3, underscoring STAT3 as an important transcription factor in MDSC differentiation. Indeed, ablation of STAT3 using conditional knockout mice reduced the expansion of MDSCs and improved T-cell responses in tumor-bearing mice.14
MDSCs have been shown to suppress T-cell responses by way of numerous mechanisms including expression of inhibitory cell surface molecules, production of regulatory cytokines, the metabolism of arginine through activation of arginase-1, production of nitric oxide, and the up-regulation of reactive oxygen species (ROS).9 Arginase-1 inhibits T-cell responses through depletion of nonessential amino acid, L-arginine, resulting in down-regulation of CD3-ζ and inhibition of T-cell proliferation.15,16 Nitric oxide (NO) production in MDSCs is induced through up-regulation of inducible nitric oxide synthase (iNOS), NO down-regulates MHC class II in APCs and leads to T-cell apoptosis.17,18 In leukocytes, ROS is primarily generated through NADPH oxidase. The oxidase is a multicomponent enzyme consisting of two membrane proteins, gp91 and p22, and at least four cytosolic components: p47phox, p67phox, p40phox, and a small G protein Rac.19 In MDSCs a number of these components have been shown to be up-regulated, including p47phox and gp91.20 Notably, the regulation of these proteins was shown to be dependent on STAT3 activation, which provides further evidence for the importance of this transcription factor.20
Here we show that HCV induces the accumulation of MDSC through extracellular core protein. Human CD33+ cells cocultured with HCV-infected hepatocytes, or treated with HCV core, suppress the activation of autologous T cells. Additionally, the suppression of T cells by HCV core-treated MDSCs is ROS-dependent. Core-treated CD33+ cells were CD14+-CD11blow/+ and HLADR−/low. Further, HCV core treatment up-regulated NOX2 component, p47phox. Lastly, CD33+ cells from chronically infected patients were CD11b+CD14+ and HLADR−/low; these cells also up-regulated p47phox compared with healthy donors. These data provide evidence that HCV core induces the accumulation of ROS producing MDSCs, thereby inhibiting host T-cell responses. Therefore, this study describes a novel mechanism for HCV-mediated immune regulation, and suggests that regulation of the MDSC population may be an attractive target for future HCV therapies.
Materials and Methods
HCV Infection
The human hepatoma cell line Huh 7.5.1 was cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and antibiotics. Huh 7.5.1 cells were seeded at 3 × 106 cells in T75 plate for 24 hours. They were then infected with 4 × 104 focus-forming unit (FFU) (multiplicity of infection [MOI] 0.01) of HCV strain JFH-1, and infected cells were cultured for 10 days in DMEM/10% FCS media. Cells were expanded 2 days following infection. Infection was confirmed by immunofluorescence. Hepatocytes were stained with monoclonal antibodies to HCV core (clone C7-50, Thermo Scientific, Rockford, IL) and subsequently stained with Alexa Fluor 488-conjugated donkey anti-mouse antibodies (Invitrogen). Nuclei were visualized using DAPI (Invitrogen). To isolate JFH-1, centrifugation using an Amicon Ultra-15 (100,000 MWCO) centrifugal filter unit was used. Briefly, 10 mL of JFH-1 infected culture media was concentrated to 1 mL. Next, peripheral blood mononuclear cells (PBMCs) were treated with indicated amounts of the concentrated virus for 7 days.
Preparation of PBMCs
Human PBMCs were isolated from healthy blood donors (Virginia Blood Services, Richmond, VA) by lympholyte gradient centrifugation (Cedarlane Laboratories, Burlington, NC).
Coculture of PBMCs with Hepatocytes
Infected hepatocytes were plated at 0.1 × 106 cells/mL in a T25 cm2 flask and cultured overnight. PBMCs were then thawed and 10 × 106 cells were cocultured with the hepatocytes for 7 days in complete media (RPMI 1640 supplemented with 10% [vol/vol] FBS) (HyClone, Logan, UT), penicillin/streptomycin (100 μg/mL), and L-glutamine (2 mM). Following 7 days of coculture, CD33+ cells were selected using magnetic beads (Miltenyi Biotec) according to the manufacturer’s instructions. CD33+ cells were cocultured with autologous magnetic bead selected (MACS) CD4 and CD8 T cells at a ratio of 1:2 (250,000 CD33+ cells to 500,000 T cells) for 3 days in the presence of 5 μg/mL anti-CD3 (OKT3; eBioscience, San Diego, CA) and 10 μg/mL anti-CD28 (CD28.6; eBioscience).
Treatment of PBMCs with HCV Core
Human PBMCs were cultured in complete media at 1 × 106 cells/mL for 7 days in the presence of 1 μg/mL recombinant HCV core protein (Virogen, Watertown, MA) or recombinant protein control, β-galactosidase (Virogen). CD33+ cells were then selected using magnetic beads and cocultured with autologous CD4 and CD8 T cells as described above. T cells and CD33+ cells were cocultured in transwell plates (Corning, Corning, NY) containing 0.4 μm pores in indicated experiments (Fig. 3).
Fig. 3.

HCV core-treated CD33+ cell mediated suppression is contact-dependent. Human PBMCs were treated with HCV core or β-gal for 7 days. CD33+ cells were then cocultured with CD4 or CD8 T cells as previously described for 3 days in transwell plates. (A,B) CFSE dilution was assessed as a measure for proliferation. (C,D) Cells were restimulated with PMA/ionomycin and intracellular expression of IFN-γ was determined. (E,F) Supernatants were collected following 3 days of CD33-T cell coculture and IFN-γ production was measured by ELISA.
Assay for T-Cell Proliferation by CFSE Dilution
Prior to coculture of CD4 and CD8 T cells with CD33+ cells, cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) according to the manufacturer’s instructions (Invitrogen). The cells were then washed in media and cocultured with CD33+ cells. Following 3 days of coculture in the presence of plate-bound anti-CD3/anti-CD28, cells were stained with APC-conjugated anti-CD4 (Leu-3a; eBiosciences) or APC-conjugated anti-CD8 (RPA-T8; eBiosciences), fixed, and collected on a FACSCanto (BD Bioscience, San Diego, CA).
Determination of Cytokine Production by Enzyme-Linked Immunosorbent Assay (ELISA)
Following 3 days of coculture of CD4 or CD8 T cells with CD33+ cells, supernatants were collected and stored at −80°C. Cytokine levels were then assessed for IFN-γ and IL-2 per the manufacturer’s instructions (eBioscience).
Intracellular Cytokine Staining
After 3 days of CD33+/T-cell coculture, cells were restimulated with 0.1 μg/mL PMA and 1 μg/mL ionomycin for 5 hours. Golgi Plug (eBiosciences) was also added during the stimulation. At the end of the stimulation, cells were permeabilized with Cytofix/Cytoperm (BD Biosciences) per the manufacturer’s instructions. The permeabilized cells were then stained with APC-conjugated anti-IFN-γ (eBiosciences) or isotype control (eBioscience). The stained cells were then collected on a FACSCanto (BD Biosciences) and analyzed using FloJo.
Analysis of Cell Surface Markers by Flow Cytometry
PBMCs were treated with HCV core for 7 days as described above. Magnetically selected CD33+ cells were then analyzed for cell surface marker expression. Cells were first blocked in 10% mouse serum on ice for 10 minutes, washed, and then stained with FITC-conjugated HLA-DR (BD Biosciences), PE-conjugated CD11b (eBiosciences), and APC-conjugated CD14 (eBiosciences) antibodies or isotype controls for 1 hour. Cells were then washed, fixed, and collected as described above.
ROS Detection
CD33+ cells were cultured in media at 37°C in the presence of 2.5 μM DCFDA (Invitrogen) and with 30 ng/mL PMA in stimulated samples for 30 minutes. Analysis by flow cytometry was then conducted as described above.
Rescue of T-Cell Function Using Inhibitors
PBMCs were treated with HCV core for 7 days as detailed above in the presence or absence of 100 U/mL of catalase where indicated. CD33+ cells were then cocultured for 3 days with autologous T cells in the presence of inhibitors of candidate suppressive molecules at the following concentrations: 100 U/mL catalase (Sigma-Aldrich, St. Louis, MO), 500 μmol/L NG-monomethyl-L-arginineacetate (Sigma-Aldrich), and 500 μmol/L N(ω)-hydroxy-nor-Larginine (Cayman Chemicals, Ann Arbor, MI).
Quantitative Polymerase Chain Reaction (PCR)
PBMCs were treated with HCV core as described above. RNA from CD33+ cells was then harvested using RNeasy mini kit (Invitrogen) and reverse transcribed into complementary DNA (cDNA) (Invitrogen) per the manufacturer’s instructions. The resultant cDNA was amplified using TaqMan Universal Master Mix II (Applied Biosystems, Carlsbad, CA). Primers for arginase-1 (HS00968979_m1), iNOS (HS01075529_ m1), p47phox (HS00165362_m1), p22phox(HS03044361_ m1), gp91phox(HS00166163_m1), and hypoxanthine-guanine phosphoribosyltransferase (HS01003267_m1) were acquired from Applied Biosystems. The samples were assessed as described.8
Western Blotting
PBMCs were treated with HCV core for 7 days as described above. Western blotting was performed as described.8 p47phox antibody was obtained from Cell Signaling Technology (Danvers, MA), and anti-actin from Santa Cruz Biotechnology (Santa Cruz, CA).
Analysis of PBMCs from Chronic HCV Patients
PBMCs were obtained under informed consent from five patients chronically infected with HCV. PBMCs from chronically infected patients, as well as healthy donor controls, were thawed in complete media and magnetically selected CD33+ cells were assessed for cell surface expression of HLA-DR, CD11b, and CD14 as described above. Additionally, RNA from these cells was harvested and qPCR was performed as previously described.
Statistical Analysis
Paired Student’s t tests were used to determine the statistical significance of the data in all figures except Fig. 6, where an unpaired Student’s t test used. Statistical analysis was performed using Prism 4 v. 4.0c. P < 0.05 was considered significant.
Fig. 6.

Chronically infected HCV patients are phenotypically similar to MDSCs and up-regulate p47phox. CD33+ cells were selected from PBMCs from chronically infected HCV patients or healthy donors. (A) The cells were assessed for expression of HLA-DR, CD11b, and CD14 by flow cytometry, where healthy donors are shown as a solid line, and chronic HCV+ patients as a dotted line. (B) RNA from these cells was harvested and expression of p47phox was assessed by qPCR. Expression levels were determined relative to HPRT. *P < 0.05.
Results
HCV-Infected Hepatocytes Induce the Generation of CD33+ Suppressor Cells
HCV clone, JFH-1, can infect Huh-derived hepatoma cells in cell culture and produce infectious virus.21–23 By using this system, we determined the effect of HCV-infected hepatocytes on the generation of suppressive CD33+ monocytes/macrophages. We first infected Huh7.5.1 cells with HCV (JFH-1) virus; infection was then confirmed by immunofluorescence (IF) for the expression of HCV core protein (Fig. 1A). HCV-infected cells (HCV+ hepatocytes) were reseeded and cultured for 24 hours. After the coculture of HCV+ or HCV− hepatocytes with PBMCs for 7 days, CD33+ cells were subsequently selected and then cocultured with autologous CD4+ and CD8+ T cells. Interestingly, CD33+ cells cocultured with HCV+ hepatocytes significantly inhibited the production of IFN-γ by both CD4+ and CD8+ T cells using ELISA (Fig. 1B). In contrast, there was no significant difference in T-cell proliferation when T cells were cocultured with HCV+ hepatocytes as compared with those with HCV− hepaotcytes (data not shown). These results suggest that HCV impairs antiviral T-cell responses through the generation of suppressive CD33+ M/Mϕ.
Fig. 1.
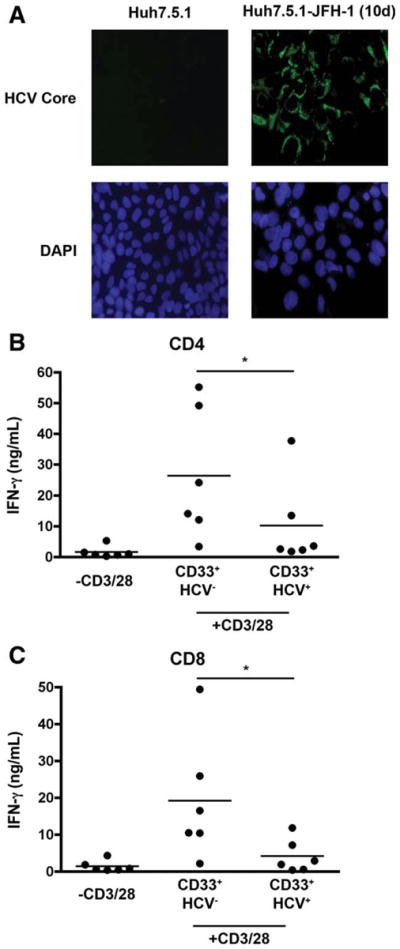
CD33+ cells cocultured with HCV (JFH-1) infected hepato-cytes suppress T cell IFN-γ production. Huh7.5.1 cells were infected with HCV strain JFH-1 for 10 days. (A) Infection was confirmed by immunofluorescence (IF) using antibodies against HCV core protein. The infected hepatocytes were then cocultured with PBMCs for 7 days and CD33+ cells were subsequently selected using magnetic beads. As a control, PBMCs were also cocultured with uninfected Huh7.5.1 cells. (B,C) CD33+ cells were then cocultured with autologous CD4+ and CD8+ T cells in the presence of CD3/28 for 3 days. IFN-γ levels were then assessed by ELISA. *P < 0.05.
To examine the effect of infectious virus on the generation of suppressive CD33+ cells, we treated PBMCs with varying doses of JFH-1 virus isolated using an Amicon filter system and CD33+ cells were then selected and cocultured with autologous CD4+ T cells. These results indicate that CD33+ cells are not altered by treatment with isolated virus (Supporting Fig. 1). Furthermore, CD33+ cells cocultured with HCV+ hepatocytes in the presence of a transwell modestly, but not significantly, suppress CD4+ T-cell activation as compared with control (Supporting Fig. 2), suggesting that close contact of CD33+ cells with HCV+ hepatocytes is required for optimal MDSC induction.
Extracellular HCV Core-Treated CD33+ Cells Suppress CD4+ and CD8+ T-Cell Activation in a Contact-Dependent Manner
Based on a report that CD33+ MDSCs, generated from human PBMCs following exposure to immunosuppressive factors for 7 days, suppress T-cell responsiveness, we assessed whether the immunomodulatory protein, extracellular HCV core, could induce MDSCs to impair T-cell responses.13 To this end, we treated human PBMCs with recombinant extracellular HCV core or control β-galactosidase (β-gal) for 7 days and then selected CD33+ cells using magnetic beads. The cells were then cocultured with CD4+ or CD8+ T cells for 3 days. HCV core-treated CD33+ cells inhibited the proliferation of both CD4 and CD8 T cells (Fig. 2A,B). Production of proinflammatory cytokines, IFN-γ and IL-2, were also inhibited in T cells cocultured with HCV core-treated CD33+ cells as determined by intracellular cytokine staining following 5-hour restimulation with PMA/ionomycin (Fig. 2C,D) and ELISA (Fig. 2E,F; Supporting Fig. 3). In contrast, both HCV core-treated and HCV+ hepatocyte cocultured with purified CD33+ cells did not suppress T cells in these culture conditions (Supporting Fig. 4), suggesting that other cells (i.e., CD33− cells) might contribute, in part, to the generation of HCV-mediated MDSCs.
Fig. 2.

HCV Core-treated CD33+ cells suppress CD4 and CD8 T-cell proliferation and IFN-γ production. Human PBMCs were treated with HCV core or β-gal for 7 days. CD33+ cells were then cocultured with CD4 or CD8 T cells as described above. (A,B) CFSE dilution was then assessed as a measure for proliferation. (C,D) Cells were restimulated with PMA/ionomycin and intracellular expression of IFN-γ was determined. (E,F) Supernatants were collected following 3 days of CD33-T cell coculture and IFN-γ production was measured by ELISA. *P < 0.05.
We next determined if HCV core-treated CD33+ cells required cell contact for T-cell suppression. To accomplish this, we cocultured CD33+ cells with T cells as described above using a transwell plate. As shown in Fig. 3, there was no longer suppression of T-cell proliferation or IFN-γ production by T cells cocultured with HCV core-treated antigen presenting cells. These results suggest that HCV core-mediated inhibition of T-cell responsiveness is dependent on cell-to-cell contact.
HCV Core-Treated CD33+ Cells Are CD14+-CD11b+/lowHLA-DR−/low, and Suppress T Cells Through the Up-regulation of ROS
Phenotypically, human MDSCs have been described as CD33+CD11b+CD14+ and HLADRlow/−.11 However, CD14 levels have varied depending on the system. We assessed the cell surface expression of CD11b, CD14, and HLA-DR in CD33 selected cells 7 days after HCV core treatment. Relative to β-gal, HCV core-treated CD33+ cells expressed equivalent levels of CD14. Notably, core-treated samples expressed only low levels of CD11b and were HLA-DRlow/− (Fig. 4). Immunomodulatory protein B7-H1 was not up-regulated in HCV core-treated samples (Supporting Fig. 5).
Fig. 4.

HCV core-treated CD33+ cells are CD14+CD11b+/lowHLA-DRlow/−. Human PBMCs were treated with 1 μg/mL HCV core or β-gal for 7 days. CD33+ cells were then selected, stained for HLADR, CD14, and CD11b, and surface expression levels were determined by flow cytometry where β-gal-treated cells are represented by a dotted line, and core-treated samples as a solid line. *P < 0.05 ***P < 0.0005.
MDSCs have been found to suppress T-cell responses through several mechanisms.9 They include metabolism of arginine by arginase-1, increased production of nitric oxide, and ROS. To delineate the mechanism by which HCV core-treated CD33+ cells suppress autologous T cells, we first assessed the expression of arginase-1, iNOS, and p47phox, a component of the nicotinamide adenine dinucleotide phosphate oxidase (NOX) complex responsible for ROS production in MDSC, by qPCR. PBMCs were treated with HCV core or β-gal for 7 days and lysates for protein and RNA analysis were harvested from CD33+ cells immediately following selection. HCV core-treated CD33+ cells do not up-regulate the expression of arginase-1 or iNOS. Strikingly, the expression of STAT3-inducible p47phox is significantly up-regulated relative to control at both the RNA and protein level (Fig. 5A,B). NOX complex members gp91phox and p22phox were also modestly up-regulated (Supporting Fig. 6). ROS levels were evaluated by loading CD33+ cells with DCFDA. HCV core-treated CD33+ cells demonstrated significantly higher ROS up-regulation following PMA stimulation compared with control (Fig. 5C). Thus, HCV core-treated CD33+ cells may use ROS to suppress T cells.
Fig. 5.

HCV core-treated CD33+ cell mediated suppression of autologous T cells is ROS-dependent. Human PBMCs were treated with 1 μg/mL HCV core or β-gal for 7 days. CD33+ cells were then selected. (A) Expression of p47phox, arginase-1, and iNOS was assessed by qPCR. Expression levels were assessed relative to hypoxanthine phosphoribosyltransferase (HPRT), and fold induction was calculated relative to untreated controls. (B) Western blot analysis was performed for p47phox. p47phox expression levels were normalized to actin. (C) CD33+ cells were collected, loaded with DCFDA, stimulated samples were treated with PMA, and fluorescence intensities were assessed by flow cytometry. CD33+ cells were cocultured with CD8 T cells as described in the presence of either ROS inactivator (catalase, 100 U/mL), or a combination of arginase inhibitor (nor-NOHA 0.5 mM) and iNOS inhibitor (L-NMMA, 0.5 mM) for 3 days. (D) CFSE dilution was assessed, and (E) cells were restimulated with PMA/ionomycin and intracellular expression of IFN-γ was determined. (F) Supernatants were collected following 3 days of CD33-T cell coculture and IFN-γ production was measured by ELISA. *P < 0.05, **P < 0.005.
Furthermore, the addition of ROS inactivating enzyme, catalase, significantly restores the proliferative capacity of CD4 and CD8 T cells upon coculture with HCV core-treated CD33+ cells (Fig. 5D; Supporting Fig. 7). The addition of catalase also significantly restores IFN-γ responses (Fig. 5E,F; Supporting Fig. 7). Importantly, catalase did not up-regulate the activation of T cells when cocultured with untreated CD33+ cells (Supporting Fig. 8). Not surprisingly, the addition of a combination of inhibitors to arginase and iNOS has no effect, as these genes were not induced following treatment with core. These results clearly demonstrate that HCV core-treated CD33+ cells suppress T-cell responses through the production of ROS.
Chronically Infected HCV Patients Are Phenotypically Similar to MDSCs and Up-regulate p47phox
CD33+ MDSCs can be detected in the peripheral blood of patients with a number of cancer varieties. Therefore, we postulated that chronically infected HCV patients might also have detectable levels of MDSCs. To test this, we first selected CD33+ cells with magnetic beads and then analyzed the expression of CD14, CD11b, and HLA-DR by flow cytometry. These data show that chronically infected persons are CD11b+, CD14+, and display a modest but not statistically significant decrease in HLA-DR expression (Fig. 6A). RNA from these CD33+ cells was also harvested and the expression of arginase-1, iNOS, and p47phox was assessed. Consistent with our results using recombinant HCV core protein, chronically infected individuals expressed significantly higher levels of p47phox compared with CD33+ cells from healthy donors (Fig. 6B). These data strongly suggest that HCV induces the accumulation of ROS producing MDSCs that are detectable in the peripheral blood, thus providing a novel mechanism for HCV-mediated immune suppression.
Discussion
MDSCs play a pivotal role in suppressing host immunity. In this report we show for the first time that HCV induces MDSCs, thus proposing a novel mechanism for HCV-mediated suppression of the host immune response. Our studies indicate that human CD33+ monocytes selected following coculture of HCV (JFH-1)-infected hepatocytes with PBMCs are capable of suppressing autologous T-cell activation. In addition, extracellular HCV core contributes to the induction and/or expansion of MDSCs, leading to the suppression of autologous T-cell proliferation and IFN-γ production following TCR stimulation. These suppressive CD33+ cells exhibit a CD14+CD11b+/low-HLADR−/low phenotype and up-regulate the expression p47phox, a component of the NOX2 complex critical for ROS production.19 The inactivation of ROS in APC-T cell cocultures reverses the suppressive function of HCV-induced MDSCs, thus underscoring ROS as a crucial immunosuppressive factor released by HCV-induced MDSCs. Importantly, CD14+CD11b+-HLADR−/low MDSCs are detectable in the circulating CD33+ monocyte subset from PBMCs of chronic HCV patients and up-regulate the expression of p47phox. Taken together, these results provide compelling evidence that HCV promotes the accumulation of CD33+ MDSCs, resulting in ROS-mediated suppression of T-cell responsiveness.
In light of the important immunoregulatory role of MDSCs, recent studies have focused on identifying factors involved in the induction and differentiation of MDSCs. STAT3 activation has been recently recognized as a key transcription factor for the promotion of MDSCs. MDSCs in melanoma patients are associated with the high level of activated STAT3 expression.25 Likewise, the inhibition of this transcription factor using conditional knockout mice reduced the expansion of MDSCs and improved T-cell responses in tumor-bearing mice.14 In humans, PBMCs treated with a number of cytokines including the combination of IL-6 and GM-CSF induces the generation of CD33+ suppressive cells.13 Importantly, STAT3 has been reported to directly regulate the expression of p47phox, a main component of the ROS-producing NOX2 complex. This study demonstrates a direct link between STAT3 activation and the production of immunosuppressive factors.20 Therefore, based on our recent report that human APCs treated with extracellular HCV core induce STAT3 activation through an IL-6 autocrine pathway, extracellular HCV core might induce MDSCs by through activation of STAT3, resulting in ROS-mediated T-cell suppression.8 Interestingly, the isolation of CD33+ cells prior to treatment with HCV core or coculture with HCV-infected hepatocytes does not result in the generation of MDSCs, suggesting that other mononuclear cells are involved in MDSC generation (data not shown).
Production of ROS by phagocytes protects the host from bacterial infection by way of its antimicrobial properties, but is also important in regulating the host immune response.26 The NADPH oxidase complex catalyzes the production of ROS and is primarily expressed in phagocytes. Functional impairment or a loss of any member of the complex causing a loss of ROS production results in the development of chronic granulomatous disease (CGD).27 Although CGD is characterized by recurrent bacterial and fungal infections and abnormal granuloma formations, CGD patients are also predisposed to autoimmune disease. Moreover, ROS-producing macrophages directly suppress T-cell responses in an arthritis model protecting animals from disease.28 These studies demonstrate a crucial role for ROS in immune regulation, in addition to the well-defined antimicrobial activity. Although the precise mechanism for MDSC-derived ROS-mediated suppression of T cells is not entirely clear, it is thought that ROS may alter the reducing milieu required for optimal T-cell activation, affect the proximal signal transduction of early T-cell signaling events, or induce T-cell apoptosis.29,30 These mechanisms all require close cell-to-cell interaction, as the half-life of ROS is extremely short. This likely explains why HCV core-treated CD33+ cells require cell-to-cell contact for suppression of T-cell responses.
Because a hallmark of chronic HCV infection is associated with poor effector T-cell responses against HCV, it is important to develop therapeutics that can reverse T-cell impairment through modulation of the immune regulatory network. One such target may include MDSCs. Here we show, for the first time, that HCV causes the accumulation of MDSCs capable of suppressing autologous T cells in an ROS-dependent fashion. Importantly, these cells are detectable in the peripheral blood of chronically infected individuals. Therefore, it is plausible that eliminating MDSCs themselves or targeting MDSC-derived suppressive factors may be beneficial in boosting T-cell responses to HCV and improving viral clearance.
Supplementary Material
Acknowledgments
Supported by Training Fellowship (5T32AI00749611 to R.T.) and NIH Grants (U19AI083024, U19AI066328 to Y.S.H.).
We thank the members of the Hahn lab for providing critical advice on this work.
Abbreviations
- APC
antigen-presenting cell
- DC
dendritic cell
- IFN-α
interferon-alpha
- IL
interleukin
- HCV
chronic hepatitis C virus
- MDSC
myeloid-derived suppressor cell
- PBMC
peripheral blood mononuclear cell
- PMN
polymorphonuclear
- ROS
reactive oxygen species
- STAT3
signal transducer and activator of transcription 3
- TLR
Toll-like receptor
Footnotes
Potential conflict of interest: Nothing to report.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Hahn YS. Subversion of immune responses by hepatitis C virus: immunomodulatory strategies beyond evasion? Curr Opin Immunol. 2003;15:443–449. doi: 10.1016/s0952-7915(03)00076-1. [DOI] [PubMed] [Google Scholar]
- 2.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 3.Liu B, Woltman AM, Janssen HL, Boonstra A. Modulation of dendritic cell function by persistent viruses. J Leukoc Biol. 2009;85:205–214. doi: 10.1189/jlb.0408241. [DOI] [PubMed] [Google Scholar]
- 4.Dustin LB, Rice CM. Flying under the radar: the immunobiology of hepatitis C. Annu Rev Immunol. 2007;25:71–99. doi: 10.1146/annurev.immunol.25.022106.141602. [DOI] [PubMed] [Google Scholar]
- 5.Kanto T, Hayashi N, Takehara T, Hagiwara H, Mita E, Naito M, et al. Density analysis of hepatitis C virus particle population in the circulation of infected hosts: implications for virus neutralization or persistence. J Hepatol. 1995;22:440–448. doi: 10.1016/0168-8278(95)80107-3. [DOI] [PubMed] [Google Scholar]
- 6.Cooper S, Erickson AL, Adams EJ, Kansopon J, Weiner AJ, Chien DY, et al. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10:439–449. doi: 10.1016/s1074-7613(00)80044-8. [DOI] [PubMed] [Google Scholar]
- 7.Dolganiuc A, Chang S, Kodys K, Mandrekar P, Bakis G, Cormier M, et al. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol. 2006;177:6758–6768. doi: 10.4049/jimmunol.177.10.6758. [DOI] [PubMed] [Google Scholar]
- 8.Tacke RS, Tosello-Trampont A, Nguyen V, Mullins DW, Hahn YS. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J Biol Chem. 2011;286:10847–10855. doi: 10.1074/jbc.M110.217653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. author reply 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010;185:2273–2284. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–191. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171:1232–1239. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 17.Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. 1998;160:5729–5734. [PubMed] [Google Scholar]
- 18.Harari O, Liao JK. Inhibition of MHC II gene transcription by nitric oxide and antioxidants. Curr Pharm Des. 2004;10:893–898. doi: 10.2174/1381612043452893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386(Pt 3):401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–5701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanda T, Basu A, Steele R, Wakita T, Ryerse JS, Ray R, et al. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J Virol. 2006;80:4633–4639. doi: 10.1128/JVI.80.9.4633-4639.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+HLA-DR−/low cells in mela-noma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010;70:4335–4345. doi: 10.1158/0008-5472.CAN-09-3767. [DOI] [PubMed] [Google Scholar]
- 26.Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J, Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomy-elitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci U S A. 2004;101:12646–12651. doi: 10.1073/pnas.0403831101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578–584. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 28.Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, et al. Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest. 2007;117:3020–3028. doi: 10.1172/JCI31935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, et al. Antigen-presenting dendritic cells provide the reducing extra-cellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A. 2002;99:1491–1496. doi: 10.1073/pnas.022630299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gringhuis SI, Papendrecht-van der Voort EA, Leow A, Nivine Levarht EW, Breedveld FC, Verweij CL. Effect of redox balance alterations on cellular localization of LAT and downstream T-cell receptor signaling pathways. Mol Cell Biol. 2002;22:400–411. doi: 10.1128/MCB.22.2.400-411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.