

Abstract
A study was made to determine if constitutively active adenosine receptors are present at mouse motor nerve endings. In preparations blocked by low Ca2+ / high Mg2+ solution, 8-cyclopentyl-1,3,dipropylxanthine (CPX, 10–100 nM), which has been reported to be both an A1 adenosine receptor antagonist and inverse agonist, produced a dose-dependent increase in the number of acetylcholine quanta released by a nerve impulse. Adenosine deaminase, which degrades ambient adenosine into its inactive congener, inosine, failed to alter the response to 100 nM CPX. 8-cyclopentyltheophylline (CPT, 3 μM), a competitive inhibitor at A1 adenosine receptors, prevented the increase in acetylcholine release produced by CPX. At normal levels of acetylcholine release, neither adenosine deaminase nor CPX affected acetylcholine release at low frequencies of nerve stimulation in (+)-tubocurarine blocked preparations. The results suggest that a proportion of the acetylcholine release process is controlled by constitutively active adenosine receptors at murine motor nerve endings, providing the first evidence for constitutive activity of G-protein-coupled receptors that modulate the function of mammalian nerve endings.
Keywords: G-protein coupled receptors, constitutive activity, A1 adenosine receptor, neuromuscular junction, end-plate potential, neurotransmitter release
1. Introduction
At the skeletal neuromuscular junction, activation of prejunctional A1 adenosine receptors has been shown to inhibit acetylcholine release. Specifically it has been shown that in amphibia, adenosine (derived from the vesicular ATP co-released with acetylcholine) acts as the predominant mediator of prejunctional neuromuscular depression at low frequencies of stimulation (Silinsky and Redman, 1996; Redman and Silinsky, 1993, 1994; Silinsky et al., 1999). Adenosine has also been shown to play a role in mediating the prejunctional depression of neuromuscular transmission in rat and mouse motor nerve endings (Hamilton and Smith, 1991; Nagano et al., 1992; Todd et al., 2010). Thus, agents that oppose the effects of adenosine on neuromuscular transmission may offer potential therapeutic benefit for diseases such as myasthenia gravis and Lambert-Eaton myasthenic syndrome where the normal processes of neuromuscular transmission are impaired.
At the mouse skeletal neuromuscular junctions, the inhibitory effects of adenosine are mediated by a reduction in nerve terminal calcium currents (Silinsky, 2004). Indeed, in wild type mice, the application of supramaximal concentrations of adenosine produce an inhibitory of acetylcholine release to approximately 50% of the control level (Hirsh et al, 2002; Silinsky, 2004, 2008); this is associated with a less than maximal inhibition of calcium currents (Silinsky, 2004, 2008). However, in our studies of murine motor nerve endings we have found that the modulation of nerve terminal calcium currents and hence acetylcholine release by adenosine is not simply limited by the number of A1 adenosine receptors in the nerve endings, but rather is limited by specific elements of the secretory apparatus. Hence, in preparations in which the vesicle protein Rab3A has been deleted, after cleavage of the vesicle protein synaptobrevin, adenosine becomes an extremely potent and efficacious inhibitor of motor nerve terminal calcium currents such that adenosine can eliminate the entire measurable nerve-terminal calcium current (Silinsky, 2008). From this observation it appears that the maximal inhibitory effects of adenosine are unlikely to be determined by the number of adenosine receptors as adenosine receptors appear to be in sufficient concentrations to completely eliminate the calcium current once the constraints of synaptobrevin and other vesicle proteins are removed (Silinsky, 2008).
One potential consequence of a high concentration of adenosine A1 receptors present at motor nerve endings might be that even in the absence of adenosine, nerve evoked acetylcholine release may be constrained by the effects of constitutively active A1 adenosine receptors. We have tested this at the mouse skeletal neuromuscular junction by comparing the effects of A1 adenosine receptor antagonists on the electrophysiological correlates of acetylcholine release at both low levels of release in which both EPPs and MEPPs can be measured and hence quantal analysis can be performed, as well as at normal physiological levels of neurotransmitter release.
2. Material and Methods
2.1 General
The methods used for anesthesia and exsanguinations are in accordance with guidelines laid down by our institutional animal welfare committee and the National Institutes of Health. Specifically mice (B6129F2J, 20–30 g in weight), were humanely anaesthetized with 5% isoflurane for 3–5 minutes, until unresponsive to touch, followed by cervical dislocation and exsanguination. Phrenic nerve-hemidiaphragm nerve-muscle preparations were dissected and pinned in a recording chamber. Solutions were delivered by superfusion with a peristaltic pump and removed by vacuum suction. Electrophysiological recordings were made at room temperature (22–24 °C) with an Axoclamp-2A amplifier, (Axon Instruments). Signals were fed into a personal computer using a Digidata 1200 A/D converter (Axon Instruments). Responses were recorded and analyzed using CDR, WCP and SCAN programs (Strathclyde University Software; John Dempster). All electrophysiological recordings were made from single endplates with each individual fiber serving as its own control. The data were further analyzed using Microsoft Excel, Corel Quattro Pro, and Sigma Plot and Sigma Stat software packages (SPSS Inc., Chicago Illinois).
2.2 Electrophysiological recording methods
Intracellular recordings using microelectrodes were made from the innervated region of the phrenic nerve-diaphragm preparations. Evoked responses were measured as end-plate potentials (EPPs) and spontaneous events measured as miniature EPPs (MEPPs). Microelectrodes were filled with 3 M KCl with resistances 3–10 MΩ. The motor nerve was stimulated at frequencies of 0.5–2Hz for recording of EPPs in the low Ca2+ high Mg2+ solution; this solution was employed to allow for a direct measure of the mean number of acetylcholine quanta released by a nerve impulse. Specifically, the mean level of quantal acetylcholine release was determined directly by calculating the ratio of the mean EPP amplitude to the mean MEPP amplitude. The number of EPPs averaged to determine the mean level of nerve-evoked quantal release ranged from 300–750 events. When EPPs were measured at normal levels of acetylcholine release using (+)-tubocurarine to reduce the EPP below threshold for muscle action potentials, the frequency of stimulation was 0.05 Hz to eliminate the effects of endogenous adenosine derivatives released from nerve ending together with acetylcholine (Redman and Silinsky, 1994).
2.3 Physiological salt solutions and drugs
Control physiological saline solution (pH 7.2 to 7.4) contained (mM) NaCl, 137; KCl, 5; CaCl2, 2; MgCl2, 2; HEPES, 10; dextrose, 11 was used unless otherwise stated. For low Ca2+ / high Mg2+ solution; CaCl2 was 0.35 mM and MgCl2 was 3.0 mM. At normal levels of acetylcholine release, (+)-tubocurarine chloride (3.5 μM) was added to the control physiological saline to reduce the EPPs below threshold for the generation of muscle action potentials. 8-cyclopentyl-1,3,dipropylxanthine (CPX); 8-cyclopentyltheophylline (CPT) and (+)-tubocurarine chloride were obtained from Sigma-Aldrich, St Louis, MO, USA; adenosine deaminase was obtained from Worthington Biochemical Corporation, Freehold, NJ, USA.
2.4 Statistical Methods
Comparisons were made by either parametric statistics (e.g. A. Student's paired t-test) or non-parametric statistics (Mann-Whitney rank sum test, see Glantz, 1992). For more than two groups, an analysis of variance for normally distributed data was followed by multiple comparisons using the Bonferroni inequality (see Glantz, 1992). For the purpose of discussion of the results, differences between groups were considered significant when P < 0.05. Unless otherwise stated, n represents the number of single experiments carried out at single end-plates on individual preparations. Data are presented as means ± 1 S.E.M.
3. Results
3.1 Effect of CPX on nerve-evoked quantal release of murine phrenic nerve endings in low Ca2+ / high Mg2+ solution
CPX is a generally employed as a highly selective competitive inhibitor of at A1 adenosine receptors at vertebrate synapses (Redman and Silinsky, 1993). However, when constitutive activity had been observed in systems where A1 adenosine receptors are expressed at high levels, CPX was found to display inverse agonist activity (Shryock et al., 1998; see also Ma and Green, 1992). Consequently our first series of experiments were designed to determine if CPX increases basal acetylcholine release. Fig. 1 shows the typical experimental data demonstrating effects of CPX on the number of acetylcholine quanta released in a preparation in which low Ca2+ / high Mg2+ solutions were used to decrease end-plate potentials (EPPs) below threshold for muscle action potentials and also allow for direct measurements of acetylcholine release. Note the control traces show fluctuating EPPs and failures of acetylcholine release. In the presence of 100 nM CPX, EPPs increased in amplitude and failures of acetylcholine release were eliminated, indicating an increase in quantal acetylcholine release. In this experiment, the mean level of quantal acetylcholine released was increased more than 2 fold by the application of 100 nM CPX. On average, 100 nM CPX increased acetylcholine release 2.3 fold over the control level (n=5 experiments). The dependence of the increase in acetylcholine release on the concentration of CPX is shown for all experiments in Fig. 2. (For further details, see figure legends).
Fig. 1.

Effect of CPX (100nM) on EPP amplitudes and the number of acetylcholine quanta released. Upper traces depict the control data, lower traces were recorded in the presence of CPX. Note the MEPPs appearing after the EPPs. The average level of acetylcholine release was 0.76 ACh quanta per impulse in control and 1.63 in the presence of CPX. Experiments were made in low Ca2+ / high Mg2+ solutions.
Fig. 2.
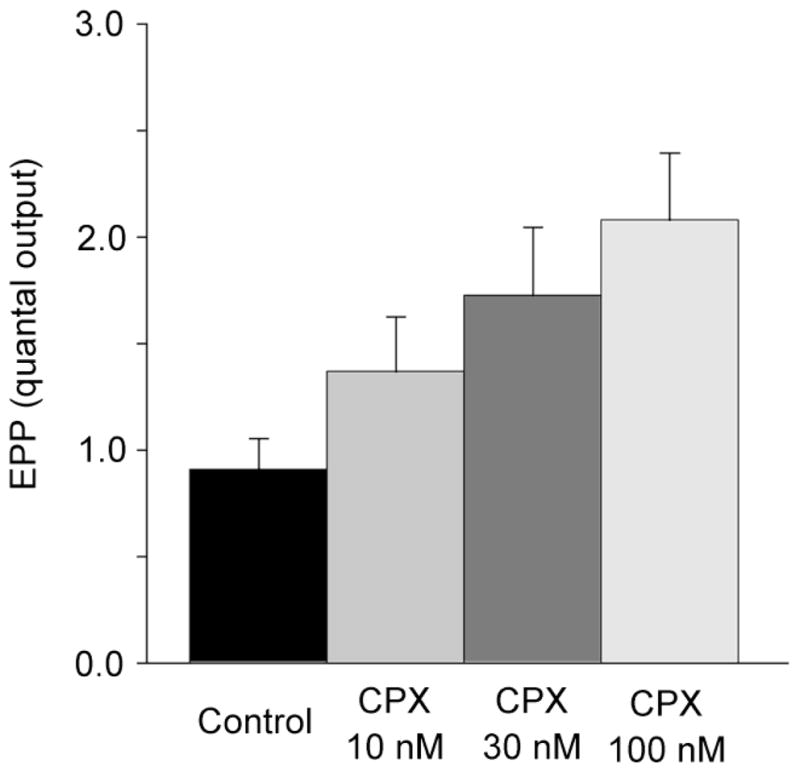
Concentration-dependent increases in the number of acetylcholine quanta released by CPX. Each bar represents the average results from 5 preparations. Increases observed at 30 and 100 nM were highly significant (P<0.05). Experiments were made in low Ca2+ / high Mg2+ solutions.
3.2 Effect of adenosine deaminase on CPX mediated increases in nerve-evoked quantal release in low Ca2+ / high Mg2+ solution
The increases in acetylcholine release produced by CPX depicted in Fig. 1 and Fig. 2 might be due to either the presence of endogenous adenosine leaching from the nerve-muscle preparation or to true negative efficacy associated with the inverse agonist action of CPX, whereby CPX produces a reduction in the proportion of adenosine receptors in the active conformation. To distinguish between these two possibilities, the effect of adenosine deaminase on the actions of CPX was examined. As Fig. 3 shows, adenosine deaminase (5 u/ml), which would degrade any endogenous adenosine to its inactive derivative, inosine (Redman and Silinsky, 1994), failed to produce a statistically-significant increase in acetylcholine release or alter the stimulatory effects of 100 nM CPX. Specifically, the mean increase in acetylcholine release produced by CPX in the absence of adenosine deaminase (2.3 fold over the control level–Fig. 2) was not different from the increase observed and in the presence of adenosine deaminase (2.2 fold over control-fig. 3).
Fig. 3.

Absence of effect of adenosine deaminase (5 u/ml) on the increases in acetylcholine release produced by CPX (100 nM). Each bar represents the average results from 6 preparations. The increase in acetylcholine release produced by CPX in the presence of adenosine deaminase was highly significant (P=0.014). Experiments were made in low Ca2+ / high Mg2+ solutions.
3.3 Effect of CPT on CPX mediated increases in nerve-evoked quantal release in low Ca2+ / high Mg2+ solution
Another competitive inhibitor of A1 adenosine receptors, CPT to our knowledge has not been implicated as an inverse agonist. The effect of CPT on acetylcholine release evoked by CPX was therefore examined. As Fig. 4 shows, CPT (3 μM) while having no significant effect on acetylcholine release, completely blocked the stimulatory effects of 100 nM CPX (for further details see figure legend).
Fig. 4.

CPT (3 μM) fails to affect acetylcholine release but occludes the stimulatory effects of CPX (100 nM) . No statistically significant differences were observed among the three groups in the figure (One Way ANOVA, P=0.75). Each bar represents the average results from 5 preparations. Experiments were made in low Ca2+ / high Mg2+ solutions.
3.4 Effects of adenosine deaminase and CPX on nerve-evoked acetylcholine release at normal physiological levels of Ca2+ and Mg2+
The results thus far suggest that a component of acetylcholine release at low levels of neurotransmitter output is regulated by endogenously active adenosine receptors. To determine if a similar effect occurs at more normal levels of acetylcholine release, the effects of adenosine deaminase and CPX were examined in preparations in which (+)-tubocurarine was used to reduce the EPPs below threshold for action potential generation. The mean number of acetylcholine packets released by a nerve impulse under these conditions has been found to be approximately 35 quanta (Hirsh et al., 2002). Under these conditions, with low frequency nerve stimulation (0.05 Hz) adenosine deaminase (5 u/ml) + CPX (100nM) had no effect on nerve evoked EPP amplitudes. Thus, EPP amplitudes were 3.25 ± 0.54 mV in control; 3.16 ± 0.55 mV in adenosine deaminase (5 u/ml); and 3.28 ± 0.59 mV in adenosine deaminase + CPX (100nM) n=5; ANOVA revealed no significant differences between the groups indicating a lack of constitutive activity at motor nerve endings under these conditions.
4. Discussion
The results presented here both confirm the previously reported inverse A1 adenosine receptor agonist activity of CPX (Shyrock et al., 1998) and provide the first demonstration of the presence of constitutive A1 adenosine receptor activity at magnesium blocked murine motor nerve terminals. Specifically, application of CPX caused a concentration dependent increase in quantal output with a similar concentration dependency to that previously reported by Shyrock et al. (1998) for human A1 adenosine receptors expressed in Chinese hamster ovary cells. In addition, pre-application of adenosine deaminase, which acts to degrade adenosine to an inactive purine (at concentrations previously shown to rapidly and completely abolish the effects of endogenous adenosine in motor nerve preparations in vitro), had no effect on the increases in secretion produced by CPX (Fig. 3). Furthermore, application of the methylxanthine A1 adenosine receptor antagonist CPT had no effect itself on quantal output (supporting the lack of effect of endogenous adenosine) but its pre-application did prevent the increases in quantal output caused by CPX indicating that the stimulatory actions of CPX are mediated through actions on A1 adenosine receptors. Thus the evidence supports the hypothesis that in this system CPT behaves as a neutral antagonist (Kenakin, 1997, 2004) that binds to A1 adenosine receptors and prevents the binding of CPX, but does not change the equilibrium between active and inactive receptors (see Fig. 4).
The reason for the observed difference in effect of CPX on basal acetylcholine release in low Ca2+ /high Mg2+ solutions versus the lack of effect seen at normal acetylcholine outputs is unknown. In previous studies we have found no evidence for any action of (+)-tubocurarine on A1 adenosine receptors (see e.g. Silinsky, 2005 Fig. 3); it thus seems possible that differences in the composition of the bathing fluid used to make direct determinations of the level of quantal release (low Ca2+ high Mg2+ solutions-see 2.2) may promote constitutive activity. This possibility is supported by the published data in other G-protein coupled receptor (GPCR) systems that reveal constitutive activity increasing as the ratio of Mg2+ to Ca2+ is increased (e.g. Glass et al., 1999), especially under conditions where receptor concentrations are not artificially high (Wang et al., 2001). It has been suggested that the effect of Mg2+ on GPCR constitutive activity may result from the Mg2+ dependency of the high-affinity interaction of GTPγS with G proteins (in this instance Gα, see Saidak et al., 2006; Gilman, 1987) which may be conferred by Mg2+ stabilization of the GTPγS G-protein complex (Zelent et al., 2001). Regardless of the explanation, the results at normal levels of acetylcholine output are consistent with previously published results in which no effect of adenosine deaminase, or competitive inhibitors at A1 adenosine receptors were observed at normal levels of release unless the motor nerve was stimulated at frequencies greater than 1Hz and endogenous adenosine emerged (Redman & Silinsky, 1994; Silinsky et al., 1999; Todd et al., 2010). Thus, in subjects that lack any abnormality in magnesium or calcium ion regulation, it appears unlikely that constitutive A1 type receptors activity contributes towards a physiological effect at the skeletal neuromuscular junction.
Conclusions
According to the mathematical framework of receptor theory, constitutive activity is more likely to occur at higher receptor concentrations (Costa and Herz, 1989; de Ligt et al., 2000), as constitutive activity is hyperbolically related to the concentration of GPCRs (Kenakin, 1997, 2004). Thus these studies support the suggestion that high concentrations of A1 adenosine receptors are present at the phrenic nerve endings innervating the murine diaphragm muscle. The results confirm the inverse agonist properties of CPX on A1 adenosine receptors and provide evidence for CPT acting as a neutral antagonist in this system. Lastly, the results presented here provide further evidence for the modulation of constitutive activities of GPCRs by changing the relative concentrations of Mg2+ and Ca2+ ions.
Acknowledgments
This work was supported by grants from the National Institutes of Health of the USPHS (NS12782; AA016513) and from Northwestern Memorial Hospital. We are deeply grateful to Dr. Jeffrey Glassroth for his support and encouragement during his all-too-brief time as Interim Dean at Northwestern University Feinberg School of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Costa T, Herz A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci USA. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ligt RAF, Kourounakis AP, IJzerman AP. Inverse agonism at G protein coupled receptors: (patho) physiological relevance and implications for drug discovery. Brit J Pharmacol. 2000;130:1–12. doi: 10.1038/sj.bjp.0703311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. G Proteins: Transducers of receptor-generated signals. Ann Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Glantz SA. Primer of Biostatistics. N.Y., NY: McGraw Hill Inc; 1992. [Google Scholar]
- Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1999;56:1362–1369. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- Hamilton BR, Smith DO. Autoreceptor-mediated purinergic and cholinergic inhibition of motor nerve terminal calcium currents in the rat. J Physiol. 1991;432:327–341. doi: 10.1113/jphysiol.1991.sp018387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh JK, Searl TJ, Silinsky EM. Regulation by Rab3A of and endogenous modulator of neuro of neurotransmitter release at mouse motor nerve endings. J Physiol. 2002;545:337–343. doi: 10.1113/jphysiol.2002.032516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Efficacy as a vector: the relative prevalence and paucity of inverse agonism. Mol Pharmacol. 2004;65:2–11. doi: 10.1124/mol.65.1.2. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Pharmacologic Analysis of Drug-Receptor Interaction. 3. Lippincott-Raven; Philadelphia, New York: 1997. [Google Scholar]
- Ma H, Green RD. Modulation of cardiac cyclic AMP metabolism by adenosine receptor agonists and antagonists. Mol Pharmacol. 1992;42:831–837. [PubMed] [Google Scholar]
- Nagano O, Földes FF, Nakatsuka H, Reich D, Ohta Y, Sperlagh B, Vizi ES. Presynaptic A1-purinoceptor-mediated inhibitory effects of adenosine and its stable analogues on the mouse hemidiaphragm preparation. Naunyn Schmiedebergs Arch Pharmacol. 1992;346:197–202. doi: 10.1007/BF00165301. Erratum 1993 Naunyn Schmiedebergs Arch Pharmacol 347 346. [DOI] [PubMed] [Google Scholar]
- Redman RS, Silinsky EM. A selective adenosine antagonist (8-cyclopentyl-1,3,- dipropylxathine) eliminates both neuromuscular depression and the action of exogenous adenosine by an effect on A1 receptors. Mol Pharmacol. 1993;44:835–840. [PubMed] [Google Scholar]
- Redman RS, Silinsky EM. ATP released together with acetylcholine as the mediator of neuromuscular depression at frog motor nerve endings. J Physiol. 1994;477:117–127. doi: 10.1113/jphysiol.1994.sp020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidak Z, Blake-Palmer K, Hay DL, Northup JK, Glass M. Differential activation of G-proteins by μ-opioid receptor agonists. Brit J Pharmacol. 2006;147:671–680. doi: 10.1038/sj.bjp.0706661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shryock JC, Ozeck MJ, Belardinelli L. Inverse agonists and neutral antagonists of recombinant human A1 adenosine receptors stably expressed in Chinese hamster ovary cells. Mol Pharmacol. 1998;53:886–893. [PubMed] [Google Scholar]
- Silinsky EM. Adenosine decreases both pre-synaptic calcium currents and neurotransmitter release at the mouse neuromuscular junction. J Physiol. 2004;558:389–401. doi: 10.1113/jphysiol.2004.061457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silinsky EM. Modulation of calcium currents is eliminated after cleavage of a strategic component of the mammalian secretory apparatus. J Physiol. 2005;566:681–688. doi: 10.1113/jphysiol.2005.090647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silinsky EM. Selective disruption of the mammalian secretory apparatus enhances or eliminates calcium current modulation at nerve endings. Proc Natl Acad Sci USA. 2008;105:6427–6432. doi: 10.1073/pnas.0708814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silinsky EM, Hirsh JK, Searl TJ, Redman RS, Watanabe M. Quantal ATP release from motor nerve endings and its role in neurally mediated depression. Prog Brain Res. 1999;120:145–158. doi: 10.1016/s0079-6123(08)63552-9. [DOI] [PubMed] [Google Scholar]
- Silinsky EM, Redman RS. Synchronous release of ATP and neurotransmitter within milliseconds of a motor nerve impulse in the frog. J Physiol. 1996;492:815–822. doi: 10.1113/jphysiol.1996.sp021348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd KJ, Darabid H, Robitaille R. Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci. 2010;30:11870–11882. doi: 10.1523/JNEUROSCI.3165-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Raehal KM, Bilsky EJ, Sadee W. Inverse agonists and neutral antagonists at mu opioid receptor (MOR): possible role of basal receptor signaling in narcotic dependence. J Neurochem. 2001;77:1590–1600. doi: 10.1046/j.1471-4159.2001.00362.x. [DOI] [PubMed] [Google Scholar]
- Zelent B, Veklich Y, Murray J, Parkes JH, Gibson S, Liebman PA. Rapid irreversible G protein alpha subunit misfolding due to intramolecular kinetic bottleneck that precedes Mg2+ “lock” after GTP/GDP exhange. Biochemistry. 2001;40:9647–9656. doi: 10.1021/bi010272u. [DOI] [PubMed] [Google Scholar]