

Abstract
Bacterial flagellin is critical to mediate NLRC4 inflammasome-dependent caspase-1 activation. However, Shigella flexneri, a non-flagellated bacterium, and a flagellin (fliC) knockout strain of Pseudomonas aeruginosa (Pa) are known to activate NLRC4 in bone marrow-derived macrophages. Furthermore, the fliC knockout strain of Pa was used in a mouse model of peritonitis to show the requirement of NLRC4. In a model of pulmonary Pa infection, flagellin was shown to be essential for the induction of NLRC4-dependent caspase-1 activation. Moreover, in all Pa studies, IL-1β production was attenuated in NLRC4−/− mice; however, the role of IL-1β in NLRC4-mediated innate immunity in the lungs against a non-flagellated bacterium was not explored. Here, we report that NLRC4 is important for host survival and bacterial clearance as well as neutrophil-mediated inflammation in the lungs following Klebsiella pneumoniae (Kp) infection. NLRC4 is essential for Kp-induced production of IL-1β, IL-17A, and neutrophil chemoattractants (KC, MIP-2, and LIX) in the lungs. NLRC4 signaling in hematopoietic cells contributes to Kp-induced lung inflammation. Furthermore, exogenous IL-1β, but not IL-18 or IL-17A, partially rescued survival, neutrophil accumulation and cytokine/chemokine expression in the lungs of NLRC4−/− mice following infectious challenge. Furthermore, IL-1R1−/− mice displayed a decrease in neutrophilic inflammation in the lungs after infection. Taken together, these findings provide novel insights into the role of NLRC4 in Kp-induced host defense.
INTRODUCTION
Lower respiratory tract infections remain a major burden of disease in the U.S. and the world as measured by disability adjusted life years (DALYs) (1). The Gram-negative bacterium, Klebsiella pneumoniae (Kp), causes severe pneumonia along with extensive lung damage even with small inoculums. In the last decade, the extensive spread of multiple drug resistant Kp strains has become a severe problem (2–4). In this regard, carbapenem-resistant/β-lactamase-producing Kp causes ≥50% mortality in the U.S. and worldwide (2–4). Furthermore, Kp is known to induce life-threatening pneumonia in alcoholics and diabetics (5–8).
The development of pneumonia depends on a complex interplay between mucosal colonization by the infectious organism and mucosal immunity (8–10). The host innate immune response against infection involves pathogen recognition by pattern recognition receptors (PRRs) expressed on host cells (11–14). In this regard, ligation of both extracellular and intracellular PRRs can induce the expression of cytokines/chemokines and neutrophil migration to the lungs during infection. The recognition of a microbe’s specific molecular structures, also called pattern associated molecular patterns (PAMPs), by PRRs leads to cascades of events ultimately resulting in neutrophil infiltration into the lungs followed by monocyte/macrophage migration to the site of infection (8, 11–14). Nucleotide-binding domain, leucine rich containing (NLR) proteins modulate host immunity via inflammation and apoptosis and are involved in the recognition of PAMPs and damage-associated molecular patterns (DAMPs) such as endogenous ligands released from infected tissues or tissues undergoing destruction (15–19). The recognition of such ligands by NLRs can lead to activation of caspase-1 (previously known as ICE) through the assembly of a cytosolic protein complex known as the inflammasome (15–19).
NLRC4 belongs to NLR family members containing an N-terminal CARD (Caspase Recruitment Domain), a central NOD domain, and a C-terminal leucine rich repeat (LRR) domain and is involved in assembly of the inflammasome complex (20). Several lines of evidence suggest an important function of NLRC4 for caspase-1 activation in response to Salmonella typhimurium (21), Shigella flexneri (22), and Legionella pneumophila (23, 24). In these investigations, animals infected with these pathogens exhibited flagellin-induced activation of NLRC4 leading to the induction of interleukin-1β, IL-18, and macrophage cell death. In particular, a C-terminal region of L. pneumophila flagellin has been shown to activate caspase-1 through NLRC4 (23, 24).
More recently, NLRC4 has been identified as a receptor for bacterial flagellin as well as type III secretion system components (25, 26). Regarding pulmonary pathogens, while a flagellin-deficient strain of P. aeruginosa caused caspase-1 activation in macrophages via NLRC4 demonstrating that NLRC4 activation can be flagellin-independent, the type III secretion system was indispensible for NLRC4-mediated caspase-1 activation (27). Furthermore, NACHT, LRR and PYD domains-containing protein 3 (NALP3) and apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) have been shown to be indispensable to protect mice against a high inoculum of Kp (7.4 × 104/mouse) (28). To this end, we studied the role of NLRC4 in neutrophil-dependent immunity in the lungs against Kp and found that NLRC4 is essential for host survival, bacterial clearance, and neutrophilic inflammation in the lungs in response to Kp infection. NLRC4 signaling in hematopoietic, but not resident cells, predominantly contributes to Kp-induced neutrophilic inflammation in the lungs. Moreover, exogenous IL-1β reversed host defense defects in NLRC4−/− mice following infectious challenge. Our data define NLRC4-dependent caspase-1 activation following Kp infection resulting in production of inflammatory cytokines, recruitment of neutrophils into the lungs, and pathogen clearance/survival.
MATERIALS AND METHODS
Animals
8- to-10-week-old female mice, genetically deficient for Nlrc4 (NLRC4−/−) (27) or Il1r1 (IL-1R1−/−; JAX Mice, Bar Harbor, ME), and weighing 20–25 grams were used for experiments. NLRC4−/− and IL-1R1−/− mice were backcrossed 10 times with age-matched C57Bl/6 mice. NLRC4−/− IL-1R1−/− mice were generated by intercrossing NLRC4−/− and IL-1R1−/− mice. Mice were kept on 12:12 hour light:dark cycle with free access to food and water, and were maintained under specific pathogen free conditions. Animal experiments were approved by the Louisiana State University Animal Research Committee.
Human monocyte-derived macrophage stimulation with Kp
Frozen human monocytes were obtained from Astarte Biologics (Redmond, WA). Cells were thawed at 37°C and resuspended in RPMI containing 5% fetal bovine serum (FBS). The population of monocytes was on average 85–90% pure as determined by flow cytometry using an anti-CD14 marker (data not shown). For monocyte/macrophage differentiation, monocytes were cultured on plates for up to 7 days in RPMI 1640 containing 5% FBS, 1% penicillin-streptomycin, and 100 ng/mL M-CSF (Peprotech, Rocky Hill, NJ). Human monocyte-to-macrophage differentiation was confirmed by changes in morphology and increased expression of macrophage mannose receptor, detected by flow cytometry. For silencing experiments, pre-validated siRNA for human NLRC4 was obtained from Santa Cruz biotechnology (Santa Cruz, CA). Cells (0.5×106) were transfected with 40 nM siRNA or a scramble control (Santa Cruz Biotechnology Inc)) using TransIT-TKO Transfection Reagent from MIRUS (Madison, WI) for 48 hours. Thereafter, cells were infected with 1 MOI of Klebsiella for 6 hours. For cytokine studies, media was collected 24 hours following infection. Cells were washed 3 times with PBS before lysing with Urea/Chaps/Tris buffer containing protease and phosphatase inhibitors. Cell lysates were used for determination of protein expression by Western blot and culture media was used to determine the levels of released cytokines including TNF-α, IL-6, IL-1β, and IL-18.
Murine alveolar macrophage stimulation with Kp
Murine alveolar macrophages were isolated from BALF fluid from NLRC4+/+ and NLRC4−/− mice as described (29–31). Mice were anesthetized by intramuscular injection of 0.02 mL ketamine cocktail (ketamine hydrochloride [80 mg/mL], acepromazine [1.76 mg/mL], and atropine [0.38 g/mL]) and then sacrificed by cardiac exsanguination. Lungs were lavaged with 0.8 mL sterile saline each time through an intratracheal catheter as described previously (31, 32), and a total of 8 mL saline was instilled and recovered from each mouse. The lavage fluid was spun at 300×g for 10 min to pellet alveolar macrophages. Cells were cultured in 12-well culture plates at 37°C with 5% CO2 at a concentration of 0.5×106 cells per well in 1 ml RPMI 1640 medium (Sigma Chemical Co., St. Louis, MO) supplemented with 10% FBS, 1 mM pyruvate, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. After 2 hours of incubation, non-adherent cells were washed off with phosphate buffered saline (PBS), and the medium was replaced. Cells were then infected with 1 MOI of Kp for designated time intervals. For cytokine studies, media was collected at 3 hours and 6 hours following infection. For Western blotting, cells were washed three times with PBS and lysed with Urea/Chaps/Tris buffer containing protease and phosphatase inhibitors.
Animal inoculation with bacteria
K. pneumoniae serotype 2 strain (ATCC 43816) was used for intratracheal inoculation because it induces substantial inflammatory responses in mice (31, 32). The bacteria were grown for 6–8 hours at 37°C in 50 mL tryptic soy broth with continuous shaking at 225 rpm until the mid-logarithmic phase was reached. Bacteria were harvested by centrifuging the culture at 1200×g for 2 min, and washed twice in sterile isotonic saline. The cells were resuspended in isotonic saline at a concentration of 103 CFUs/50 μL/mouse. Ketamine/xylazine mixture was injected i.p. to anesthetize WT and NLRC4−/− mice before surgery. A mid-ventral incision was made, muscles were isolated, and the trachea was exposed. The Kp suspension (103 CFUs in 50 μL) in 0.9% saline (pH 7.4) was inoculated i.t. The CFUs were validated by serially diluting the suspension of initial inoculums and subsequently plating 50 μL aliquots of each dilution onto a tryptic soy agar (TSA) plate and onto a MacConkey agar plate. Similarly, for enumerating bacterial CFUs in lungs and spleen, whole tissues were homogenized in PBS for 15 and 30 sec, respectively, and 20 μL of homogenates were plated in 10-fold serial dilutions onto TSA and onto MacConkey agar plates. After inoculation of NLRC4−/− and NLRC4 +/+ mice with Kp, their survival was monitored for up to 15 days.
Collection of BALF
The animals were euthanized and exsanguinated by cardiac puncture at the designated time points. The trachea was exposed and cannulated with a 20-gauge catheter as described earlier (31–34). BAL fluid was collected by instilling 0.8 mL of PBS containing heparin and dextrose four times. Total leukocytes in BAL fluid were enumerated by counting on a hemocytometer. BAL cells were subsequently subjected to cytospin, stained with Diff-Quick, and differential leukocyte cell counts were determined by standard light microscopy. The remaining (2 mL) of the undiluted cell-free BAL fluid was passed through a 0.22 μm filter and used for the estimation of cytokines/chemokines.
Harvesting lungs
At the designated time points after infection, the whole (non-lavaged) lungs were excised from mice and snap frozen. Further, for long-term storage these lung tissues were stored at −70°C and used for cytokine/chemokine determination, western blots, and MPO activity assay. Briefly, lung tissue was homogenized in 2 mL PBS supplemented with 0.1% triton X-100 and complete protease inhibitor (1 tablet/50 mL media), and the resulting homogenates were centrifuged at 12,000×g/20 min. The supernatants were harvested, passed thorough a 0.22 μm filter and used as required.
Measuring MPO activity
MPO is an enzyme found in the cells of the myeloid lineage and has been used largely as a marker for neutrophil migration into the lungs. Briefly, the lung samples were weighed, homogenized, centrifuged, and the pellet obtained was resuspended in 50 mM potassium phosphate buffer (pH 6.0) supplemented with 0.5% hexadecyltrimethylammonium bromide (HTAB) as described earlier (31–38). Samples were then sonicated, incubated at 60°C for 2 hours, and assayed for MPO activity in a hydrogen peroxide/O-dianisidinebuffer at 460 nm. Change in absorbance was measured every 5 min at 460 nm using a spectrophotometer. The activity was calculated between 0 sec and 90 sec.
Mouse cytokine ELISA
Cytokines/chemokines in the BALF, lung homogenates, and culture media of alveolar macrophages were determined at different time points by sandwich ELISA as described earlier (31–38). The minimum detection limit of the assay was 2 pg/mL of protein. For mouse lungs, TNF-α, IL-6, LIX, and MIP-2 concentrations were normalized to the total protein concentration in the samples measured by Bradford assay (Bio-Rad, Hercules, CA). Data are expressed as pg/mg of total protein for lung tissue and in pg/mL for BALF.
Lung histology
The lungs were perfused from the right ventricle of heart with 10 mL isotonic saline 24 hours post-infection and harvested. For hematoxylin and eosin staining, lungs were fixed in 4% phosphate-buffered formalin, processed in paraffin blocks, and fine sections (5 μm in thickness) were cut. Semiquantative histology was performed by a Veterinary Pathologist in blinded fashion according to the following scoring scale: 0, no inflammatorycells (macrophages or neutrophils) present in section; 1, <5%of section is infiltrated by inflammatory cells; 2, 5–10% of section is infiltrated by inflammatory cells; and 3, >10% of section is infiltrated by inflammatory cells as indicated in our earlier publications (32, 33).
NF-κB DNA binding assay
An ELISA-based NF-κB DNA binding assay in the lungs of saline treated and Kp-infected NLRC4−/− and NLRC4 +/+ mice was performed to detect the activation of the p65 subunit of NF-κB in the nucleus as per the manufacturer’s protocol (Active Motif, Carlsbad, CA). Nuclear extracts were prepared using the Nuclear Extraction Kit (Active Motif). Nuclear extracts containing equal amounts of protein from each lung sample were added to the pre-coated (NF-κB-specific oligonucleotide) 96-well plate. The plate was incubated for 1 hour at RT. After washing the plate three times, a primary Ab specific for NF-κB/p65 was added and the plate was incubated for 1 hour at RT. After three washings to remove excess primary Ab, an anti-HRP conjugate was added to the plate and the optical density was measured at 450 nm (31–34).
Bone marrow chimeras
BM transplantation experiments were performed as described in earlier publications (31, 35). BM was flushed from tibias and femurs from donor mice and a total of 8 × 106 BM cells were injected into the tail veins of lethally irradiated (two 525-rad doses separated by 3 hours) recipient mice. Reconstituted mice were treated with 0.2% neomycin sulfate for the first 3 weeks post-transplantation. Experiments were performed 8 weeks after BM reconstitution. We found that more than 84% of blood leukocytes were derived from donor marrow at the time the mice were used for experiments (6–8 weeks post-transplantation).
Immunoblotting
The harvested lungs from NLRC4+/+ and NLRC4−/− mice were homogenized in 1 mL of PBS containing 0.1 % Triton X-100 and complete protease and phosphatase inhibitor cocktail (Roche Co., IN) in chilled conditions for 2.5 min. TissueLyser II (Qiagen, Valencia, CA) was used for the homogenization and the samples were subsequently centrifuged at maximum speed in a microfuge for 20 min at 4°C to remove cellular debris. The resulting supernatant was used for immunoblotting. Bradford protein assay (Bio-Rad) was performed to ensure equal quantity loading of protein on the gel. The samples were resolved on 8–15% tris-glycine gels and the proteins were transferred on to a PVDF membrane using standard protocols. Antibodies recognizing phospho-IKKα/β (Ser 176/180), phospho-NF-κB/p65 (Ser 536), phospho-Iκ-Bα (Ser 32), IKKβ, NF-κB/p65, Iκ-Bα, VCAM-1, ICAM-1, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), phospho-p38 MAPK (Thr180/Tyr182), phospho-SAPK/JNK (Thr183/Tyr185), cleaved caspase-1, cleaved IL-1β, and cleaved IL-18 were added at a 1:1000 dilution, whereas, Abs to total p38 and GAPDH were added at 1:5000 (Cell Signaling, Danvers, MA; Santa Cruz Biotech Inc., Santa Cruz, CA). Secondary antibodies consisted of horseradish peroxidase-conjugated goat anti-mouse (for total and phospho Iκ-Bα; GE Healthcare, Waukesha, WI), anti-rabbit (for phospho antibodies, IKKβ, NF-κB/p65, total p38 and GAPDH; GE Healthcare) and rabbit anti-goat (for VCAM-1 and ICAM-1; eBiosciences, San Diego, CA). The membranes were processed with ECL chemiluminescence kit (ThermoFisher Scientific, Waltham, MA), and the signal was detected by exposing the processed blots to X-ray film (Biomax Films, Kodak, NY) (31–34).
Cell surface and intracellular staining
The method to detect IL-17A-producing cells in the lungs was described in earlier publications (34, 39, 40). Briefly, lungs were minced, digested with collagenase for 90 min and made into a single cell suspension. Cells were stimulated with PMA and ionomycin in the presence of GolgiStop (BDBiosciences, San Diego, CA) for 5 hours. Following stimulation, cells were washed with PBS, surface stained with markers for IL-17A-producing T cells (γδ, NK1.1, CD4 and CD8α) for 30 mins. Cells were washed, fixed, and permeabilized for intracellular staining with IL-17A and IFN-γ Abs. Finally, cells were washed and resuspended for flow cytometric analysis. FlowJo software (Tree Star, Ashland, OR) was used to analyze the data.
Administration of recombinant murine IL-1β, IL-17A, IL-18 and IL-1α
A single dose of 1 μg of active recombinant murine proteins in 50 μl [IL-1β, IL-1α, IL-17A and IL-18 (R&D Systems, Minneapolis, MN)] in 0.1% BSA containing saline or vehicle alone (0.1% BSA in saline) was administered 1 h after i.t. administration with K. pneumoniae (1 × 103 CFUs/50 μl) to NLRC4−/− mice. This i.t. dose of rIL-1β (1 μg/50 μl) was previously shown in other models to be biologically active in vivo (41–43).
Determination of pyroptosis by flow cytometry
Lung or spleen digests from C57BL/6 or NLRC4−/− mice challenged with K. pneumoniae for 24 or 48 h were used to determine cells undergoing pyroptosis. Briefly, lungs were minced, digested with collagenase filtered through 0.70 μfilter to make single cell suspensions. Furthermore, spleens were mashed and the resulting cell suspension was passed through 0.70 μ filter. Following 2 PBS washings, cells were FcR blocked and aliquoted for surface staining with conjugated PerCP anti-mouse Gr-1/Ly6G or EMR1 and APC anti-mouse CCR2 or CXCR2. Red blood cells (RBCs) were lysed by adding NH4Cl lysing buffer. Samples were centrifuged and washed twice with 1xPBS. Cells were resuspended in 1x binding buffer and 5 μl of FITC Annexin V and 5 μl of PI were added according to the manufacturer’s protocol (Annexin V apoptosis detection kit from BD Pharmingen). The cell suspension was vortexed and incubated for 15 mins in the dark at room temperature. A total of 100 μl 1x binding buffer was added and the cells were analyzed by flow cytometry. CCR2 or CXCR2 positive Gr-1/Ly6G (neutrophils) and EMR1 (macrophages) that were positive for FITC Annexin V and negative for PI are shown in histograms.
Statistical analysis
Data are expressed as mean ± SE. The intensity of immunoreactive bands was determined using Gel Digitizing Software (UN-SCAN-IT gel™) from Silk Scientific, Inc (Orem, Utah). Data were analyzed by ANOVA followed by Bonferroni’s post hoc analysis for multiple comparisons. All statistical calculations were performed using InStat software and GraphPad Prizm 4.0. Differences were considered statistically significant at *P<0.05 when compared with control. Survival curves were compared by Wilcoxon rank sign test.
RESULTS
NLRC4 regulates Kp-induced proinflammatory mediators in human and murine macrophages
Macrophages are the sentinel cells of pathogen recognition in the lung and are a critical component of the innate immune response to microbes (44, 45). The NLRC4 inflammasome has been implicated in influencing the production of proinflammatory mediators in bone marrow-derived macrophages (23, 25). We first compared the differences in the levels of inflammatory cytokines in human blood monocyte-derived macrophages following NLRC4 siRNA knockdown and in murine alveolar macrophages obtained from NLRC4−/− mice following infection with the Gram-negative bacterium, Kp. In siRNA-transfected human monocyte-derived macrophages, we found reduced IL-6, IL-1β, and IL-18 levels (Fig. 1A), along with attenuated activation of NF-κB and MAPKs 6 hours post-Kp infection (Fig. 1B–C). Similarly, in NLRC4−/−murine alveolar macrophages, we observed decreased cytokine (TNF-α, IL-6, and IL-1β) and neutrophil chemokine (KC and MIP-2) levels (Fig. 1D), as well as reduced activation of NF-κB and MAPKs 1 hour and 3 hours post-Kp infection compared to NLRC4+/+ macrophages (Fig 1E–F). Together, these data suggest that NLRC4 is an important factor in the Kp-mediated inflammatory response in macrophages.
Figure 1. Effect of NLRC4 on cytokine/chemokine production and NF-κB and MAPK activation in vitro after Kp infection.
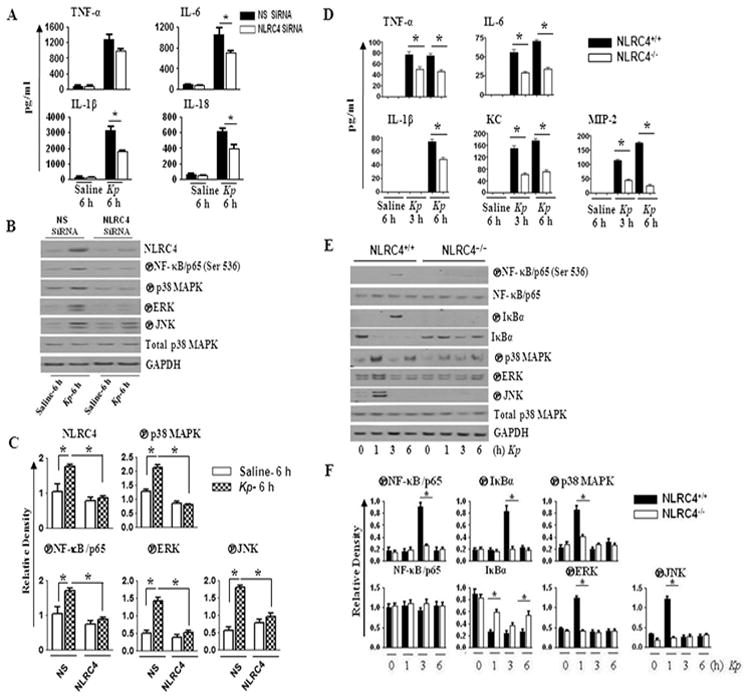
A. Human monocyte-derived macrophages (0.5 x106) were transfected with 40 nM NLRC4 siRNA or scrambled siRNA (nonspecific; NS) for 48 h. Cells were then infected with 1 MOI of Kp for 6 h and cytokine/chemokine levels in supernatants were measured by sandwich ELISA. Means ± SE values were obtained from three independent experiments. *P < 0.05 comparing NLRC4 siRNA- with scrambled (control) siRNA-treated cells. B. Human macrophages (0.5 x106) were transfected with siRNA and then infected with 1 MOI of Kp for 6 h and cell lysates were used for the measurement of NF-κB and MAPK activation by western blotting. Representative blots are shown from three blots/experiments with identical results. C. Densitometry was performed from three separate blots using Gel Digitizing Software and normalized to GAPDH (for NF-κB activation) or total p38 (for MAPK activation). Means ± SE values were obtained from three separate experiments. *P < 0.05 comparing NLRC4 siRNA-transfected with control siRNA-transfected macrophages. D. Mouse alveolar macrophages (0.5 X 106 cells/mL) were challenged with 1 MOI of Kp for different time intervals, and culture supernatants were harvested for cytokine/chemokine determination by ELISA. Means ± SE values were obtained from three independent experiments. *P < 0.05 comparing NLRC4−/− with NLRC4+/+ macrophages. E. Mouse alveolar macrophages infected with 1 MOI of Kp were used for the assessment of NF-κB and MAPK activation by western. Representative blots are shown from three blots/experiments with identical results. F. Immunoreactive bands were quantified by densitometry and normalized to GAPDH (for NF-κB activation) or total p38 (for MAPK activation). Data are expressed as means ± SE (n = 3). NS, non-specific
NLRC4−/− mice show reduced survival, enhanced bacterial burden in the lungs, as well as attenuated neutrophil influx and cytokine production following Kp infection
To assess the importance of NLRC4 in the lungs during Gram-negative bacterial infection, NLRC4−/− and NLRC4+/+ (WT) mice were infected intratracheally (i.t.) with two doses of Kp (103 or 104 CFUs/mouse) and survival was monitored up to 15 days post-infection. The NLRC4−/− group showed reduced survival to both of the infectious doses. In this regard, only 16.7% of NLRC4−/−animals survived to day 15 after infection with the higher dose of Kp, compared to 50% of WT mice. Similarly, in response to the lower dose of Kp, only 40% of NLRC4−/− animals survived whereas 70% of WT mice survived to day 15 (Figs 2A–B).
Figure 2. Role of NLRC4 in host defense against pulmonary Kp infection.

A–B. NLRC4 deficiency enhances mortality induced by intratracheal K. pneumoniae. NLRC4−/− and C57BL/6 (NLRC4+/+) mice were inoculated i.t. with 1,000 (A) or 10,000 CFUs (B) of Kp. Mortality was monitored (*, P < 0.05 by log rank test). Data shown represent n = 12 mice/group from one of two representative independent experiments. C–D. NLRC4−/− mice show higher bacterial burden in the lungs and dissemination in response to Kp infection. Mice were treated with 1 × 103 CFUs of Kp i.t. and lung and spleen homogenates were cultured 24 h or 48 h. Data shown represent mean parenchymal CFUs ± SE (*P < 0.05 comparing NLRC4+/+ with NLRC4−/− mice). E–G. NLRC4 deficiency reduces influx of PMNs to the Kp-exposed lung. NLRC4−/− or C57BL/6 (NLRC4+/+) mice were exposed to 1 × 103 CFUs of Kp i.t. BALF total white blood cells (E) and +/+ neutrophils (F) were enumerated 24 h and 48 h after exposure (*, P < 0.05 between NLRC4 with NLRC4−/− mice; n = 4–7/group). MPO activity, a quantitative measure of PMNs, was measured in lung homogenates 24 h and 48 h (G) after Kp challenge (*, P < 0.05). Data shown are representative of three independent experiments (n = 4–6/group). H. Reduced lung pathology in NLRC4−/− mice following Kp inoculation. Mice were inoculated with 1 × 103 CFUs of Kp/mouse i.t. Lungs were obtained 24 h and 48 h post-infection and stained with H&E, and inflammatory changes in histological sections were scored. Shown are representative sections from four mice under each condition with identical results (magnification, × 200). Semiquantitative Inflammation score is a quantification of four lung sections in each group. I. NLRC4 deficiency inhibits Kp-induced airspace cytokine and chemokine expression. TNF-α, IL-1β, IL-17A, KC, MIP-2, and LIX in BALF (pg/ml) and lung homogenates (pg/mg) were measured by ELISA 24 h and 48 h after Kp challenge (*, P < 0.05 between NLRC4+/+ with NLRC4−/− mice; n = 4–7 mice/group).
To determine whether survival of NLRC4-deficient mice was associated with a defect in bacterial clearance in the lungs and/or bacterial dissemination, we determined bacterial counts 24 hours and 48 hours post-Kp infection. NLRC4−/− mice demonstrated higher lung and spleen CFUs compared with their WT counterparts at 48 hours (Figs 2C–D). We also observed enhanced dissemination of Kp in blood, kidneys, and liver of NLRC4−/− mice as compared with their littermate controls (data not shown). Thus, NLRC4−/− mice have a defect in bacterial clearance and dissemination, which likely explains the decreased survival of NLRC4−/− mice following Kp infection.
In order to elucidate the mechanisms associated with enhanced bacterial CFUs in the lungs and spleens, we assessed pulmonary neutrophil recruitment following Kp challenge. Total WBC and neutrophil accumulation in the airspaces of NLRC4−/− mice was reduced at 48 hours compared with WT controls (Figs. 2E–F). In control (saline-challenged) groups, neutrophil accumulation in the lungs was not observed either in NLRC4−/− or in NLRC4+/+ groups (Figs. 2E–F). Consistent with reduced neutrophil recruitment into the lungs, we also observed attenuated MPO activity in lung homogenates from NLRC4−/− mice (Fig. 2G). NLRC4+/+ mice demonstrated severe suppurative bronchopneumonia (score of 3.0), whereas NLRC4−/− mice displayed moderate suppurative pneumonia (score of 2.0) 48 hours after Kp infection. In contrast, no pathological changes were observed in saline-challenged (control) lungs obtained from either NLRC4−/− or NLRC4+/+ animals (Fig. 2H).
Since the impairment in Kp-induced neutrophil accumulation observed in NLRC4−/− mice likely reflects a decrease in the production of cytokines/chemokines in the lungs upon infection, we quantified the expression of cytokines (TNF-α, IL-1β, and IL-17A) and neutrophil chemoattractants (KC, MIP-2, and LIX) in bronchoalveolar lavage (BAL) fluid and lung homogenates 24 hours and 48 hours after Kp challenge. We found that the levels of TNF-α, KC, MIP-2, and LIX were attenuated in NLRC4−/− mice 48 hours following Kp infection. Strikingly, IL-1β and IL-17A levels in NLRC4−/− mice were attenuated even as early as 24 hours following Kp challenge (Fig. 2I).
NLRC4−/− mice exhibit decreased IL-17A-producing cells in the lung
Because we observed a reduction in IL-17A in the lungs of NLRC4−/− mice following Kp infection, we decided to look for the presence of IL-17A producing-T cell subsets in the lungs during Kp infection by flow cytometry using intracellular and surface staining. In an earlier report, we found that natural killer cells (NK) and gamma delta (γδ) T cells were the predominant sources of IL-17A production in the lungs 6 hours post E. coli infection (34). In contrast, upon Kp infection we found that γδ T cells (A), CD4+ T cells (B), CD8+ T cells (C), and NK/NKT cells (D) all produce IL-17A in the lung 48 hours post-infection (E), however, the proportion of T cell subsets that produce IL-17A and/or IFN-γ was reduced in the lungs of NLRC4−/− mice as compared to controls (Fig. 3).
Figure 3. Importance of NLRC4 in IL-17A producing cells in the lung in response to Kp infection.

A. Shown are representative FACS plots of lung homogenates after Kp infection showing IFN-γ- and IL-17A-producing γδ, CD4, CD8, and NK T cells. Lung T cells were isolated 48 h after Kp infection and immunostained for surface CD8, and intracellular IFN-γ and IL-17A. Shown is a representative of three separate experiments. B. NLRC4 deficiency decreases the number of of IL-17A-expressing T cells. Data shown are representative of three separate experiments (*P < 0.05; n = 4–6/group).
NLRC4 deficiency impairs activation of NF-κB and MAPK as well as expression of ICAM-1 and VCAM-1
To investigate the cause of the reduced cytokine/chemokine expression in NLRC4−/− mice, we examined the activation of transcription factors, including NF-κB/p65, in the lungs following Kp infection. In this regard, we focused on the activation of NF-κB, as this is the most extensively studied transcription factor known to regulate a variety of genes encoding proinflammatory factors (46, 47). To determine activation of NF-κB/p65 in NLRC4−/− mice after Kp infection, we determined p65 DNA binding using nuclear extracts from the lungs. Our findings revealed a decrease in NF-κB/p65 DNA binding in cells isolated from NLRC4−/− mice as compared to their WT counterparts 48 hours after Kp infection (Fig. 4A). We utilized western blotting to further examine NF-κB activation in the lungs following Kp challenge. In NLRC4−/−mice, we observed a decrease in NF-κB/p65 phosphorylation at Ser536 24 hours and 48 hours post-Kp infection. The decrease in p65 phosphorylation correlated with a decrease in the phosphorylation of IκBα (Ser32) in the lungs of NLRC4−/− mice starting at 24 hours post-infection and persisted up to 48 hours after Kp infection. Concomitantly, we observed increased accumulation of IκBα protein in NLRC4−/− mice 48 hours after Kp infection. In control (saline-challenged) mice, however, we detected extremely low levels of phosphorylated IKKα/β or NF-κB/p65 in NLRC4−/− and NLRC4+/+ (WT) animals (Figs. 4B–C).
Figure 4. Effect of NLRC4 on activation of NF-κB, expression of cell adhesion molecules, and activation of MAPK in vivo after Kp infection.

A. NLRC4 ablation decreases NF-κB DNA binding in lung homogenates following Kp infection. NLRC4+/+ and NLRC4−/− mice were infected with 1 × 103 CFUs of Kp/mouse i.t. and lungs were obtained 24 h and 48 h post-infection. Nuclear fractions obtained from lung homogenates were used for NF-κB binding ELISA. The values are means ± standard errors. Values significantly different between the Kp- and saline-treated groups are indicated by asterisks (P < 0.05; 4–6 mice/group). OD450 nm, optical density at 450 nm. B. NLRC4 deficiency reduces NF-κB activation and expression of ICAM-1 and VCAM-1 in the lung after Kp exposure. NLRC4+/+ and NLRC4−/− mice were infected with 1 × 103 CFUs of Kp/mouse i.t. and lungs were obtained 24 h and 48 h post-infection. Lung homogenates were used for activation of NF-κB and expression of ICAM-1 and VCAM-1 by Western blotting. Representative blot is shown from three blots/experiments with identical results. C. Protein immunoblot bands were quantified by densitometry and normalized to GAPDH. Data are expressed as means ± SE (n = 3). D. NLRC4 ablation reduces MAPK activation in the lung following Kp exposure. NLRC4+/+ and NLRC4−/− mice were infected with 1 × 103 CFUs of Kp/mouse i.t. and lungs were obtained 24 h and 48 h post-infection. Lung homogenates were used to determine activation of MAPKs by western blotting. A representative blot is shown from three blots/experiments with identical results. E. The intensity of immunoreactive bands was quantified by densitometry and normalized to total p38. Data are expressed as means ± SE (*, P < 0.05 between NLRC4+/+ with NLRC4−/− mice; n = 3/group).
Neutrophil recruitment from the bloodstream into lungs involves numerous sequential steps mediated by interactions of cell adhesion molecules on leukocytes and endothelium (8–10, 48). Since it has been shown that VCAM-1 and ICAM-1 play key roles in neutrophil extravasation, we determined the expression levels of VCAM-1 and ICAM-1 in lung homogenates after Kp infection. In NLRC4−/− mice, the expression of both ICAM-1 and VCAM-1 was reduced 48 hours following Kp infection compared to control mice (Figs. 4B–C).
Decreased neutrophil accumulation in the lungs observed in response to Kp in NLRC4−/− mice could also be the result of attenuated activation of MAPKs, which regulate transcription factors such as AP-1 and STAT-1 (49, 50). In this context, we studied the activation of ERK, JNK, and p38. Unlike WT controls, NLRC4−/− mice displayed attenuated activation of ERK and JNK at both 24 hours and 48 hours after Kp infection. Not surprisingly, activation of MAPKs was not observed in WT or in NLRC4−/− mice following saline challenge (Figs. 4D–E).
NLRC4−/− mice demonstrate reduced cleavage of caspase-1, IL-1β, and IL-18 following Kp infection
In general, inflammasome activation triggers autocatalytic activation or cleavage of caspase-1 (16, 19). Caspase-1, in turn, cleaves pro-IL-1β and pro-IL-18, producing mature IL-1β and IL-18 (16, 19, 27). In an effort to clarify if NLRC4 has functions in Kp-induced cleavage of caspase-1, pro-IL-1β or pro-IL-18, we measured their cleavage products in the lungs following Kp infection and observed reduced cleavage of caspase-1, IL-1β, and IL-18 in NLRC4-deficient mice as compared to control mice (Figs. 5A–B). This suggests that activation of caspase-1 and cleavage of IL-1β and IL-18 are dependent upon NLRC4 activation following infection with Kp. dependent neutrophil-mediated host innate immunity against Kp.
Figure 5. Importance of NLRC4 on cleavage of caspase-1, IL-1β, and IL-18 in vivo after Kp infection.

A. NLRC4−/− mice demonstrate reduced cleavage of caspase-1, IL-1β, and IL-18 in lung homogenates following Kp infection. NLRC4+/+ and NLRC4−/− mice were infected with 1 × 103 CFUs of Kp/mouse i.t. and lungs were obtained 24 h and 48 h after infection. Lung homogenates were used to determine activation of caspase-1 and cleavage of IL-1β and IL-18. A representative blot is shown from three blots/experiments with identical results. B. Protein immunoblot bands were quantified by densitometry and normalized to GAPDH. Data expressed as means ± SE (*, P < 0.05 between NLRC4+/+ with NLRC4−/− mice; n = 3). Cl, cleaved.
NLRC4 does not induce pyroptosis in pulmonary macrophages and neutrophils following Kp infection
NLRC4 has been shown to mediate pyroptosis in flagellated bacterial infection models, caused by Burkholderia pseudomallei (51) and Salmonella typhimurium (22). Caspase-1 activation can trigger a form of cell death called pyroptosis that requires the proteolytic cleavage of caspase-1 (52). Pyroptosis is characterized by the insertion of pores into the plasma membrane of myeloid cells, including macrophages and neutrophils, which can be detected by annexin V binding. In order to investigate the role of NLRC4-mediated caspase-1 dependent pyroptosis in host defense following Kp-mediated caspase-1 activation, we used flow cytometry in cells isolated from the lung and spleen. Our results suggest that caspase-1 activation by NLRC4 in response to Kp infection in vivo does not induce pyroptosis in CCR2 or CXCR2 positive macrophages and neutrophils of the lung and spleen (Fig. 6).
Figure 6. Role of NLRC4 in the induction of pyroptosis in macrophages and neutrophils in the lung and spleen following Kp infection.
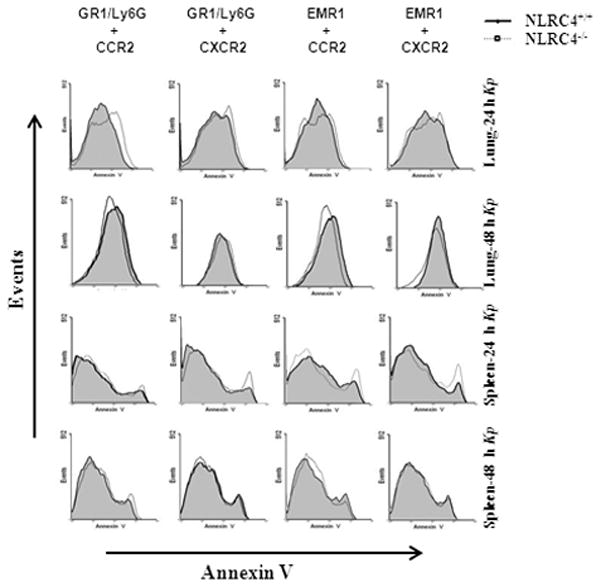
A. Shown are representative FACS plots of CCR2 or CXCR2 positive neutrophils and macrophages obtained from lung and spleen homogenates following Kp infection. Infected lungs and spleens were collagenase digested and used to determine propidium iodide negative neutrophils and macrophages which were Annexin V positive by Flow cytometry at 24 or 48 h post-infection to determine cells undergoing pyroptosis. Representative histograms from CCR2 or CXCR2 expressing Annexin V positive neutrophils and macrophages are shown from three independent experiments (n = 3/group).
Neutrophil recruitment to the lungs in response to Kp infection is dependent upon NLRC4 signaling by BM-derived cells and not resident lung cells
Because lung inflammation in Kp-infected mice could be due to recruited bone marrow cells or resident alveolar cells, we determined which population of cell-derived NLRC4 signaling was important in promoting neutrophil recruitment and host immunity. To address this issue, we lethally-irradiated WT or NLRC4−/− mice were reconstituted with bone marrow from donor WT or NLRC4−/− mice to generate four groups: 1) WT mice reconstituted with WT marrow (WT→WT); 2) WT mice reconstituted with NLRC4−/− marrow (NLRC4−/−→WT); 3) NLRC4−/− mice reconstituted with WT marrow (WT→NLRC4−/−); and 4) NLRC4−/− mice reconstituted with NLRC4−/− marrow (NLRC4−/−→NLRC4−/−). Eight weeks post-reconstitution, these bone marrow chimera mice were i.t. inoculated with Kp and neutrophil accumulation in the BAL fluid was determined. We found that Kp-induced neutrophil influx into the lungs was attenuated in NLRC4−/−→NLRC4−/− and NLRC4 −/−→WT chimera mice as compared with WT→WT chimera animals (Fig. 7A). Neutrophil recruitment to the lung was not observed in any chimeric mice in response to saline challenge (data not shown). Together, these data suggest that NLRC4 signaling by bone marrow cells is required for neutrophil recruitment and innate immunity against Kp infection.
Figure 7. Relative contribution of NLRC4-expressing bone marrow-derived versus resident cells to Kp-induced neutrophil recruitment.

A. Bone marrow chimeras were generated with WT and NLRC4−/− mice. Mice were then infected with 1 × 103 CFUs of Kp/mouse i.t. and BALF neutrophils were enumerated 48 h after exposure (*, P < 0.05 between WT/NLRC4+/+ with NLRC4−/− mice; n = 4–6/group). B. Administration of rIL-7A, rIL-1β, or rIL-18 after Kp infection rescues NLRC4 deficiency. NLRC4−/− mice and WT controls were inoculated i.t. with Kp (1 × 103 CFUs in 50 μl of PBS) and administered a single dose of recombinant murine IL-17A, IL-1β, IL-18, or vehicle. BALF neutrophils (B) were enumerated 48 h after exposure (*, P < 0.05 between NLRC4+/+ with NLRC4−/− mice; n = 5–7/group). C. Survival was monitored up to 15 days post-exposure (*, P < 0.05 between NLRC4+/+ with NLRC4−/− mice; n = 20/group). D–F. NLRC4−/− mice and WT controls were inoculated i.t. with Kp (1 × 103 CFUs in 50 μL of PBS) followed by administration of different doses of recombinant murine IL-1β or vehicle (saline) alone to NLRC4−/− mice. BALF total white blood cells (D), neutrophils (E), and cytokine/chemokines levels (F) were measured 48 h after Kp exposure (*, P < 0.05 between NLRC4+/+ with NLRC4−/− mice; n = 4–6/group).
Administration of rIL-1β but not rIL-17A or rIL-18 following Kp infection rescues neutrophil recruitment in NLRC4−/− mice
Since reduced IL-1β, IL-17A, and IL-18 levels were observed in NLRC4−/− mice following Kp infection, we wanted to determine if exogenous IL-1β, IL-17A, or IL-18 could rescue neutrophil accumulation and cytokine/chemokines production in the lungs of NLRC4−/− mice following Kp infection. To this end, NLRC4−/− mice were administered a single dose of 1 μg of IL-1β, IL-17A, or IL-18 i.t. 1 hour post-Kp infection. Administration of IL-1β in NLRC4−/− mice after Kp infection resulted in increased neutrophil recruitment to the lungs (Figs. 7B) as compared to administration of IL-18 or IL-17A, which did not augment neutrophil recruitment (Figs. 7B). Notably, unlike IL-1β, administration of exogenous IL-1α in NLRC4−/− mice after Kp challenge did not enhance neutrophil recruitment to the lungs (data not shown). We also performed survival experiments in response to Kp administration after i.t. IL-1β treatment. Our data indicated that administration of IL-1β in NLRC4−/− mice after Kp infection resulted in enhanced survival (Figs. 7C) Given the critical role of IL-1β in promoting neutrophil accumulation against i.t. Kp challenge, we administered three concentrations of IL-1β in NLRC4−/− mice to demonstrate concentration-dependent rescue after Kp infection. In this regard, rIL-1β partially rescued neutrophil influx in NLRC4−/− mice in a dose-dependent manner (Fig. 7D–E). In addition, we found that the production of KC, MIP-2, LIX, and IL-17A in the lungs of NLRC4−/− mice was augmented in Kp-challenged mice following administration of exogenous IL-1β (Fig. 7F). Together, these findings demonstrate that rIL-1β plays an important role in NLRC4-dependent neutrophil-mediated host innate immunity against Kp
IL-1R1−/− deficient mice exhibit defects in bacterial clearance, neutrophil recruitment, and chemokine/cytokine production after exposure to Kp
To investigate the contribution of NLRC4-dependent IL-1β in promoting neutrophil accumulation against i.t. Kp challenge, we used IL-1β receptor deficient (IL-1R1−/−) mice in our Kp infection model and evaluated survival, bacterial CFUs, neutrophil recruitment, and neutrophil chemoattractant levels. As compared with WT controls, IL-1R1−/− mice displayed reduced survival, enhanced bacterial burden and dissemination in the lungs, along with reduced neutrophil chemoattractant expression (KC and MIP-2) and neutrophil accumulation in the lungs following Kp infection (Figs. 8A-G). Similarly, activation of NF-κB and MAPKs, and expression of ICAM-1 and VCAM-1 were reduced in IL-R1−/− mice following Kp infection (Figs. 8H–I). To determine if exogenous IL-1β alone could reconstitute neutrophil recruitment, NLRC4/IL-1R1−/− (DKO) and IL-1R1−/− (KO) mice were administered a single dose of 1 μg of IL-1β i.t. 1 hour post-infection, however, we found that IL-1β administration did not rescue neutrophil influx in DKO mice (Fig. 8J). Collectively these results support the role of NLRC4-IL-β axis in Kp-induced innate immunity.
Figure 8. Role of IL-1R1 in host defense against pulmonary Kp infection.

A. IL-1R1−/− mice are unable to protect mice and control bacterial growth during acute Kp infection. Mice treated with 1 × 103 CFUs of Kp i.t., and survival was monitored up to 15 days (*, P < 0.05 between NLRC4+/+ with NLRC4−/− mice; n = 20/group). In the other set of experiments, lung homogenates were cultured 24 h or 48 h later. B. Data shown represent mean parenchymal CFUs ± SE (*P < 0.05 comparing NLRC4+/+ with NLRC4−/− mice; n = 4–6/group). C–G. Reduced inflammatory cell recruitment (C–D), and cytokines/chemokines (E–G) in BALF of IL-1R1−/− mice in response to Kp infection. Infiltrating leukocytes from the BALF of IL-1R1−/− and wild-type (C57Bl/6) mice were enumerated on day 1 and 2 after Kp infection. (*, P < 0.05 between NLRC4+/+ with NLRC4−/− mice; n = 4–6/group). H. IL-1R ablation results in reduced NF-κB and MAPK activation as well as expression of ICAM-1 and VCAM-1 in the lung after Kp exposure. IL-1R1+/+ and IL-1R1−/− mice were infected with 1 × 103 CFUs of Kp/mouse i.t. and lungs were obtained 24 h and 48 h post-infection. Lung homogenates were used to assess activation of NF-κB and MAPK as well as expression of ICAM-1 and VCAM-1 by immunoblotting. A representative blot is shown from 3 blots/experiments with identical results. I. Protein immunoblot bands were quantified by densitometry and normalized to GAPDH or total p38. Data are expressed as means ± SE (n = 3). J. Administration of rIL-1β after Kp infection did not rescue neutrophil recruitment in IL-1R1−/− mice. IL-1R1−/− (KO) and IL-1R1/NLRC4−/− (DKO) mice were inoculated i.t. with Kp (1 × 103 CFUs in 50 μL of PBS) and then administered a single dose of recombinant murine IL-1β or vehicle (0.1% BSA). Total neutrophils in BALF (C) were enumerated 48 h after exposure (n = 4–6/group).
DISCUSSION
The NLRC4 inflammasome is important for the host immune response against intracellular pathogens, such as S. typhimurium (21) and L. pneumophila (23, 24). Further, these studies demonstrated that cytosolic bacterial flagellin is the major trigger that activates the NLRC4 inflammasome (21–24). Although NLRC4 is essential for host immunity against extracellular pathogens such as P. aeruginosa (53, 54), P. aeruginosa flagellin is not required to signal via NLRC4 despite the fact that proteins secreted by the type III secretion system are essential to induce activation of caspase-1 via NLRC4 (27). For Kp, a non-flagellated extracellular bacterium, ASC and NALP3 have been shown to be important in promoting caspase-1 activation and IL-1β release in murine macrophages following an extremely high dose of Kp challenge (7.4 × 104 CFUs/mouse) (28). In this investigation (28), survival between WT and NLRC4−/− mice following an extremely high dose of Kp infection was not different. Furthermore, no detailed mechanistic studies were performed to understand the role of NLRC4 in host defense against Kp infection. In the current study we explore the functional consequence of NLRC4 loss on host immunity in the lung following infection with Kp using inoculum (1 × 103 and 1 × 104 CFUs/mouse) based on the LD50 concentration.
The antibacterial defenses of the lung include activity of resident alveolar macrophages (55, 56). Any defect in host defense functions in macrophages ultimately result in infectious complications in the host. For example, we show via siRNA knockdown of NLRC4 in human macrophages that NLRC4 contributes to IL-18, IL-6, and TNF-α secretion following Kp infection. These findings underscore the importance of NLRC4 in the human immune response to infection with a non-flagellated bacterium.
The initial host response to bacterial infection is the recruitment of leukocytes from the bloodstream to the site of infection where they phagocytose and eliminate the infectious microbes (8, 10). Here, we show that absence of NLRC4 results in enhanced susceptibility to intrapulmonary Kp infection. We also show that NLRC4 limits bacterial colonization and dissemination of Kp, which is associated with neutrophil recruitment and the production of neutrophil chemoattractants. Until the current study, NLRC4 was not previously known to be associated with the expression of KC, MIP-2, or LIX during bacterial infections. In this regard, involvement of NLRC4 is supported by the observation that NLRC4 regulates NF-κB and MAPK activation in the lungs (Figs. 4A–E). Thus, our data suggest that NLRC4 regulates the production of neutrophil chemokines as well as TNF-α and the expression of ICAM-1 and VCAM-1 via activation of NK-κB and MAPKs, all of which contribute to neutrophil recruitment to the lung during Kp infection. However, our data cannot rule out the possibility that decreased activation of NK-κB and MAPKs in the lungs following Kp infection could be due to attenuated neutrophil influx following Kp infection in NLRC4−/− mice. It is possible that the production of cytokines/chemokines, and adhesion molecules via activation of NF-κB and MAPKs could be secondary to the reduced IL-1β production.
IL-17A has been shown to recruit lung neutrophils both directly and indirectly by stimulating the production of chemokines such as KC, MIP-2, and G-CSF (55, 57). Although IL-23 was shown to induce IL-17A and IL-17F expression in CD4+ T cells (57, 58) and is required for the maximum and sustained differentiation of Th17 cells (58, 59), additional mediators, including IL-6, IL-21 and IL-22, have been shown to promote Th17 differentiation as well (56, 60, 61). Hyper IgE Syndrome (HIES) patients have low levels of IL-17 and have recurrent bacterial infections due to attenuated neutrophil recruitment to tissues (62). Importantly, mice with a constitutively-active NALP3 inflammasome display enhanced IL-1β activity, increased Th17 cell numbers, and neutrophil-dominated inflammation in skin (63). Our data show that NLRC4 is important for the differentiation or recruitment of IL-17A in T cells the lung following Kp challenge (Fig. 3). The contribution of other cells to IL-17 production cannot be ruled out by our studies as a recent study demonstrated that neutrophils produce IL-17A in the lung in a dectin-1 and IL-23-dependent manner during Aspergillus fumigates infection (64).
Our finding that IL-1β is important for neutrophil-mediated host immunity against Kp infection in the lung is consistent with earlier findings showing the requirement of IL-1R activation in host immunity against systemic infections, brain abscesses, septic arthritis, and skin infections (65–67). It has been demonstrated in acute inflammatory arthritis models in rabbits that IL-1β is required for the production of neutrophil CXC chemokines, such as IL-8 (41). In a murine model of cutaneous infection caused by Staphylococcus aureus, it has been shown that IL-1β is required for neutrophil recruitment to the skin and for the production of KC and MIP-2 (41).
Recent studies have provided evidence that both intracellular and extracellular bacteria can activate the NLRC4 inflammasome (23, 24, 27, 68). Although WT S. typhimurium activates caspase-1 in an NLRC4-dependant fashion, flagellin-deficient mutants showed impaired caspase-1 activation except at high MOI, which can induce caspase-1-dependent IL-1β secretion in macrophages (22). Furthermore, L. pneumophila can signal through the NLRC4 inflammasome to activate caspase-1 in the presence of a co-activator, NAIP-5 (69, 70). The activation of caspase-1 was evident in WT L. pneumophila, whereas a flagellin knockout mutant of L. pneumophila failed to cause caspase-1 activation. In studies involving P. aeruginosa, both the non-flagellated strain, PA103ΔU, and the flagellin-deficient mutant strain, PAKΔfic, induced activation of caspase-1 as well as secretion of IL-1β (27). Notably, the translocon proteins of the type III secretion system (TTSS), such as PopB and PopD were required for the induction of caspase-1 activation and secretion of IL-1β (27). Caspase-1 activation requires intact TTSS of other bacterial pathogens as well, including S. typhimurium and Shigella flexneri (21, 22). Although structurally different, the functionally analogous type IV secretion system is necessary to activate caspase-1 in murine macrophages following L. pneumophila infection because the Dot mutants of L. pneumophila, which lack a functional type IV secretion system, failed to cause caspase-1 activation (71). We, however, found that a non-flagellated bacterium, Kp, can induce caspase-1 activation and secretion of IL-1β in macrophages via NLRC4 although Kp-induced caspase-1 activation via NLRC4 does not induce pyroptosis. Since IL-1R1−/− mice show much more pronounced survival phenotype than NLRC4−/− mice upon Kp infection, these findings suggest the role of other NLRs/inflammasomes that could be determined by future investigations.
While hematopoietic cells in the lung produce several neutrophil chemotactic substances such as KC (72, 73) and MIP-2 (74, 75), resident cells, including alveolar epithelial type II cells, produce other neutrophil chemoattractants, such as LIX (38). Intriguingly, we found that a requirement for NLRC4 signaling predominantly via hematopoietic cells for neutrophil accumulation in the lung in response to Kp infection. Furthermore, our findings are consistent with earlier reports of the role of hematopoietic cells or resident cells in lung inflammation. First, MyD88 derived from hematopoietic cells is more important for LPS-induced expression of TNF-α and IL-12p40 (76), although both hematopoietic and resident cell-derived MyD88 signaling are essential for LPS-induced neutrophil influx (77–79). In addition, MD-2 signaling in both hematopoietic and resident cells is essential for neutrophil-mediated inflammation, and the expression of MIP-2, TNF-α and IL-6 is mediated by both cell types in the lungs after LPS challenge (35). Finally, KC produced by both hematopoietic and resident cells is important for bacterial clearance and neutrophil recruitment to the lung upon Kp infection (31).
The mechanism(s) by which NLRC4 senses Kp and the components of Kp that induce NLRC4 activation are two unknowns highlighted by our studies. Several structurally and functionally diverse stimuli can activate the NLRP3 inflammasome, such as bacteria, virus, pore-forming toxins, extracellular ATP, silica crystals, and amyloid (80–85), although it is not clear how a single inflammasome, such as NLRP3 can recognize multiple stimuli. Thus, it is possible that NLRC4 can recognize endogenous ligands during bacteria-induced inflammation in addition to directly recognizing pathogens; however, the endogenous/bacterial ligands that can activate NLRC4 and the mechanisms responsible for NLRC4 activation during Kp infection in the lung remain to be elucidated. Based upon the complete genome sequencing of a Kp isolate, the genome of Kp342 has ten of eleven established protein secretion systems, including Type I, Type II, Type IV, Type V, and Type VI secretion systems (86). It is not currently known whether these protein secretion systems are important for bacterial virulence and whether the serotype 2 used in this investigation encodes these protein secretion systems.
Acknowledgments
Supported by a Scientist Award from the Flight Attendant Medical Research Institute (YCSA-062466); and grants from the NIH (R01 HL-091958 and R01 HL-091958S1 via ARRA) to SJ
The authors thank Millennium Pharmaceuticals for providing NLRC4−/− mice. We thank the lung biology members G. Balamayooran, T. Balamayooran, Liliang Jin and K. Jeyagowri for helpful discussions.
ABBREVIATIONS
- NLRC4
ICE Protease Activating Factor
- ICAM-1
Intracellular cell-adhesion molecule-1
- LIX
Lipopolysaccharide-induced CXC chemokines
- KC
Keratinocyte cell derived chemokines
- MIP-2
macrophage inflammatory protein-2
- VCAM-1
Vascular cell-adhesion molecule-1
- LIX
LPS-induced CXC chemokine
- BALF
Bronchoalveolar lavage fluid
- MPO
Myeloperoxidase
References
- 1.Mizgerd JP. Lung infection--a public health priority. PLoS Med. 2006;3:e76. doi: 10.1371/journal.pmed.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Endimiani A, Depasquale JM, Forero S, Perez F, Hujer AM, Roberts-Pollack D, Fiorella PD, Pickens N, Kitchel B, Casiano-Colon AE, Tenover FC, Bonomo RA. Emergence of blaKPC-containing Klebsiella pneumoniae in a long-term acute care hospital: a new challenge to our healthcare system. J Antimicrob Chemother. 2009;64:1102–1110. doi: 10.1093/jac/dkp327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mataseje LF, Boyd DA, Willey BM, Prayitno N, Kreiswirth N, Gelosia A, Poutanen SM, Low DE, Jenkins SG, Katz K, Mulvey MR. Plasmid comparison and molecular analysis of Klebsiella pneumoniae harbouring bla(KPC) from New York City and Toronto. J Antimicrob Chemother. 2011;66:1273–1277. doi: 10.1093/jac/dkr092. [DOI] [PubMed] [Google Scholar]
- 4.Schwaber MJ, Lev B, Israeli A, Solter E, Smollan G, Rubinovitch B, Shalit I, Carmeli Y. Containment of a country-wide outbreak of carbapenem-resistant Klebsiella pneumoniae in Israeli hospitals via a nationally implemented intervention. Clin Infect Dis. 2011;52:848–855. doi: 10.1093/cid/cir025. [DOI] [PubMed] [Google Scholar]
- 5.Jong GM, Hsiue TR, Chen CR, Chang HY, Chen CW. Rapidly fatal outcome of bacteremic Klebsiella pneumoniae pneumonia in alcoholics. Chest. 1995;107:214–217. doi: 10.1378/chest.107.1.214. [DOI] [PubMed] [Google Scholar]
- 6.Mackowiak PA, Martin RM, Smith JW. The role of bacterial interference in the increased prevalence of oropharyngeal gram-negative bacilli among alcoholics and diabetics. Am Rev Respir Dis. 1979;120:589–593. doi: 10.1164/arrd.1979.120.3.589. [DOI] [PubMed] [Google Scholar]
- 7.Nelson S, Kolls JK. Alcohol, host defence and society. Nat Rev Immunol. 2002;2:205–209. doi: 10.1038/nri744. [DOI] [PubMed] [Google Scholar]
- 8.Balamayooran G, Batra S, Fessler MB, Happel KI, Jeyaseelan S. Mechanisms of neutrophil accumulation in the lungs against bacteria. Am J Respir Cell Mol Biol. 2010;43:5–16. doi: 10.1165/rcmb.2009-0047TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med. 2008;358:716–727. doi: 10.1056/NEJMra074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun. 2009;77:568–575. doi: 10.1128/IAI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 14.Balamayooran T, Balamayooran G, Jeyaseelan S. Review: Toll-like receptors and NOD-like receptors in pulmonary antibacterial immunity. Innate Immun. 2010;16:201–210. doi: 10.1177/1753425910366058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chamaillard M, Girardin SE, Viala J, Philpott DJ. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol. 2003;5:581–592. doi: 10.1046/j.1462-5822.2003.00304.x. [DOI] [PubMed] [Google Scholar]
- 16.Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- 17.Pedra JH, Cassel SL, Sutterwala FS. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol. 2009;21:10–16. doi: 10.1016/j.coi.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126:659–662. doi: 10.1016/j.cell.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Sutterwala FS, Ogura Y, Flavell RA. The inflammasome in pathogen recognition and inflammation. J Leukoc Biol. 2007;82:259–264. doi: 10.1189/jlb.1206755. [DOI] [PubMed] [Google Scholar]
- 20.Sutterwala FS, Flavell RA. NLRC4/IPAF: a CARD carrying member of the NLR family. Clin Immunol. 2009;130:2–6. doi: 10.1016/j.clim.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pereira MS, Morgantetti GF, Massis LM, Horta CV, Hori JI, Zamboni DS. Activation of NLRC4 by Flagellated Bacteria Triggers Caspase-1-Dependent and -Independent Responses To Restrict Legionella pneumophila Replication in Macrophages and In Vivo. J Immunol. 2011;187:6447–6455. doi: 10.4049/jimmunol.1003784. [DOI] [PubMed] [Google Scholar]
- 24.Pereira MS, Marques GG, Dellama JE, Zamboni DS. The Nlrc4 Inflammasome Contributes to Restriction of Pulmonary Infection by Flagellated Legionella spp. that Trigger Pyroptosis. Front Microbiol. 2011;2:33. doi: 10.3389/fmicb.2011.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 26.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willingham SB, I, Allen C, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, Duncan JA, Ting JP. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183:2008–2015. doi: 10.4049/jimmunol.0900138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Xu F, Barrett E. Metalloelastase in lungs and alveolar macrophages is modulated by extracellular substance P inmice. Am J Physiol Lung Cell Mol Physiol. 2008;295:L162–170. doi: 10.1152/ajplung.00282.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagatani K, Dohi M, To Y, Tanaka R, Okunishi K, Nakagome K, Sagawa K, Tanno Y, Komagata Y, Yamamoto K. Splenic dendritic cells induced by oral antigen administration are important for the transfer of oral tolerance in an experimental model of asthma. J Immunol. 2006;176:1481–1489. doi: 10.4049/jimmunol.176.3.1481. [DOI] [PubMed] [Google Scholar]
- 31.Cai S, Batra S, Lira SA, Kolls JK, Jeyaseelan S. CXCL1 regulates pulmonary host defense to Klebsiella Infection via CXCL2, CXCL5, NF-kappaB, and MAPKs. J Immunol. 2010;185:6214–6225. doi: 10.4049/jimmunol.0903843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai S, Batra S, Shen L, Wakamatsu N, Jeyaseelan S. Both TRIF-and MyD88-dependent signaling contribute to host defense against pulmonary Klebsiella infection. J Immunol. 2009;183:6629–6638. doi: 10.4049/jimmunol.0901033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balamayooran G, Batra S, Balamayooran T, Cai S, Jeyaseelan S. Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infect Immun. 2011;79:2567–2577. doi: 10.1128/IAI.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balamayooran T, Batra S, Balamayooran G, Cai S, Kobayashi KS, Flavell RA, Jeyaseelan S. RIP2 Controls Pulmonary Host Defense to E. coli Infection via the Regulation of IL-17A. Infect Immun. 2011 doi: 10.1128/IAI.05641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai S, Zemans RL, Young SK, Worthen GS, Jeyaseelan S. Myeloid differentiation protein-2-dependent and -independent neutrophil accumulation during Escherichia coli pneumonia. Am J Respir Cell Mol Biol. 2009;40:701–709. doi: 10.1165/rcmb.2008-0152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeyaseelan S, Chu HW, Young SK, Freeman MW, Worthen GS. Distinct roles of pattern recognition receptors CD14 and Toll-like receptor 4 in acute lung injury. Infect Immun. 2005;73:1754–1763. doi: 10.1128/IAI.73.3.1754-1763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeyaseelan S, Chu HW, Young SK, Worthen GS. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect Immun. 2004;72:7247–7256. doi: 10.1128/IAI.72.12.7247-7256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, Worthen GS. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am JRespir Cell Mol Biol. 2005;32:531–539. doi: 10.1165/rcmb.2005-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Passos ST, Silver JS, O’Hara AC, Sehy D, Stumhofer JS, Hunter CA. IL-6 promotes NK cell production of IL-17 during toxoplasmosis. J Immunol. 2010;184:1776–1783. doi: 10.4049/jimmunol.0901843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, Kolls JK, Khader SA, Randall TD. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. 2011;12:639–646. doi: 10.1038/ni.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, O’Connell RM, Iwakura Y, Cheung AL, Cheng G, Modlin RL. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol. 2007;179:6933–6942. doi: 10.4049/jimmunol.179.10.6933. [DOI] [PubMed] [Google Scholar]
- 42.Ozawa H, Nakagawa S, Tagami H, Aiba S. Interleukin-1 beta and granulocyte-macrophage colony-stimulating factor mediate Langerhans cell maturation differently. J Invest Dermatol. 1996;106:441–445. doi: 10.1111/1523-1747.ep12343589. [DOI] [PubMed] [Google Scholar]
- 43.Antonopoulos C, Cumberbatch M, Dearman RJ, Daniel RJ, Kimber I, Groves RW. Functional caspase-1 is required for Langerhans cell migration and optimal contact sensitization in mice. J Immunol. 2001;166:3672–3677. doi: 10.4049/jimmunol.166.6.3672. [DOI] [PubMed] [Google Scholar]
- 44.Schultz MJ, van der Poll T. Modulation of pulmonary innate immunity during bacterial infection: animal studies. Arch Immunol Ther Exp (Warsz) 2002;50:159–167. [PubMed] [Google Scholar]
- 45.Speert DP. Bacterial infections of the lung in normal and immunodeficient patients. Novartis Found Symp. 2006;279:42–51. disussion 51–45, 216–219. [PubMed] [Google Scholar]
- 46.Batra S, Balamayooran G, Sahoo MK. Nuclear Factor-kappaB: a Key Regulator in Health and Disease of Lungs. Arch Immunol Ther Exp (Warsz) 2011;59:335–351. doi: 10.1007/s00005-011-0136-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quinton LJ, Mizgerd JP. NF-kappaB and STAT3 signaling hubs for lung innate immunity. Cell Tissue Res. 2011;343:153–165. doi: 10.1007/s00441-010-1044-y. [DOI] [PubMed] [Google Scholar]
- 48.Mizgerd JP. Molecular mechanisms of neutrophil recruitment elicited by bacteria in the lungs. Semin Immunol. 2002;14:123–132. doi: 10.1006/smim.2001.0349. [DOI] [PubMed] [Google Scholar]
- 49.Kwon D, Fuller AC, Palma JP, Choi IH, Kim BS. Induction of chemokines in human astrocytes by picornavirus infection requires activation of both AP-1 and NF-kappa B. Glia. 2004;45:287–296. doi: 10.1002/glia.10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soriano SF, Serrano A, Hernanz-Falcon P, Martin de Ana A, Monterrubio M, Martinez C, Rodriguez-Frade JM, Mellado M. Chemokines integrate JAK/STAT and G-protein pathways during chemotaxis and calcium flux responses. Eur J Immunol. 2003;33:1328–1333. doi: 10.1002/eji.200323897. [DOI] [PubMed] [Google Scholar]
- 51.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS Pathog. 2011;7:e1002452. doi: 10.1371/journal.ppat.1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 53.Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Nunez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol. 2007;37:3030–3039. doi: 10.1002/eji.200737532. [DOI] [PubMed] [Google Scholar]
- 54.Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci U S A. 2008;105:2562–2567. doi: 10.1073/pnas.0712183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldstein E, Lippert W, Warshauer D. Pulmonary alveolar macrophage. Defender against bacterial infection of the lung. J Clin Invest. 1974;54:519–528. doi: 10.1172/JCI107788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gordon SB, Read RC. Macrophage defences against respiratory tract infections. Br Med Bull. 2002;61:45–61. doi: 10.1093/bmb/61.1.45. [DOI] [PubMed] [Google Scholar]
- 57.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reibman J, Hsu Y, Chen LC, Kumar A, Su WC, Choy W, Talbot A, Gordon T. Size fractions of ambient particulate matter induce granulocyte macrophage colony-stimulating factor in human bronchial epithelial cells by mitogen-activated protein kinase pathways. Am J Respir Cell Mol Biol. 2002;27:455–462. doi: 10.1165/rcmb.2001-0005OC. [DOI] [PubMed] [Google Scholar]
- 59.Taylor PR, Brown GD, Reid DM, Willment JA, Martinez-Pomares L, Gordon S, Wong SY. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol. 2002;169:3876–3882. doi: 10.4049/jimmunol.169.7.3876. [DOI] [PubMed] [Google Scholar]
- 60.Balch SG, Greaves DR, Gordon S, McKnight AJ. Organization of the mouse macrophage C-type lectin (Mcl) gene and identification of a subgroup of related lectin molecules. Eur J Immunogenet. 2002;29:61–64. doi: 10.1046/j.1365-2370.2002.00266.x. [DOI] [PubMed] [Google Scholar]
- 61.Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009;21:274–280. doi: 10.1016/j.coi.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 62.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30:860–874. doi: 10.1016/j.immuni.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Werner JL, Gessner MA, Lilly LM, Nelson MP, Metz AE, Horn D, Dunaway CW, Deshane J, Chaplin DD, Weaver CT, Brown GD, Steele C. Neutrophils produce interleukin 17A (IL-17A) in a dectin-1-and IL-23-dependent manner during invasive fungal infection. Infect Immun. 2011;79:3966–3977. doi: 10.1128/IAI.05493-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hultgren OH, Svensson L, Tarkowski A. Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J Immunol. 2002;168:5207–5212. doi: 10.4049/jimmunol.168.10.5207. [DOI] [PubMed] [Google Scholar]
- 66.Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-alpha play a pivotal role in the host immune response ina mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- 67.Verdrengh M, Thomas JA, Hultgren OH. IL-1 receptor-associated kinase 1 mediates protection against Staphylococcus aureus infection. Microbes Infect. 2004;6:1268–1272. doi: 10.1016/j.micinf.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 68.Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozoren N, Brady G, Meshinchi S, Jagirdar R, Gewirtz A, Akira S, Nunez G. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem. 2006;281:35217–35223. doi: 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- 69.Lightfield KL, Persson J, Trinidad NJ, Brubaker SW, Kofoed EM, Sauer JD, Dunipace EA, Warren SE, Miao EA, Vance RE. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect Immun. 2011;79:1606–1614. doi: 10.1128/IAI.01187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fortier A, Doiron K, Saleh M, Grinstein S, Gros P. Restriction of Legionella pneumophila replication in macrophages requires concerted action of the transcriptional regulators Irf1 and Irf8 and nod-like receptors Naip5 and Nlrc4. Infect Immun. 2009;77:4794–4805. doi: 10.1128/IAI.01546-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Case CL, Shin S, Roy CR. Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun. 2009;77:1981–1991. doi: 10.1128/IAI.01382-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bozic CR, Kolakowski LF, Jr, Gerard NP, Garcia-Rodriguez C, von Uexkull-Guldenband C, Conklyn MJ, Breslow R, Showell HJ, Gerard C. Expression and biologic characterization of the murine chemokine KC. J Immunol. 1995;154:6048–6057. [PubMed] [Google Scholar]
- 73.Frevert CW, Farone A, Danaee H, Paulauskis JD, Kobzik L. Functional characterization of rat chemokine macrophage inflammatory protein-2. Inflammation. 1995;19:133–142. doi: 10.1007/BF01534386. [DOI] [PubMed] [Google Scholar]
- 74.Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, Cerami A. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci U S A. 1989;86:612–616. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Driscoll KE, Hassenbein DG, Howard BW, Isfort RJ, Cody D, Tindal MH, Suchanek M, Carter JM. Cloning, expression, and functional characterization of rat MIP-2: a neutrophil chemoattractant and epithelial cell mitogen. J Leukoc Biol. 1995;58:359–364. doi: 10.1002/jlb.58.3.359. [DOI] [PubMed] [Google Scholar]
- 76.Noulin N, V, Quesniaux F, Schnyder-Candrian S, Schnyder B, Maillet I, Robert T, Vargaftig BB, Ryffel B, Couillin I. Both hemopoietic and resident cells are required for MyD88-dependent pulmonary inflammatory response to inhaled endotoxin. J Immunol. 2005;175:6861–6869. doi: 10.4049/jimmunol.175.10.6861. [DOI] [PubMed] [Google Scholar]
- 77.Quinton LJ, Jones MR, Simms BT, Kogan MS, Robson BE, Skerrett SJ, Mizgerd JP. Functions and regulation of NF-kappaB RelA during pneumococcal pneumonia. J Immunol. 2007;178:1896–1903. doi: 10.4049/jimmunol.178.3.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol. 2004;287:L143–152. doi: 10.1152/ajplung.00030.2004. [DOI] [PubMed] [Google Scholar]
- 79.Quinton LJ, Jones MR, Robson BE, Simms BT, Whitsett JA, Mizgerd JP. Alveolarepithelial STAT3, IL-6 family cytokines, and host defense during Escherichia coli pneumonia. Am J Respir Cell Mol Biol. 2008;38:699–706. doi: 10.1165/rcmb.2007-0365OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 83.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Nunez G. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 84.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 85.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 86.Fouts DE, Tyler HL, DeBoy RT, Daugherty S, Ren Q, Badger JH, Durkin AS, Huot H, Shrivastava S, Kothari S, Dodson RJ, Mohamoud Y, Khouri H, Roesch LF, Krogfelt KA, Struve C, Triplett EW, Methe BA. Complete genome sequence of the N2-fixing broad host range endophyte Klebsiella pneumoniae 342 and virulence predictions verified in mice. PLoS Genet. 2008;4:e1000141. doi: 10.1371/journal.pgen.1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]