

Abstract
BACKGROUND
Men and women with type 1 long QT syndrome (LQT1) exhibit time-dependent differences in the risk for cardiac events.
OBJECTIVE
We hypothesized that sex-specific risk for LQT1 is related to the location and function of the disease-causing mutation in the KCNQ1 gene.
METHODS
The risk for life-threatening cardiac events (comprising aborted cardiac arrest [ACA] or sudden cardiac death [SCD]) from birth through age 40 years was assessed among 1051 individuals with LQT1 (450 men and 601 women) by the location and function of the LQT1-causing mutation (prespecified as mutations in the intracellular domains linking the membrane-spanning segments [ie, S2–S3 and S4–S5 cytoplasmic loops] involved in adrenergic channel regulation vs other mutations).
RESULTS
Multivariate analysis showed that during childhood (age group: 0–13 years) men had >2-fold (P < .003) increased risk for ACA/SCD than did women, whereas after the onset of adolescence the risk for ACA/SCD was similar between men and women (hazard ratio = 0.89 [P = .64]). The presence of cytoplasmic-loop mutations was associated with a 2.7-fold (P < .001) increased risk for ACA/SCD among women, but it did not affect the risk among men (hazard ratio 1.37; P = .26). Time-dependent syncope was associated with a more pronounced risk-increase among men than among women (hazard ratio 4.73 [P < .001] and 2.43 [P = .02], respectively), whereas a prolonged corrected QT interval (≥500 ms) was associated with a higher risk among women than among men.
CONCLUSION
Our findings suggest that the combined assessment of clinical and mutation location/functional data can be used to identify sex-specific risk factors for life-threatening events for patients with LQT1.
Keywords: Cytoplasmic-loop mutations, Sex, Long QT syndrome, Sudden cardiac death
Introduction
Long QT syndrome type 1 (LQT1) is the most commonly occurring of the congenital long QT syndromes (LQTS).1 It is caused by mutations in the KCNQ1 gene that impair the slow-acting potassium channel that gives rise to IKs. The resulting prolongation of ventricular repolarization increases the potential for cardiac arrhythmogenic events that can cause syncope or sudden cardiac death (SCD). Patients with LQT1 experience the majority of their events during exercise, possibly because the phase 3 IKs repolarizing current activates during increased heart rate and is essential for QT-interval adaptation during tachycardia.1,2 Prior studies have shown that mutations located at the membrane-spanning (MS) region and missense vs nonmissense mutations are associated with a greater risk for cardiac events in patients with LQT1.3 The MS region includes the MS domains and the MS linkers. Mutations in the intracellular linkers that connect the MS domains of the KCNQ1 (Kv7.1) channel subunit (defined herein as the S2–S3 and S4–S5 cytoplasmic [C]-loop mutations) were shown to affect adrenergic channel regulation by protein kinase A4 and may therefore predispose to increased risk for life-threatening events in this population.5
The phenotypic expression of LQT1 is affected by sex and age, wherein men with LQT1 experience increased risk for cardiac events, mainly during the childhood period.6 Prior studies, however, did not relate sex-specific risk in this population to the location and function of the disease-causing mutation in the KCNQ1 gene. Furthermore, sex differences in the clinical course of LQT1 were related previously to a cardiac event composite end point, which comprised mostly nonfatal syncope. Accordingly, the present study was designed to evaluate whether the combined assessment of clinical and mutation location/functional data can identify sex-specific risk factors for life-threatening cardiac events in men and women with LQT1.
Methods
Study population
The study population comprised 1051 LQT1-positive subjects from 259 proband identified families. Patients were drawn from the Rochester, NY, enrolling center (center 1) of the International LQTS Registry (n = 755), the Netherlands LQTS Registry (n = 85), and the Japanese LQTS Registry (n = 83), as well as from data submitted by other investigators specifically for this collaborative mutation analysis project: Denmark (n = 43), Israel (n = 34), Sweden (n = 4), and Salt Lake City, UT (n = 47). The proband in each family had otherwise unexplained, diagnostic corrected QT-interval (QTc) prolongation or experienced LQTS-related symptoms. Patients with congenital deathness were excluded from the study.
Data collection and management
For each patient, information on personal history, including cardiac events, electrocardiograms, and therapies, as well as family history was obtained at enrollment. Clinical data were then collected yearly on prospectively designed forms with information on demographic characteristics, personal and family medical history, electrocardiogram findings, medical therapies, left cardiac sympathetic denervation, implantation of a pacemaker or an implantable cardioverter defibrillator (ICD), and the occurrence of LQT1-related cardiac events. The QT interval was corrected for heart rate (QTc) by using Bazett's formula.7 Data common to all LQTS registries involving genetically tested individuals were merged electronically into a common database for the present study.
Genotype characterization
The KCNQ1 mutations were identified with the use of standard genetic tests conducted in academic molecular genetic laboratories including the Functional Genomics Center, University of Rochester Medical Center, Rochester, NY; Baylor College of Medicine, Houston, TX; Windland Smith Rice Sudden Death Genomics Laboratory, Mayo Clinic, Rochester, MN; Boston Children's Hospital, Boston, MA; Laboratory of Molecular Genetics, National Cardiovascular Center, Suita, Japan; Department of Clinical Genetics, Academic Medical Center, Amsterdam, The Netherlands; and Molecular Cardiology Laboratory, Policlinico S. Matteo and University of Pavia, Pavia, Italy.
Mutations were defined as any nonsynonym rare variants (<1% of the healthy population) identified in a proband with a prolonged QT interval. Based on prior data regarding mutation location/function and arrhythmic risk in LQT1,3–5,8 mutations were categorized by their location and type in the KCNQ1-encoded channel subunit as follows: (1) missense mutations in the MS region: defined as amino acid residues from 120 to 355, excluding mutations within the MS linkers; (2) missense mutations in the C loops: defined as the coding sequence involving amino acid residues from 174 to 190 (S2–S3 linker) and from 242 to 259 (S4–S5 linker); (3) missense mutations in the N-terminus region, defined as amino acid residues before 120, and the C-terminus region, defined as amino acid residues after residue 355, were combined into one category labeled as the other region for this analysis (hence called the N/C terminus); and (4) other LQT1 mutations as the reference group (ie, splice sites, in-frame insertions, in-frame deletions, nonsense, and frameshift).
The specific mutations included in the present study, by location, type, and number of patients, are detailed in the Supplementary Appendix Table 1, and the distribution of the mutations in the KCNQ1 gene by their frequency among study patients is shown in Figure 1.
Figure 1.
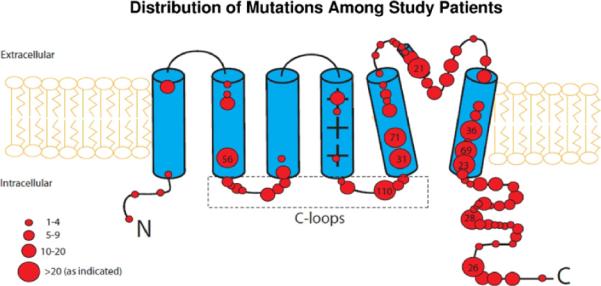
Distribution of mutations in the KCNQ1 (Kv7.1) potassium channel subunit among study patients. Numbers in larger circles denote the number of patients with the mutation. C loops = cytoplasmic loops.
End point
The primary end point of the study was the occurrence of a first life-threatening cardiac event, comprising aborted cardiac arrest (ACA) (requiring defibrillation as part of resuscitation), or LQT1-related SCD (abrupt in onset without evident cause, if witnessed, or death that was not explained by any other cause if it occurred in a nonwitnessed setting such as sleep). To further validate the consistency of the results among patients who received an ICD during follow-up, we also assessed a secondary end point comprising the first occurrence of ACA, SCD, or appropriate ICD shock during follow-up.
Statistical analysis
The baseline and follow-up clinical characteristics of the study population were evaluated by using the chi-square test for categorical variables and the Mann-Whitney-Wilcoxon test for continuous variables. The cumulative probability of a first ACA or SCD by sex and by mutation location was assessed by using the Kaplan-Meier method, and significance was tested by using the log-rank test. Follow-up was censored at age 40 years to avoid confounding by acquired cardiovascular disease. Multivariate Cox proportional-hazards regression models were used to evaluate the independent contribution of clinical and genetic factors to the first occurrence of ACA or SCD. Prespecified covariates in the total population model included sex, QTc duration (categorized as a 3-level covariate: >500 ms, 500–550 ms, <500 ms [reference]), mutation location and type (as defined above), the occurrence of syncope during follow-up, and medical therapy with beta-blockers. Syncope and beta-blocker therapy were assessed as time-dependent covariates in the multivariate models. To avoid violation of the proportional hazards assumption due to sex-risk crossover during adolescence, we employed an age-sex interaction term in the total population multivariate model. The effect of each covariate in men and women was assessed by interaction-term analysis (ie, by including a sex-by-risk factor interaction term in the multivariate models), with interactions tested one at a time. Patients without available baseline QTc data (n = 151) were included as a separate (QTc missing) covariate in the multivariate models.
Because almost all the subjects were first- and second-degree relatives of probands, the effect of lack of independence between subjects was evaluated in the Cox model with grouped jackknife estimates for family membership.9 All grouped jackknife standard errors for the covariate risk factors fell within 3% of those obtained from the unadjusted Cox model, and therefore only the Cox model findings are reported. The statistical software used for the analyses was SAS version 9.20 (SAS Institute Inc, Cary, NC). A 2-sided .05 significance level was used for hypothesis testing.
Results
The clinical characteristics of the study patients by sex are shown in Table 1. Baseline QTc was similar between men and women during childhood and significantly higher among women after the onset of adolescence. During follow-up, similar numbers of men and women were treated with beta-blockers, but a higher proportion of women were treated with an ICD. There were no significant sex differences in the distribution of the mutation by location (Table 1). However, patients with missense mutations localizing to the C loops exhibited a significantly longer baseline QTc (503 ± 58 ms) than did patients with other mutations (480 ± 51 ms; P < .001).
Table 1.
Characteristics of the study population
| Characteristic | Men (n = 450) | Women (n = 601) | P |
|---|---|---|---|
| ECG parameters | |||
| Overall QTc (ms) | 473 ± 55 | 486 ± 55 | <.001 |
| Age ≤ 13 y | 486 ± 53 | 485 ± 54 | .21 |
| Age > 13 y | 460 ± 53 | 487 ± 56 | <.001 |
| QTc > 500 ms (%) | 22 | 27 | .09 |
| RR (ms) | 842 ± 220 | 837 ± 199 | .55 |
| Mutation location (%) | |||
| Cytoplasmic loop (S2–S3 or S4–S5 IDL) | 19 | 19 | .92 |
| MS | 53 | 54 | .67 |
| N/C terminus | 28 | 26 | .58 |
| Mutation type (%) | |||
| Missense | 83 | 79 | .13 |
| LQTS therapies during follow-up (%) | |||
| Beta-blockers | 45 | 46 | .74 |
| Pacemaker | 1.6 | 1.5 | .94 |
| ICD | 4 | 9 | .003 |
| LCSD | 1.1 | 0.5 | .30 |
| Cardiac events during follow-up (%) | |||
| Syncope | 35 | 36 | .84 |
| ACA | 3 | 4 | .22 |
| SCD | 13 | 8 | .007 |
| Appropriate ICD shocks | 0.2 | 1.7 | .03 |
| ACA or SCD* | 15 | 11 | .07 |
Values are mean ± SD unless otherwise indicated.
ACA = aborted cardiac arrest; ECG = electrocardiogram; ICD = implantable cardioverter defibrillator; LCSD = left cervical sympathetic denervation; LQTS = long QT syndrome; MS = membrane spanning; QTc = corrected QT interval; SCD = sudden cardiac death; SD = standard deviation.
Only the first event for each patient was considered.
Risk factors for ACA or SCD in the total LQT1 population
During follow-up, 138 study patients (13%) experienced the primary end point of a first ACA or SCD. Kaplan-Meier event rates were significantly higher among men than among women throughout follow-up (P = .008 for the overall difference; Figure 2). Notably, life-threatening cardiac events among men occurred predominantly during childhood, whereas among women event rates increased after this time period. Thus, by age 14 years, the cumulative probability of ACA or SCD was 10% among men as compared with only 3% among women, and by age 40 years, the respective events rates were 19% and 15% (Figure 2).
Figure 2.
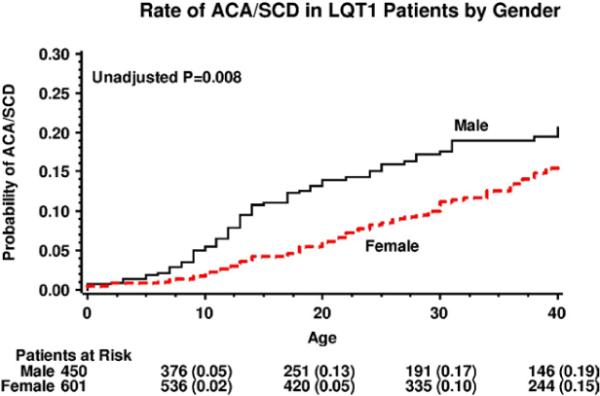
Kaplan-Meier estimates of the cumulative probability of aborted cardiac arrest or sudden cardiac death in patients with LQT1 by sex. ACA = aborted cardiac arrest; LQT1 = long QT syndrome type 1; SCD = sudden cardiac death.
Consistent with those findings, multivariate analysis in the total study population showed that during childhood men had >2-fold (P = .003) increase in the risk for ACA or SCD as compared with women whereas after the onset of adolescence, there was no statistically significant difference in the risk for ACA or SCD between men and women (hazard ratio [HR] 0.89; P = .64; Table 2). Additional risk factors within the total study population included the presence of missense mutations localizing to the C loops (1.9-fold risk increase [P = .005]), QTc 500–550 and >550 ms (>3-fold and >4-fold risk increase, respectively [P < .001 for both]), and the occurrence of syncope during follow-up (3.4-fold risk increase [P < .001]; Table 2). Results were similar when the secondary end point of a first ACA, SCD, or appropriate ICD shock was assessed.
Table 2.
Multivariate analysis: Risk factors for ACA/SCD among all patients with LQT1*
| Relative risk |
|||
|---|---|---|---|
| Risk factor | Hazard ratio | 95% Confidence interval | P |
| Sex | |||
| Men vs women ≤ 13 y | 2.31 | 1.41–3.92 | .003 |
| Men vs women >13 y | 0.92 | 0.61–1.51 | .72 |
| Mutation location (vs nonmissense mutations) | |||
| Cytoplasmic loop (S2–S3/S4–S5 linkers) | 1.93 | 1.37–2.75 | .005 |
| MS (S1, S2, S3, S4, S5, P-loop, S6) | 1.02 | 0.71–1.85 | .51 |
| N/C terminus | 0.96 | 0.52–1.57 | .72 |
| QTc duration (ms) | |||
| >550 vs <500 | 4.18 | 2.06–8.46 | <.001 |
| 500–550 vs <500 | 3.35 | 1.83–6.11 | <.001 |
| Time-dependent syncope | |||
| Syncope vs no syncope | 3.40 | 2.22–5.21 | <.001 |
ACA = aborted cardiac arrest; LQT1 = long QT syndrome type 1; MS = membrane spanning; QTc = corrected QT interval; SCD = sudden cardiac death.
Models were further adjusted for missing QTc values, time-dependent beta-blocker therapy.
Sex-specific risk factors for life-threatening cardiac events in patients with LQT1
Kaplan-Meier survival analysis showed that women with LQT1 with missense C-loop mutations exhibited a significantly higher rate of ACA or SCD than did women whose LQT1-causative mutation localized elsewhere (P < .001 for the overall difference during follow-up; Figure 3A). In contrast, among men, the respective rates of ACA or SCD remained high, predominantly during the childhood period, regardless of mutation location/type (P = .33 for the overall difference during follow-up [Figure 3B]).
Figure 3.

Kaplan-Meier estimates of the cumulative probability of aborted cardiac arrest or sudden cardiac death in (A) women with LQT1 and (B) men with LQT1, by mutation location. ACA = aborted cardiac arrest; C-loop mutations = cytoplasmic-loop mutations; LQT1 = long QT syndrome type 1; SCD = sudden cardiac death.
Sex-specific multivariate analysis (Table 3) showed that women with C-loop mutations exhibited nearly a 3-fold (P = .01) increased risk for ACA or SCD than did women with other mutation types, whereas the risk for ACA or SCD among men was not significantly different by mutation location/type (P value for mutation-location-by-sex interaction = .07). Similar results were observed in an additional analysis in which the large subset of patients with the V254M C-loop mutation was excluded from the multivariate models (risk associated with C loop vs other mutations among women: HR 2.55, 95% confidence interval [CI] 1.02–5.94; among men: HR 1.03, 95% CI 0.33–4.11).
Table 3.
| Men with LQT1 |
Women with LQT1 |
|||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Mutation location (vs nonmissense mutations) | ||||
| Cytoplasmic loop (S2–S3/S4–S5 linkers) | 1.21 (0.72–2.04 | 0.48 | 2.62 (1.59–4.26) | <.001 |
| MS | 1.02 (0.63–1.97) | 0.54 | 1.01 (0.62–1.89) | .56 |
| N/C terminus | 0.89 (0.52–1.91) | 0.87 | 1.14 (0.51–2.37) | .43 |
| QTc duration (ms) | ||||
| 500–550 vs <500 | 1.70 (0.63–4.57) | 0.29 | 6.85 (2.74–17.10) | <.001 |
| >550 vs < 500 | 3.11 (1.19–8.15) | 0.02 | 5.93 (1.89–18.62) | .002 |
| Time-dependent syncope | ||||
| Syncope vs no syncope | 4.06 (2.22–7.41) | <0.001 | 2.43 (1.43–4.85) | .002 |
ACA = aborted cardiac arrest; CI = confidence interval; HR = hazard ratio; LQT1 = long QT syndrome type 1; QTc = corrected QT interval; SCD = sudden cardiac death; MS = membrane spanning.
Findings were further adjusted for missing QTc values, time-dependent beta-blocker therapy.
Models were carried out in the total population by using interaction-term analysis, with interactions tested one at a time; cytoplasmic loop-by-sex interaction = .07; all other interaction P values were >.10.
Additional risk factors for ACA or SCD among both men and women included a prolonged QTc and the occurrence of time-dependent syncope (Table 3). Notably, women with prolonged QTc, both in the range of 500–550 ms and >550 ms, experienced a pronounced increase (approximately 6-fold) in the risk of ACA or SCD, whereas the risk associated with a prolonged QTc in men was more modest and evident only in those with QTc >550 ms (Table 3 and Figures 4A and B, respectively). The occurrence of syncope during follow-up was associated with a 4-fold (P < .001) increase in the risk for subsequent ACA or SCD among men and with a 2.4-fold (P = .002) increase in the risk for subsequent ACA or SCD among women.
Figure 4.
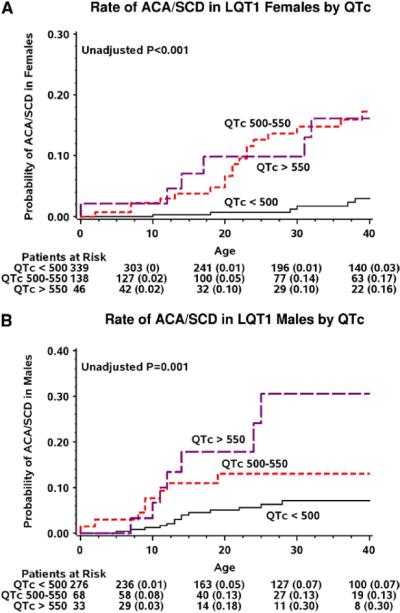
Kaplan-Meier estimates of the cumulative probability of aborted cardiac arrest or sudden cardiac death in (A) women with LQT1 and (B) men with LQT1, by QTc duration. ACA = aborted cardiac arrest; C-loop mutations = cytoplasmic-loop mutations; LQT1 = long QT syndrome type 1; QTc = corrected QT interval; SCD = sudden cardiac death.
The combined assessment of clinical and genetic data identified a very low rate of life-threatening events (0.03 events per 100 patient-years) among women aged 13 years or younger without C-loop mutations, no history of prior syncope, and QTc <500 ms.
Time-dependent medical therapy with beta-blockers was associated with a significant 61% reduction in the risk for ACA or SCD in the total study population (HR 0.39; 95% CI 0.22–0.70; P = .001), with beta-blocker protection seen similarly between men and women (P values for beta-blocker-by-sex interaction = .56). Notably, this analysis showed that the risk associated with C-loop mutations in women was even more pronounced among those who did not receive beta-blocker therapy (HR 4.51; 95% CI 2.57–7.23; P < .001).
Discussion
In the present study, we assessed for the first time sex-specific risk factors for life-threatening cardiac events in a large population of 1051 genetically confirmed patients with LQT1. Our findings show that among probands and relatives with LQT1, (1) men exhibit a significantly higher rate of life-threatening cardiac events than do women, especially prior to puberty, and (2) mutation location shows a sex-specific association with the risk for ACA or SCD. Thus, the risk for life-threatening events was shown to be increased among women with LQT1 with mutations localizing to C-loop domains (S2–S3 and S4–S5) of the KCNQ1-encoded protein, whereas the risk for ACA or SCD among men with LQT1 was high even among those who harbored mutations localizing to other regions of the channel that had been ascribed previously as lower-risk mutations. These findings suggest that a combined approach that incorporates clinical and genetic data can be used for improved risk assessment and management of men and women with LQT1.
A previous study from the International LQTS Registry has shown that men with LQT1 have an increase in the risk for any LQT1-related cardiac event, including syncope, during childhood, whereas after the onset of adolescence the risk for events in this population is attenuated without a significant sex difference.6 Because of a limited sample of 243 patients with LQT1, the study did not assess sex-related differences in the risk for only life-threatening cardiac events (ACA or SCD) in this population. Thus, our findings of the present study extend prior observations and show that men with LQT1 display a higher rate of ACA or SCD than do women from birth through age 40 years, with a predominant risk increase during the childhood period.
Patients with LQT1 experience ventricular tachyarrhythmias more frequently during physical effort,2 possibly due to the lack of adaptive QT shortening with decreasing RR intervals during tachycardia.10 Thus, the early predominance of life-threatening cardiac events among men may be related to sex differences in the level of physical activity during childhood among registry patients. After the onset of adolescence, an increase in the levels of testosterone, which was shown to shorten action potential duration and ventricular repolarization,11–13 may result in a reduction in the risk for arrhythmic events in men. This mechanism is supported by the fact that the risk for life-threatening events during childhood was higher among men despite the fact that the average QTc was similar between men and women during this time period, whereas after the onset of adolescence the QTc was significantly reduced in men and remained virtually unchanged in women (Table 1).
Mutations located in the MS region, including the MS domains and the C loops, of the KCNQ1 protein have been associated with greater prolongation in the QTc during exercise14 and an increase in the risk for cardiac events in patients with LQT1.3 Importantly, the C loops were shown to modify the function of KCNQ1 channel subunit, including functional interaction with the auxiliary beta subunits encoded by KCNE1 and modulation of the channel's PKA-dependent, adrenergic regulation.4 Thus, patients who harbor C-loop mutations may be sensitive to even mild degrees of adrenergic stimulation, resulting in arrhythmic events that may occur during less intense physical activity. This mechanism may explain the sex-specific association of mutation location to arrhythmic risk shown in the present study, as women who carry mutations in the adrenergic-sensitive C loops may have an increased risk for life-threatening events even during milder degrees of physical activity. In contrast, participation in more intense physical activity among men with LQT1, especially during childhood, may predispose them to arrhythmic events even among those who carry non–C-loop mutations (which are less sensitive to sympathetic activation). It is also possible that sex differences in the regulation of the ion channel contribute to the differential effect of mutation location/function on arrhythmic risk between men and women.
Similar to prior studies,15–17 we have also shown that QTc is a major risk factor for cardiac events in patients with LQTS. However, our data suggest that in LQT1 the risk associated with QTc is more pronounced among women (who exhibited >6-fold risk with QTc exceeding 500 ms, whereas a significant risk increase among men was evident only among those with QTc >550 ms).
It was suggested recently that patients with LQT1 experience a very low rate of cardiac events during beta-blocker therapy.18 In the present study, medical therapy with beta-blockers was associated with a pronounced reduction in the risk for ACA or SCD in the total LQT1 population, without a statistically significant difference between men and women. However, our findings suggest that sex-specific risk factors should be taken into account in the management of patients with LQT1. These clinical and mutation-related risk factors are shown in Figure 1 of the Supplementary Appendix and include (1) the preadolescence period in men, especially among those who experience syncope during childhood and those with QTc >550 ms, and (2) following the onset of adolescence in women, especially among those with C-loop mutations, QTc ≥500 ms, and/or history of syncope.
Study limitations
Although we have shown recently that the S2–S3 and S4–S5 C-loop linkers have an important functional role in adrenergic channel regulation through PKA,4,5 further studies are needed to relate the functional expression of discrete KCNQ1 mutations to sex-specific risk in LQT1, and their interaction with possible hormonal modulation of cardiac risk in this population.
The present study shows that beta-blocker therapy is associated with a significant reduction in the risk for life-threatening events in both men and women with LQT1. However, because of sample size limitations, we did not evaluate sex-specific differences in response to beta-blocker therapy between patients with LQT1 with higher-risk (and adrenergic-sensitive) C-loop mutations and those with mutations localizing elsewhere. In addition, we did not carry out comprehensive analysis of the relationship between all functional regions of the KCNQ1-encoded protein (including functional areas within the C-terminus or N-terminus domains) and sex-specific risk.
We excluded patients with congenital deafness from the study. However, 12 patients (1%) had 2 different mutations in the KCNQ1 gene. To validate the consistency of the results for patients with single mutations, all multivariate models were repeated after excluding the 12 patients with >1 mutation. This confirmatory analysis yielded virtually identical results regarding the risk associated with clinical factors and mutation location as in the primary analysis. However, because of a small sample of patients with >1 mutation, the current results should be interpreted with caution in the risk assessment of this subset.
Conclusions and clinical implications
Our data extend prior knowledge regarding genotype-specific risk assessment in LQTS.16,17 The present results suggest that the functional effects of mutations in the KCNQ1-encoded channel subunit may explain differences in the risk for life-threatening cardiac events between men and women with LQT1. Here, both men and women with LQT1-causative mutations localizing to the C loops (S2–S3 and S4–S5 linkers), the intracellular domains that connect the MS domains of the KCNQ1-encoded protein, have increased risk for not only LQT1-triggered syncope but also LQT1-triggered life-threatening cardiac events of ACA and SCD, possibly due to the increased sensitivity of these functional domains to adrenergic stimulation. In contrast, men with LQT1 were shown to have an increased risk for ACA or SCD even in the presence of mutations localizing elsewhere predicted at the molecular/cellular level to be associated with lower risk. These findings suggest that a genotypespecific approach, incorporating clinical and mutation location/functional data, might further improve the risk assessment and management of patients with the most common genetic subtype of LQTS.
Acknowledgments
This work was supported by research grants HL-33843 and HL-51618 from the National Institutes of Health, Bethesda, MD, and by a research grant from GeneDx to the Heart Research Follow-Up Program in support of the LQTS Registry. Dr Ackerman is a consultant for Transgenomic (approved by Mayo Clinic's Medical-Industry Relations Office and Conflict of Interests Review Board). In addition, “cardiac channel gene screen” and “know-how relating to long QT genetic testing” license agreements, resulting in consideration and royalty payments, were established between Genaissance Pharmaceuticals (then PGxHealth and now Transgenomic) and Mayo Medical Ventures (now Mayo Clinic Health Solutions) in 2004. Dr Ackerman is also a consultant for Biotronik, Boston Scientific Corporation, Medtronic, and St Jude Medical. However, none of these entities provided financial support for this study.
ABBREVIATIONS
- ACA
aborted cardiac arrest
- C-loop mutations
cytoplasmic-loop mutations
- HR
hazard ratio
- ICD
implantable cardioverter defibrillator
- LQTS
long QT syndrome
- LQT1
long QT syndrome type 1
- MS
membrane spanning
- QTc
corrected QT interval
- SCD
sudden cardiac death
Footnotes
Jason Costa, Coeli M. Lopes, Alon Barsheshet, and Ilan Goldenberg contributed equally to this article.
Appendix Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.hrthm.2012.01.020.
References
- 1.Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51:2291–2300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 3.Moss AJ, Shimizu W, Wilde AAM, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matavel A, Medei E, Lopes CMB. PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels (Austin) 2010;4:3–11. doi: 10.4161/chan.4.1.10227. [DOI] [PubMed] [Google Scholar]
- 5.Barsheshet A, Goldenberg I, O-Uchi J, et al. Mutation Specific Risk and Response to Therapy in Type 1 Long QT Syndrome. American Heart Association Scientific Sessions; Chicago, IL: 2010. [Google Scholar]
- 6.Zareba W, Moss AJ, Locati EH, et al. International Long QT Syndrome Registry. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol. 2003;42:103–109. doi: 10.1016/s0735-1097(03)00554-0. [DOI] [PubMed] [Google Scholar]
- 7.Bazett H. An analysis of the time relations of electrocardiograms. Heart. 1920;7:14. [Google Scholar]
- 8.Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 9.Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. Springer-Verlag; New York, NY: 2000. [Google Scholar]
- 10.Swan H, Viitasalo M, Piippo K, Laitinen P, Kontula K, Toivonen L. Sinus node function and ventricular repolarization during exercise stress test in long QT syndrome patients with KvLQT1 and HERG potassium channel defects. J Am Coll Cardiol. 1999;34:823–829. doi: 10.1016/s0735-1097(99)00255-7. [DOI] [PubMed] [Google Scholar]
- 11.Hara M, Danilo P, Jr, Rosen MR. Effects of gonadal steroids on ventricular repolarization and on the response to E4031. J Pharmacol Exp Ther. 1998;285:1068–1072. [PubMed] [Google Scholar]
- 12.Brouillette J, Trépanier-Boulay C, Fiset C. Effect of androgen deficiency on mouse ventricular repolarization. J Physiol. 2003;546:403–413. doi: 10.1113/jphysiol.2002.030460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ridley JM, Shuba YM, James AF, Hancox JC. Modulation by testosterone of an endogenous hERG potassium channel current. J Physiol Pharmacol. 2008;59:395–407. [PubMed] [Google Scholar]
- 14.Shimizu W, Horie M, Ohno S, et al. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: multicenter study in Japan. J Am Coll Cardiol. 2004;44:117–125. doi: 10.1016/j.jacc.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 15.Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: prospective longitudinal study of 328 families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- 16.Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341–1344. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 17.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 18.Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of β-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of β-blocker treatment “failures”. Circulation. 2009;119:215–221. doi: 10.1161/CIRCULATIONAHA.108.772533. [DOI] [PubMed] [Google Scholar]