

Abstract
Extracellular adenosine has an important role in regulating the severity of inflammation during an immune response. While there are four adenosine receptor (AR) subtypes, the A2AAR is both highly expressed on lymphocytes and known as a prime mediator of adenosine’s anti-inflammatory effects. To define the importance of A2AAR signaling during neuroinflammatory disease progression, we utilized the experimental autoimmune encephalomyelitis (EAE) animal model for multiple sclerosis. In EAE induction experiments, A2AAR antagonist treatment protected mice from disease development and its associated CNS lymphocyte infiltration. However, A2AAR−/− mice developed a more severe acute EAE phenotype characterized by more proinflammatory lymphocytes and activated microglial/macrophages. Interestingly, very high levels of A2AAR were expressed on the choroid plexus, a well-established CNS lymphocyte entry point. To determine the contribution of A2AAR signaling in lymphocytes and the CNS during EAE, we utilized bone marrow chimeric mice. Remarkably, A2AAR−/− donor hematopoietic cells potentiated severe EAE, while lack of A2AAR expression on non-hematopoietic cells protected against disease development. While no defect in the suppressive ability of A2AAR−/− regulatory T cells was observed, A2AAR−/− lymphocytes where shown to proliferate more and produced more IFNγ following stimulation. Despite this more proinflammatory phenotype, A2AAR antagonist treatment still protected against EAE when A2AAR−/− lymphocytes were adoptively transferred to T cell deficient A2AAR+/+ mice. These results indicate that A2AAR expression on non-immune cells (likely in the CNS) is required for efficient EAE development, while A2AAR lymphocyte expression is essential for limiting the severity of the inflammatory response.
Introduction
Adenosine is an endogenous purine nucleoside that modulates a wide range of physiological functions (1). Most notable amongst its many roles is its importance in controlling inflammation (2). While adenosine is typically produced inside a cell, extracellular levels of adenosine rise as a consequence of the catabolism of adenosine triphosphate (ATP) that is released from stressed or damaged cells (3, 4). This extracellular ATP is converted to adenosine diphosphate (ADP) and adenosine monophosphate (AMP) by CD39 and then to adenosine by CD73 (5). The half-life of extracellular adenosine is on the order of seconds, as it is removed from the extracellular space either by adenosine deaminase (ADA), which converts it to inosine, or by cellular reuptake via equilibrative or concentrative nucleoside transporters (6, 7). Therefore, acute increases in extracellular adenosine levels tend to only have local and tissue limited effects. During inflammation, increases in extracellular adenosine levels effectively turn off the local inflammatory response to protect against excessive cellular damage to the surrounding tissue (2). Cells that express one or more of the four G-protein coupled adenosine receptors (AR) subtypes (A1, A2A, A2B, and/or A3) have the capacity to respond to extracellular adenosine. Since adenosine has such potent effects on inflammation, modulators of adenosine signaling are being evaluated as potential therapeutic options for diseases that have an inflammatory component (8). One such disease is multiple sclerosis (MS).
MS is an autoimmune inflammatory disease of the central nervous system (CNS) that affects more than 2.5 million people worldwide. During MS, infiltrating autoreactive immune cells attack and destroy myelin surrounding the axons of nerve cells in the brain and spinal cord resulting in loss of neurological function (9). The damage caused by this neuroinflammation can give rise to problems with vision, cognition, sensation, and coordination and balance (10). MS disease progression can present in patients in several forms, with new symptoms either occurring in discrete attacks (relapsing) or accumulating slowly over time (progressive). While the cause of MS is unknown, a majority of MS treatment research is focused on the prevention of disease relapse and progression (11). Therapeutically this can be accomplished in two general ways: 1) preventing/limiting the inflammatory response of lymphocytes, or 2) preventing lymphocyte infiltration in the CNS. Extracellular adenosine has been shown to be involved in both.
It is well documented that extracellular adenosine has potent anti-inflammatory properties (2). The inhibitory effects of extracellular adenosine and AR signaling have been observed in lymphocytes (5, 12–14), neutrophils (15–17), monocytes/macrophages (18–20), and dendritic cells (21, 22). For example, CD73−/− mice (which lack the ability to synthesize extracellular adenosine) have been shown to undergo a more severe form of inflammatory bowel disease (23). Likewise in the experimental autoimmune encephalomyelitis (EAE) animal model for MS, adoptively transferred lymphocytes from CD73−/− mice cause more severe EAE (as compared to those transferred from CD73+/+ wild type mice) when given to T cell deficient recipients (24). The anti-inflammatory effects of adenosine on immune cells are thought to be mainly mediated by A2AAR signaling (2, 8, 25–27).
In addition to extracellular adenosine’s role in down-modulating inflammation, it also has the potential to stimulate the migration of immune cells. For example, increases in extracellular adenosine and AR signaling promote the chemotaxis of neutrophils (28, 29) and immature dendritic cells (30) and induce cell migration into the lungs following injury to augment tissue repair (31). Additionally, AR signaling has been shown to be required for efficient lymphocyte migration into the CNS during EAE. CD73−/− mice or wild type animals that are given either broad spectrum AR antagonists (such as caffeine) or A2AAR specific antagonists are protected from EAE development due to a lack of lymphocyte migration into the brain and spinal cord (24, 32, 33). A2AAR signaling in the brain at the choroid plexus, which is the blood to cerebral spinal fluid barrier and the initial entry point into the CNS for lymphocytes during EAE (34–36), may promote this lymphocyte migration (24).
Since A2AAR signaling on lymphocytes has been suggested to have both anti-inflammatory (2) and pro-migratory effects on lymphocytes (24), we sought to define the role of A2AAR during the development of EAE. Utilizing A2AAR−/− mice, we determined that absence of A2AAR expression led to the development of a more severe form of EAE compared to wild type mice. This heightened disease course was due to the increased proinflammatory phenotype of A2AAR−/− immune cells, which overcame any protection that was imparted by the lack of A2AAR signaling on non-immune cells within the CNS. Our results demonstrate for the first time the differential role for the A2AAR in inflammation versus its role in CNS barrier function.
Materials and Methods
Mice
C57BL/6 mice (Jackson Laboratories) were used as wild type. A2AAR−/− mice (37) were a gift from Dr. Jiang-Fan Chen (Boston University School of Medicine, Boston, MA). Tcrα−/−;mice for transfer EAE experiments were purchased from The Jackson Laboratories. MOG-specific T cells from 2d2 TCR transgenic mice (38) were utilized for suppression assay experiments. All genetically modified mice were on the C57BL/6 background. Animals were bred and housed under specific pathogen-free conditions at Cornell University, Ithaca, NY. All procedures were done in accordance with approved IACUC protocols.
EAE Induction and Scoring
EAE was induced as previously described (39). Briefly, a 1:1 emulsion of MOG35–55 peptide (1 mg/ml in PBS) (Anaspec) and complete Freund’s adjuvant (CFA, Sigma) was injected subcutaneously (50 μl) into each flank. Pertussis toxin (PTX, 20 ng) (Biological Laboratories Inc.) was given intravenously (200 μl in PBS) at the time of immunization and again two days later. To induce EAE in Tcrα−/− mice, wild type and A2AAR−/− mice were primed with CFA:MOG peptide, and after 7 days, CD4+ T cells were isolated from spleen and lymph nodes by magnetic negative separation. CD4+ cells (106) were transferred intravenously to Tcrα−/− mice in a total of 200 μL in sterile PBS, with concomitant MOG:CFA subcutaneous injection and PTX intravenous injection. For SCH58261 (Tocris Bioscience) treatments, mice were given 5 mg/kg in olive oil via subcutaneous injection every 3–4 days, starting at day 1 post immunization. Mice were scored daily for EAE based on disease symptom severity; 0 = no disease, 0.5 – 1.0 = weak/limp tail, 2.0 = limp tail and partial hind limb paralysis, 3.0 = total hind limb paralysis, 4.0 = both hind limb and fore limb paralysis, 5.0 = death. Mice with a score of 4.0 were euthanized.
Immunostaining
Anesthetized mice from EAE experiments (post Day 30) were perfused with PBS, the brains, spinal cords, and spleens were isolated and snap frozen in Tissue Tek-OCT medium. Five micron sections (brains in a sagittal orientation) were affixed to Supefrost/Plus slides (Fisher Scientific), fixed in acetone, and stored at −80°C. For immunostaining, slides were thawed, washed in PBS, blocked with Casein (Vector) in normal goat serum (Zymed), and then incubated with antibodies against CD45 (YW62.3, AbD Serotec), CD4 (L3T4, BD Biosciences), CD11b (M1/70.15, Caltag), F480 (RM2915, Caltag), IBA-1 (polyclonal, Wako Pure Chemical Industries) or FoxP3 (FJK-16s, Ebioscience). For brightfield microscopic visualizations, slides were stained with a goat anti-rat biotin antibody (Jackson ImmunoResearch, Inc.) and then incubated with streptavidin-HRP (Invitrogen), developed with an AEC kit (Invitrogen), and counterstained with hematoxylin (Fisher Scientific). Images were obtained on a Zeiss Axio Imager M1 microscope utilizing AxioVision software.
Fluorescence In Situ Hybridization (FISH)
For detection of AR mRNA in the brain, we performed FISH using Biotin-labeled A2AAR DNA oligonucleotide probes (5′-ATACCCGTCACCAAGCCATTGTACCGGAGTGGAATTCGGATGGCG-3′, Integrated DNA Technologies) (40). Anesthetized mice were perfused with PBS and brains were isolated and snap frozen in Tissue Tek-OCT medium. Twelve micron cryosections were mounted on Superfrost/Plus slides (Fisher Scientific) and then fixed (4% neutral buffered paraformaldehyde) and rinsed (1× PBS). Next, the sections were equilibrated in 0.1 M triethanolamine and acetylated in 0.1 M triethanolamine with 0.25% acetic anhydride. The sections were dehydrated through an ascending ethanol series, and stored at room temperature. For hybridization, the sections were rehydrated in PBS, equilibrated in 5× SSC (NaCl 0.75M, Na-Citrate 0.075M), and prehybridized for 1 h at 42°C in hybridization buffer (50% deionized formamide, 4× SSC, salmon sperm DNA 40 μg/ml, 20% (w/v) dextran sulphate, 1× Denhardt’s solution). The probes (300 ng/ml) were denatured for at 80°C and added to the pre-warmed (42°C) buffer (hybridization mix). The hybridization reaction was carried out at 42°C for 38 h with 250 μl of hybridization mix. The sections were washed in 2× SSC (room temperature), 0.2× SSC 0.1% SDS (65°C), and then equilibrated in PBS. Sections were incubated with Texas-Red X conjugated streptavidine (Molecular Probes, S6370, 1 μg/ml) and then washed in PBS followed by 0.2× SSC 0.1% SDS (65°C) and PBS washes. Sections were coverslipped with Vectashield mounting medium with DAPI (Vector Laboratories). Images were acquired using a Zeiss Axio Imager M1 fluorescent microscope.
Suppression Assay
Spleen and lymph node cells were isolated from naïve wild type, A2AAR−/−, and 2d2 transgenic mice. Lymphocytes were incubated with ACK buffer (0.15M NH4Cl, 1 mM KHCO3, 0.1mM EDTA, pH 7.3) to lyse red blood cells. For wild type and A2AAR−/− cells, CD4+ cells were enriched by negative magnetic selection by incubating the cells with antibodies to CD8 (TIB-105), IAb,d,v,p,q,r (212.A1), FcR (2.4-G2), B220 (TIB-164), NK1.1 (HB191) and then with BioMag goat anti-mouse IgG, IgM, and goat anti-rat IgG (Qiagen), and then by removing the antibody/bead bound cells. These enriched CD4+ cells were incubated with an antibody against biotinylated-CD25 (PC61.5, eBioscience) and followed by anti-biotin conjugated beads (Miltenyi Biotec Inc.). The CD25+ fraction was magnetically isolated and utilized as the suppressor populations. Effector cells from 2d2 mice were first labeled with CFSE and then cultured (3.5 × 106) with MOG (10 μg/ml) and suppressor cells at varying concentrations. Proliferation was measured after 72 hours via CFSE loss analyzed on a FACSCanto II (BD Biosciences) with BD FACSDiva software (BD Biosciences).
Bone Marrow Radiation Chimeric Mice
For chimeras, the bone marrow recipients were irradiated (Mark I model 68 gamma irradiator, caesium source) with 2 × 650 rads spaced at a 4-h interval, which is effectively lethal to hematopoietic/immune cells (41–43). Twenty-four hours later, bone marrow was aseptically obtained from donor mice by removing the femurs/tibias from both legs and flushing out the bone marrow with a 27g needle and syringe. The bone marrow containing stem cells was washed and transferred intravenously (106 cells/mouse) to irradiated recipients. After 8–10 weeks of reconstitution, mice were utilized for EAE studies.
Proliferation Assay
Isolated lymphocytes (5 × 106 cells/ml) from the spleens and lymph nodes of MOG immunized mice were treated with varying concentrations of MOGpeptide (0, 1, 5, and 25 μg/well) or the mitogen concanavalin A (ConA). After 48 h of culture, 1 μCi 3H-thymidine was added to each well. Eighteen hours after the addition of thymidine, cells were harvested using a Tomtec Mach III harvester and quantified using the LS6500 Multi-purpose Scintillation Counter (Beckman Coulter).
Cytokine ELISA
Isolated lymphocytes (5 × 106 cells/ml) from the spleens and lymph nodes of MOG immunized mice were treated with varying concentrations of MOGpeptide (0, 1, 5, and 25 μg/well). Supernatants were collected at 48 hrs and analyzed utilizing IFNγ, IL-17, TNFα, and IL1β ELISA kits (eBioscience) according to manufacturer’s instructions. Cytokine measurements on samples were performed on a BioTek Synergy 4 and concentrations were derived from a standard curve utilizing Gen5 data analysis software.
Statistical analyses
Statistical differences between groups over a time course, as in EAE studies, were determined utilizing GraphPad Prism and Microsoft Excel software. Statistical differences between EAE treatment groups were determined by two-way ANOVA analysis, while difference between time points was determined utilizing the Mann-Whitney U test. The Student’s T-test was utilized for other comparisons unless stated within the figure legends. Statistical differences were determined where P ≤ 0.05.
Results
A2AAR−/− mice develop more severe EAE
Extracellular adenosine and AR signaling have been previously shown to be involved in both the development and progression of EAE (24, 32). For example, mice unable to produce extracellular adenosine (CD73−/− mice) (24) or given the A2AAR specific antagonist SCH58261 [Figure 1A and (24)] are protected against EAE induction and lack CNS lymphocyte infiltrates that are associated with disease progression (24). To fully investigate the importance of A2AAR signaling during EAE progression, we first actively induced EAE in wild type and A2AAR−/− mice (37) by the MOG35–55 immunization method (see materials and methods) and monitored and scored them daily for clinical signs of EAE. A2AAR−/− mice developed more severe EAE than wild type control mice (Figure 1B). While A2AAR−/− mice exhibited significantly more severe paralysis at days 12–16 post EAE induction (Figure 1B), no difference between the mean day of disease onset and average maximum EAE score between wild type and A2AAR−/− mice was observed (Table 1). These studies indicate that genetic disruption of the A2AAR does not confer protection against EAE, but instead promotes a more severe acute disease.
Figure 1. A2AAR antagonism protects against EAE while A2AAR−/− mice are susceptible to EAE.

(A) EAE was induced in wild type mice that were given SCH58261 A2AAR antagonist (open triangles, n=12) or vehicle (closed triangles, n=13) treatment, disease activity was monitored daily and the mean EAE score was calculated. The results shown are from 3 separate experiments. Error bars represent the standard error of the mean. (B) EAE was induced in wild type (closed squares, n=12) and A2AAR−/− mice (open squares, n=13), disease activity was monitored daily and the mean EAE score was calculated. The results shown are representative of 2 separate experiments. Error bars represent the standard error of the mean. Statistically different mean EAE scores at each time point are indicated (*, p ≤ 0.05).
Table I.
A2AAR−/− mice are susceptible to EAE development.A
| IncidenceB | Mean Dayof OnsetC | Mean Max EAE ScoreD | |
|---|---|---|---|
| WT | 12/12 | 11.6 +/−1.1 | 2.3 +/−0.3 |
| A2AAR−/− | 13/13 | 10.5 +/−0.9 | 2.7 +/−0.2 |
Wild type and A2AAR−/− mice were induced to develop EAE and scored daily for EAE severity based on the 5 point scale assessing ascending paralysis.
. Indicates the number of mice that achieved a score of 0.5 (weak tail) in the experimental group.
Indicates the average day of onset (an EAE score of 0.5, +/− the s.e.m.).
Indicated the average of the maximum EAE score for each individual mouse (+/− the s.e.m.).
A2AAR−/− mice have more CNS lymphocyte infiltrates compared to wild type during EAE
EAE is mediated by the infiltration of autoreactive immune cells into the CNS. The inflammatory response directed against myelin, which insulates neuronal axons in the brain and spinal cord, induces paralysis in mice as a result of motor neuron demyelination (34, 44). To assess CNS lymphocyte infiltration during EAE in A2AAR−/− mice, brain and spinal cord sections were examined for presence of CD45+ (general leukocyte marker) and CD4+ T cells by immunohistochemistry (Figure 2). Following EAE induction, both wild type and A2AAR−/−mice had distinct patches of immune cell infiltration in the cerebellum (Figure 2A, 2E, 2I, 2M), hippocampus (Figure 2B, 2F, 2J, 2N), and spinal cord (Figure 2C–D, 2G–H, 2K–L, 2O–P). However, A2AAR−/− mice had visually and quantitatively significantly more CD45+ (Figure 2A–H) and CD4+ (Figure 2I–Q) cells in their brains and spinal cords compared to wild type mice. These results indicate that the more severe disease observed in A2AAR−/− mice (Figure 1B) is associated with increased immune cell numbers in the CNS.
Figure 2. Increased numbers of lymphocytes are observed in the brain and spinal cord of A2AAR−/− mice following EAE.

Representative pictures taken from frozen tissue sections of the cerebellum (A, E, I, M), hippocampus (B, F, J, N) and spinal cord (C–D, G–H, K–L, O–P) from post-EAE induced wild type (A–D, I–L) and A2AAR−/− (E–H, M–P) mice were labeled with either CD45 (A–H) or CD4 (I–P) antibodies to detect lymphocyte infiltration in the CNS follow disease induction. Immunoreactivity was detected with HRP anti-rat Ig plus AEC (red) against a hematoxylin stained nuclear background (blue). Scale bars represent 100 μm. (Q) In post-EAE induced wild type and A2AAR−/− mice, six anatomically similar fields per brain [cerebellum (2x), hippocampus (2x), frontal lobe, and brain stem] and 4 fields per spinal cord per mouse were analyzed at 10x magnification for CD4 cell infiltration. Error bars represent the standard error of the mean (n ≤ 10). Statistically different infiltrating CD4 cells mean values counted per field between wild type and A2AAR−/− mice in each tissue are displayed (*, p ≤ 0.05; **, p ≤ 0.01).
A2AAR−/− mice have increased dissemination and retention of CD11b+/F480+ and IBA1+ cells in the CNS during EAE
Macrophage/microglia migration and activation in the CNS is critical for the demyelination and clinical signs of EAE (45, 46). To assess the frequency of macrophages/microglia in the CNS of wild type and A2AAR−/− mice with MOG35–55 induced EAE, brain and spinal cord sections were stained with CD11b and F480 and analyzed by immunohistochemistry (Figure 3A–P). While both wild type and A2AAR−/− mice had a large number of CD11b+ (Figure 3A–H) and F480+ (Figure 3I–P) cells present in their CNS following EAE, the frequency of CD11b+/F480+ cells was dramatically higher in A2AAR−/− (Figure 3E–H, M–P) compared to wild type mice (Figure 3A–D, I–L). The most noticeable differences in frequency between the groups was observed in the spinal cord (Figure 3C-D, G-H, K–L, O–P), with a majority of the A2AAR−/− mice displaying heavy patches of CD11b+/F480+ cells (Figure 3G–H, O–P). These data suggest that the absence of A2AAR signaling results in increased microglia/macrophage migration to the spinal cord during EAE.
Figure 3. Increased macrophage/microglial infiltration and activation are observed in CNS of A2AAR−/− mice following EAE.

Representative pictures taken from frozen tissue sections of the cerebellum (A, E, I, M), hippocampus (B, F, J, N) and spinal cord (C–D, G–H, K–L, O–P, Q–T) from EAE induced wild type (A–D, I–L, Q–R) and A2AAR−/− (E–H, M–P, S–T) mice were labeled with either CD11b (A–H), F480 (I–P), or IBA1 (Q–T) antibodies to detect microglial/microphage infiltration and activation in the CNS follow disease induction. Immunoreactivity was detected with HRP anti-rat Ig plus AEC (red) against a hemotoxylin stained nuclear background (blue). Inset figures show increased magnification. Scale bars represent 100 μm. (U) In EAE induced wild type and A2AAR−/− mice, seven fields per spinal cord per mouse were analyzed at 10x magnification for IBA1 staining. Error bars represent the standard error of the mean (n ≤ 5). Statistically different IBA1+ mean spinal cord counts per field between EAE induced wild type and A2AAR−/− mice are displayed (***, p ≤ 0.001).
We next determined the activation state of the macrophages/microglia in the CNS following EAE induction in spinal cord sections stained for the presence of Iba1 (Figure 3Q–T), a marker for macrophage and microglial cell activation (47). Spinal cord sections from A2AAR−/−mice with EAE had many areas that reacted strongly with the Iba1 antibody and displayed a greater preponderance of ameboid shaped cells (Figure 3S–U), which are representative of activated microglia (48). Conversely, while spinal cord sections from wild type mice with EAE did have a few areas depicting microglia with an activated phenotype (data not shown), a majority of their spinal cord had microglial cells that displayed a predominantly ramified morphology with less Iba1 staining (Figure 3Q–R, U), which is consistent with that of resting microglial cells (48). This suggest that lack of A2AAR expression on microglia cells confers a more inflammatory microglia population in the CNS microenvironment, which is consistent with the more severe disease observed A2AAR−/− mice (Figure 1B).
A2AAR−/− lymphocytes produce more IFNγ than those from wild type mice
Proinflammatory cytokines, such as IFNγ and IL-17, have been shown to have a prominent role in the inflammatory response mediated by the infiltrating CNS lymphocytes during EAE (49). To determine if A2AAR−/− lymphocytes are intrinsically more proinflammatory than those from wild type mice, lymphocytes from MOG-immunized wild type and A2AAR−/− mice were restimulated with MOG in vitro and cytokine production was assessed (Figure 4A–D). Lymphocytes activated with MOG from A2AAR−/− mice produced significantly more IFNγ in a dose dependent manner compared to those from wild type mice (Figure 4A). No significant difference in IL-17 (Figure 4B), TNFα (Figure 4C), IL1β (Figure 4D), IL4 (data not shown), or IL10 (data not shown) production was detected. High production of IFNγ suggests that the lack of A2AAR expression on lymphocytes promotes a more proinflammatory phenotype.
Figure 4. A2AAR−/− lymphocytes produce higher levels of IFNγ.

Splenocytes from wild type (black bars) and A2AAR−/− (white bars) post-EAE induced mice were isolated and stimulated in culture with varying concentrations of MOG. (A) IFNγ, (B) IL-17, (C) TNFα and (D) IL1β levels were measured by ELISA. Error bars represent the standard error of the mean (n ≤ 5). Statistically different values of wild type compared A2AAR−/− mice for each condition are displayed (*, p ≤ 0.05).
A2AAR−/− lymphocytes have a higher proliferative potential while T regulatory (Treg) cell frequency and function are unaltered
Adenosine has been shown to inhibit lymphocyte proliferation (50). To determine if A2AAR−/−lymphocytes are intrinsically more proliferative than those from wild type mice, lymphocytes were isolated from the spleen and lymph nodes of wild type and A2AAR−/− mice, stimulated with the mitogen ConA, and proliferation was measured based on thymidine incorporation (Figure 5A). While there was no difference in the baseline proliferation within control non-stimulated splenocytes, A2AAR−/− lymphocytes that were stimulated with ConA proliferated significantly more compared to those from wild type mice (Figure 5A). To determine if lymphocytes from A2AAR−/− mice also proliferated more in response to antigenic stimuli, lymphocytes from mice immunized with MOG35–55 were isolated and their recall response to MOG was tested in culture (Figure 5B). Similar to the results for ConA stimulation, lymphocytes reactivated with MOG from A2AAR−/− mice proliferated significantly more than those from wild type mice (Figure 6B). These results indicate that lymphocytes from A2AAR−/− mice have an enhanced proliferative potential.
Figure 5. A2AAR−/− lymphocytes proliferate more despite normal regulatory T cells frequency and function.

(A–B) Splenocytes from wild type (black bars) and A2AAR−/− (white bars) post-EAE induced mice were isolated and stimulated in culture with (A) ConA or (B) varying concentrations of MOG. (A–B) Cell proliferation was assessed by thymidine incorporation and measured utilizing a scintillation counter. Error bars represent the standard error of the mean (n ≤ 5). (C–E) Frozen tissue sections of (C–D) spleen and (D) brain and spinal cord from EAE induced (C) wild type and (D) A2AAR−/− mice were labeled with an antibody against FoxP3 to detect Treg cells. Immunoreactivity was detected with HRP anti-rat Ig plus AEC (red) against a hemotoxylin stained nuclear background (blue). Scale bars represent 100 μm. (E) Wild type and A2AAR−/− mice from EAE mice were analyzed for total FoxP3 staining in their brain and spinal cord. Error bars represent the standard error of the mean (n ≤ 6). (F) Lymphocytes from naïve wild type and A2AAR−/− CD4+CD25+ were used as suppressor T cells for the effector T cells isolated from 2d2 MOG-specific TCR transgenic mice stimulated in vitro with MOG. Percent of 2d2 T cell proliferation in response to MOG stimulation is displayed for varying effector 2d2 cell to suppressor cell ratios. Error bars represent the standard error of the mean (n ≥ 2). Statistically different values of wild type compared A2AAR−/− mice for each condition are displayed (*, p ≤ 0.05).
Figure 6. A2AAR expression on hematopoietic and non-hematopoietic subsets in bone marrow chimeric mice influences EAE susceptibility.

(A–D) Fluorescence in situ hybridization of A2AAR expression (red) in the brain (A) choroid plexus, (B) meninges, (C) hippocampus, and (D) cerebellum of naïve wild type mice (DAPI stained nuclei, blue). Scale bars represent 100 μm. (E–F) Gamma-irradiated recipient wild type and A2AAR−/− mice given bone marrow from wild type or A2AAR donor mice, were induced to develop EAE eight weeks after the irradiation/reconstitution. Disease activity was monitored daily and the mean EAE score was calculated. Plot legends are read as “bone marrow donor” into “irradiated recipient” mice. (E) Error bars represent the standard error of the mean (n ≤ 8). Statistical comparison between the total EAE disease course for the chimera groups in each plot was performed via two-way ANOVA analysis, with the resulting p values displayed. (F) In post-EAE induced wild type and A2AAR−/− mice, five anatomically similar fields per brain [cerebellum (2x), hippocampus (2x), and frontal lobe] and 4 fields per spinal cord per mouse were analyzed at 10x magnification for CD4 cell infiltration. Error bars represent the standard error of the mean (n ≤ 5). Statistically different infiltrating CD4 cells mean values counted per field compared to the wild type into A2AAR−/− group are displayed (*, p ≤ 0.05).
Treg cells have a critical role in autoimmune suppression and regulation of inflammation (51). Decreases in Treg cell number or function have been shown to leave mice susceptible to severe EAE (52). To determine if the Treg cell population is altered in A2AAR−/− mice, we analyzed their frequency (FoxP3+) and functionality (ability to suppress effector cell proliferation). Immunohistochemistry staining of FoxP3+ cells in the spleen and CNS showed no difference in the frequency of FoxP3+ cells between wild type and A2AAR−/− mice with EAE (Figure 5C–E). Additionally, no difference was observed in the ability of A2AAR−/− mice Treg cells (CD4+CD25+ from naïve mice) to suppress effector cell proliferation compared to Tregs from wild type mice (Figure 5F). At all Treg to T effector cell ratios, A2AAR−/− Treg cells suppressor function was similar to that of wild type Treg cells (Figure 5F). These findings indicate that the severe EAE observed in A2AAR−/− mice was not due to a significant alteration in the Treg cell population.
A2AAR expression on immune cells limits the severity of EAE, whereas A2AAR expression on non-immune cells promotes more severe EAE
It is well established that the A2AAR is expressed on a variety cells, including lymphocytes (5, 12, 53) and cells in the CNS (54, 55). Within the brain, high expression of the A2AAR was found throughout the choroid plexus (Figure 6A), a site shown to be involved in immune cell migration into the CNS (34–36). A2AAR expression, while less than that at the choroid plexus, was also observed in the meninges (Figure 6B) and near the hippocampus (Figure 6C) and cerebellum, albeit to a lesser degree (Figure 6D). Since A2AAR−/− mice are susceptible to severe acute EAE (Figure 1B), we next wanted to determine where the A2AAR must be expressed (i.e., on immune cells or the CNS) in order to prevent this aggravated disease phenotype. Therefore, we utilized the radiation bone marrow chimera experimental model system (41–43), in which the animal’s immune cells (which are both sensitive to radiation and derived from stem cells in the bone marrow) are replaced following irradiation and subsequent bone marrow transplant from donor animals. Gamma-irradiated wild type mice were reconstituted with bone marrow from either naïve wild type or A2AAR−/− donor mice and then immunized with MOG35–55 to induce EAE (Figure 6E). Wild type mice reconstituted with A2AAR−/− cells developed significantly more severe EAE compared to those that received bone marrow from wild type donors (Figure 6E and Table 2). This exacerbated disease development suggests that A2AAR expression on bone marrow derived immune cells is required to control the severity of EAE.
Table II.
A2AAR−/− hematopoietic cells produce more severe EAE while A2AAR−/− recipient mice are more resistant to EAE development.A
| IncidenceB | Mean Dayof OnsetC | Mean Max EAE ScoreD | |||
|---|---|---|---|---|---|
| WTinto WT | 7/7 | 7.7+/−0.4 | 3.0+/−0.2 |  |
|
| A2AAR−/−into WT | 8/8 | 8.0+/−0.4 | 3.9+/−0.4 | ||
| WT into A2AAR−/− | 7/7 | 9.6+/−1.6 | 1.1+/−0.4 | ||
Bone marrow chimeric mice were induced to develop EAE and scored daily for EAE severity based on the 5 point scale assessing ascending paralysis.
Indicates the number of mice that achieved a score of 0.5 (weak tail) in the experimental group.
Indicates the average day of onset (an EAE score of 0.5, +/− the s.e.m.).
Indicated the average of the maximum EAE score for each individual mouse (+/− the s.e.m.).
WT into WT compared to A2AAR−/− into WT Mean Max EAE Score; p = 0.0566.
WT into WT compared to WT into A2AAR−/− Mean Max EAE Score; p = 0.00073.
To determine the influence of A2AAR on non-immune cells (i.e., radiation resistant cells), irradiated A2AAR−/− mice were reconstituted with bone marrow from wild type donor mice and then induced to develop EAE (Figure 6E). A2AAR−/− mice reconstituted with wild type cells were protected from EAE and developed only mild disease compared to wild type donor cells transferred into wild type recipient mice (Figure 6E and Table 2). This protection from disease development suggests that A2AAR expression on radiation resistant cells (such as those in the CNS) is required for EAE progression, which is similar to the protective EAE effects of the A2AAR antagonist SCH58261 observed in wild type mice (Figure 1A).
To assess CNS lymphocyte infiltration during EAE in the chimeric mice, brain and spinal cord sections were examined for presence of CD4+ T cells by immunohistochemistry (Figure 6F). Similar to EAE severity (Figure 6E), A2AAR−/− recipient mice had virtually no CD4+ T cell infiltration in the spinal cord, while mice that received A2AAR−/− donor cells had significant spinal cord infiltration (Figure 6F). Overall, these results indicate that lack of A2AAR expression on hematopoietic cells (such as lymphocytes) promotes severe EAE, while lack of A2AAR on non-hematopoietic cells (most likely in the CNS) is protective during EAE development.
A2AAR antagonism protects against EAE mediated by A2AAR−/− adoptively transferred cells
We have shown that the SCH58261 A2AAR antagonist can protect against EAE development [Figure 1A and (24)], despite the fact that genetic disruption of the A2AAR leaves mice prone to developing a severe acute form of EAE (Figure 1B), which is likely due to the more proinflammatory nature of A2AAR−/− lymphocytes (Figures 4 and 5). Therefore, we next asked whether SCH58261 treatment can prevent EAE when A2AAR−/− effector T cells are utilized to induce disease. To test this, primed CD4+ T cells from the spleen and lymph nodes of MOG immunized wild type and A2AAR−/− mice were transferred into Tcrα−/− (A2AAR+/+) recipient mice and given SCH58261 or vehicle control treatments following EAE induction. Tcrα−/− mice lack endogenous T cells and cannot develop EAE on their own [data not shown and (56)]. In the vehicle control groups, A2AAR+/+tcrα−/− recipient mice that received CD4+ T cells from A2AAR−/− donors developed a more severe disease progression compared to those that received wild type CD4+ T cells (Figure 7). However, mice that received SCH58261 treatment, regardless of whether wild type or A2AAR−/− CD4+ T cells were transferred, were protected from EAE development (Figure 7). These results suggest that A2AAR antagonist mediated blockade is effective at preventing EAE in a lymphocyte-independent manner. Overall, our results indicate that the A2AAR has an important role in mediating both the proinflammatory potential of lymphocytes and the susceptibility to CNS disease progression/lymphocyte infiltration during EAE development.
Figure 7. The A2AAR antagonist SCH58261 protects against EAE mediated by adoptively transferred A2AAR−/− cells.
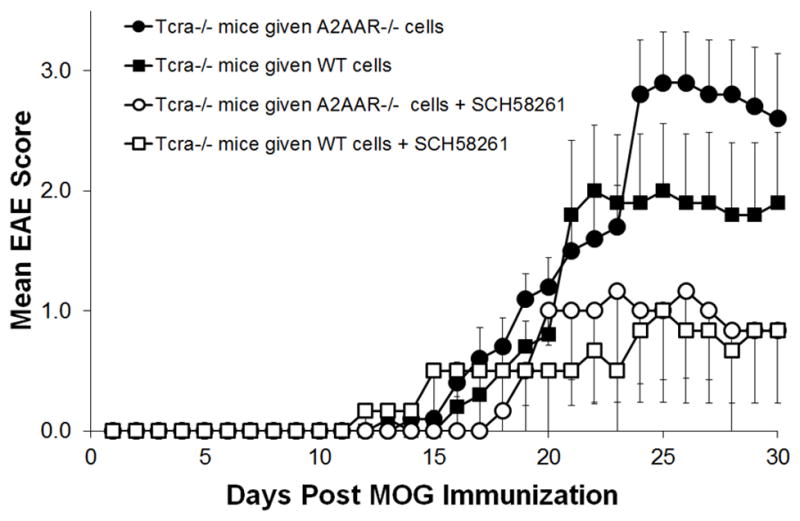
EAE was induced in T cell deficient Tcrα−/− mice that received adoptively transferred lymphocytes from wild type (squares) or A2AAR−/− (circles) mice and given SCH58261 A2AAR antagonist (open shape) or vehicle (closed shape) treatment (n ≤ 5). Disease activity was monitored daily and the mean EAE score was calculated.
Discussion
In this study and previously (24), we have demonstrated that blockade of the A2AAR with an A2A specific AR antagonist, protected mice from EAE by hindering lymphocyte entry into the brain and spinal cord of wild type mice. This finding was unexpected as this function of adenosine, that is, mediating lymphocyte migration into the CNS, was previously unknown. Further, the fact that adenosine suppresses the immune response and resolves inflammation (2) are at odds with the finding that blockade of the A2AAR, which mediates the preponderance of adenosine‘s suppressive and anti-inflammatory functions (2), protects mice from EAE (24). The purpose of this study was to delineate adenosine’s role in the immune response from its function in mediating immune cell migration into the CNS via the A2A receptor.
We show that A2AAR−/− mice developed more severe EAE than their wild type counterparts. This severe disease was characterized in the CNS by increased numbers of lymphocytes and activated macrophage/microglia cells in the CNS parenchyma, especially in the spinal cord. Furthermore, we found that the highly pro-inflammatory nature of the A2AAR−/−immune cells appears to be directly responsible for the severe EAE in the A2AAR−/− mice [similar observations were made at both day 16 during the peak of disease (not shown) and day 30 post-EAE induction]. For example, the transfer of hematopoietic cells lacking the A2AAR into irradiated wild type recipients not only caused more severe EAE than wild type cells transferred into irradiated wild type mice, but the disease was more severe than the EAE observed in the parent A2AAR−/− mice (3.9 vs. 2.7, mean maximum EAE score). More importantly, A2AAR−/− (recipient) mice that received wild type (donor) bone marrow were protected from EAE development. This suggests that A2AAR−/− mice are susceptible to a severe acute form of EAE due to the more proinflammatory immune cells. This is further supported by data showing that A2AAR−/− lymphocytes are more proliferative and produced more IFNγ than their wild type counterparts. These results are consistent with the role of the A2AAR in extracellular adenosine mediated regulation of inflammation and of the immune response (2, 8, 25–27). Also important, our chimeric data suggest that the lack of A2AAR signaling in the CNS (most likely on CNS barrier cells such as the choroid plexus and the blood-brain barrier) confers protection against EAE development. Therefore, the A2AAR has two apparent roles during EAE progression: 1) to control the magnitude of the inflammatory response (via expression on lymphocytes), and 2) to allow for efficient lymphocyte entry/infiltration into the CNS (via expression at the choroid plexus). These studies show that the heightened proinflammatory potential of A2AAR−/− immune cells can mask the protective effects imparted by the absence of A2AAR signaling on CNS barriers.
Our findings showing a role for the A2AAR in controlling the severity of EAE is consistent with its role described in other experimental model systems demonstrating the importance of A2AAR signaling in limiting inflammation and tissue injury. For example, mice lacking the A2AAR have increased liver damage in ischemia-reperfusion liver injury (25) and are more sensitive to bronchiolitis obliterans (26). Additionally, A2AAR expression on bone marrow derived cells is important in protecting against damaged caused by inflammation in LPS induced acute lung injury (57), myocardial infarction (13), spinal cord injuries (58) cerebral ischemia (37), and other various neuroinflammatory injuries (59). Furthermore, the use of A2AAR specific agonists have been shown to be beneficial in the treatment of inflammatory bowel disease (8) and ischemia-reperfusion liver (25) and lung (60) injuries. These protective effects of A2AAR stimulation have been attributed to its ability to inhibit the production of the proinflammatory cytokines IL-12, INFγ, IL-6, and TNFα (12, 27, 53, 61, 62). Indeed, we also show that lymphocytes from A2AAR−/− mice with EAE produce more IFNγ that those from wild type mice. We also observed that A2AAR−/− lymphocytes have a higher proliferative capacity than those expressing the A2AAR. Therefore, it was not surprising we observed that A2AAR−/−bone marrow derived cells (i.e. lymphocytes) were able to cause more severe EAE compared to those from A2AAR+/+ wild type mice.
Our results also suggest that A2AAR signaling plays a major role in regulating lymphocyte migration into the CNS. For instance, mice treated with an A2AAR antagonist (24) or that lack A2AAR expression in their CNS have significantly fewer lymphocytes in their CNS during EAE compared to control mice. Consistent with our observations, other studies have also identified extracellular adenosine signaling as having an important role in regulating cell migration, albeit in a cell and tissue specific manner. For example, extracellular adenosine has been shown to induce chemotaxis in immature dendritic cells (30), endothelial cells (63, 64), oligodendrocytes (65), and bronchial epithelial cells (31). Conversely, it has also been reported that extracellular adenosine can inhibit the migration of eosinophils (66), mast cells (67) and microglia/monocytes (68). Furthermore, with other cell types such as neutrophils, adenosine signaling has been shown to have the capacity to both induce (28, 29) and inhibit (17, 69) cell migration depending on the experimental system. Therefore, the effects of extracellular adenosine on cell migration may not only vary among cell types, but also depend on the circumstance in which the AR signaling occurs.
During EAE progression within the CNS, our results suggest that extracellular adenosine may be functioning as a major “danger/damage” signal (70–72). ATP released from stressed or damaged cells in the CNS is hydrolyzed into extracellular adenosine (3, 4). Since the CNS is an immune privileged site (73), we believe this “danger” signal is a trigger which promote lymphocyte infiltration into the CNS (24). Our results suggest that this extracellular ATP/adenosine danger signal is modified (ATP hydrolysis to adenosine), interpreted (AR signaling), and executed by the choroid plexus (lymphocyte entry), which possess the enzymes (CD39 and CD73) and receptors (A2AAR) required to synthesize and bind extracellular adenosine and has been shown to be a CNS entry point for lymphocytes during EAE progression (34–36). Our bone marrow chimera data provides further evidence of this, as the lack of the A2AAR on non-bone marrow derived cells (such as those comprising the choroid plexus) confers a degree of protection against lymphocyte infiltration its subsequent EAE development. Similar results are observed during lung injury, where AR signaling has been shown to induce cell migration to repair damaged tissue (31).
Extracellular adenosine signaling has also been shown to play a role in inflammation during hypoxia and ischemia/reperfusion injuries. Hypoxia that results from ischemia typically leads to vascular leakage, the accumulation of inflammatory cells and elevated serum cytokine levels (74). Additionally, just as hypoxia can induce inflammation, inflamed tissues often become severely hypoxic (74). Interestingly, hypoxic conditions have been reported to promote increases in extracellular adenosine levels by both blocking adenosine’s uptake into cells (75) and its catalysis into AMP by adenosine kinase (76). Therefore, hypoxic states promote ideal conditions for stimulating the A2AAR, which requires high concentrations of extracellular adenosine to become activated (1). The increased extracellular adenosine levels during hypoxia have been associated with tissue protection against inflammation, which is in part regulated by the induction of netrin-1 that effectively attenuates neutrophil transmigration (77). Interestingly, netrin-1 expression has also been found in EAE lesions in rats (78). However, a direct link between netrin-1 and extracellular adenosine signaling during EAE has yet to be established.
Overall, it is evident that extracellular adenosine plays a vital and complex role in the development of EAE. A2AAR signaling, while important for controlling the magnitude of an inflammatory response, is also involved in lymphocyte entry into the CNS. Additionally, complete ablation of the A2AAR does not protect mice from developing EAE, whereas A2AAR blockade with drug treatments directly inhibiting signaling is beneficial in EAE (24). Furthermore, both acute and chronic-relapsing remitting EAE can be prevented/reversed by the drug methylthioadenosine (79, 80), which acts both as an agonist for the A1AR and an antagonist for the A2AAR (81). Therefore, the data presented here strongly suggests that since extracellular adenosine and A2AAR signaling are so highly involved in lymphocyte infiltration and inflammation in the CNS, therapeutic strategies targeting extracellular ATP/adenosine metabolism and signaling pathways may be beneficial in the treatment of many diseases such as MS that have a major neuroinflammatory component.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS063011 (to M.S.B.) and F32 NS 066682 (to J.H.M.).
Works Cited
- 1.Fredholm BB, APIJ, Jacobson KA, Linden J, Muller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharmacological reviews. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blackburn MR, Vance CO, Morschl E, Wilson CN. Adenosine receptors and inflammation. Handbook of experimental pharmacology. 2009:215–269. doi: 10.1007/978-3-540-89615-9_8. [DOI] [PubMed] [Google Scholar]
- 3.Cook SP, McCleskey EW. Cell damage excites nociceptors through release of cytosolic ATP. Pain. 2002;95:41–47. doi: 10.1016/s0304-3959(01)00372-4. [DOI] [PubMed] [Google Scholar]
- 4.Motte S, Communi D, Pirotton S, Boeynaems JM. Involvement of multiple receptors in the actions of extracellular ATP: the example of vascular endothelial cells. The international journal of biochemistry & cell biology. 1995;27:1–7. doi: 10.1016/1357-2725(94)00059-x. [DOI] [PubMed] [Google Scholar]
- 5.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. The Journal of experimental medicine. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mistry G, Drummond GI. Adenosine metabolism in microvessels from heart and brain. Journal of molecular and cellular cardiology. 1986;18:13–22. doi: 10.1016/s0022-2828(86)80978-6. [DOI] [PubMed] [Google Scholar]
- 7.Redzic ZB, Biringer J, Barnes K, Baldwin SA, Al-Sarraf H, Nicola PA, Young JD, Cass CE, Barrand MA, Hladky SB. Polarized distribution of nucleoside transporters in rat brain endothelial and choroid plexus epithelial cells. Journal of neurochemistry. 2005;94:1420–1426. doi: 10.1111/j.1471-4159.2005.03312.x. [DOI] [PubMed] [Google Scholar]
- 8.Odashima M, Bamias G, Rivera-Nieves J, Linden J, Nast CC, Moskaluk CA, Marini M, Sugawara K, Kozaiwa K, Otaka M, Watanabe S, Cominelli F. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology. 2005;129:26–33. doi: 10.1053/j.gastro.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 9.Boppana S, Huang H, Ito K, Dhib-Jalbut S. Immunologic aspects of multiple sclerosis. The Mount Sinai journal of medicine, New York. 2011;78:207–220. doi: 10.1002/msj.20249. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Zacharia AB. Therapeutics for multiple sclerosis symptoms. The Mount Sinai journal of medicine, New York. 2011;78:176–191. doi: 10.1002/msj.20245. [DOI] [PubMed] [Google Scholar]
- 11.Buck D, Hemmer B. Treatment of multiple sclerosis: current concepts and future perspectives. Journal of neurology. 2011;258:1747–1762. doi: 10.1007/s00415-011-6101-2. [DOI] [PubMed] [Google Scholar]
- 12.Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol. 2005;174:1073–1080. doi: 10.4049/jimmunol.174.2.1073. [DOI] [PubMed] [Google Scholar]
- 13.Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, French BA, Linden J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–2064. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 14.Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: involvement of protein kinase A isozyme I (PKA I) Immunologic research. 2006;36:91–99. doi: 10.1385/IR:36:1:91. [DOI] [PubMed] [Google Scholar]
- 15.Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 16.Harada N, Okajima K, Murakami K, Usune S, Sato C, Ohshima K, Katsuragi T. Adenosine and selective A(2A) receptor agonists reduce ischemia/reperfusion injury of rat liver mainly by inhibiting leukocyte activation. The Journal of pharmacology and experimental therapeutics. 2000;294:1034–1042. [PubMed] [Google Scholar]
- 17.Wakai A, Wang JH, Winter DC, Street JT, O’Sullivan RG, Redmond HP. Adenosine inhibits neutrophil vascular endothelial growth factor release and transendothelial migration via A2B receptor activation. Shock (Augusta, Ga. 2001;15:297–301. doi: 10.1097/00024382-200115040-00008. [DOI] [PubMed] [Google Scholar]
- 18.Salmon JE, Brogle N, Brownlie C, Edberg JC, Kimberly RP, Chen BX, Erlanger BF. Human mononuclear phagocytes express adenosine A1 receptors. A novel mechanism for differential regulation of Fc gamma receptor function. J Immunol. 1993;151:2775–2785. [PubMed] [Google Scholar]
- 19.Eppell BA, Newell AM, Brown EJ. Adenosine receptors are expressed during differentiation of monocytes to macrophages in vitro. Implications for regulation of phagocytosis. J Immunol. 1989;143:4141–4145. [PubMed] [Google Scholar]
- 20.Zhang JG, Hepburn L, Cruz G, Borman RA, Clark KL. The role of adenosine A2A and A2B receptors in the regulation of TNF-alpha production by human monocytes. Biochemical pharmacology. 2005;69:883–889. doi: 10.1016/j.bcp.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Chen L, Fredholm BB, Jondal M. Adenosine, through the A1 receptor, inhibits vesicular MHC class I cross-presentation by resting DC. Molecular immunology. 2008;45:2247–2254. doi: 10.1016/j.molimm.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, Girolomoni G, Norgauer J. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood. 2003;101:3985–3990. doi: 10.1182/blood-2002-07-2113. [DOI] [PubMed] [Google Scholar]
- 23.Louis NA, Robinson AM, MacManus CF, Karhausen J, Scully M, Colgan SP. Control of IFN-alphaA by CD73: implications for mucosal inflammation. J Immunol. 2008;180:4246–4255. doi: 10.4049/jimmunol.180.6.4246. [DOI] [PubMed] [Google Scholar]
- 24.Mills JH, Thompson LF, Mueller C, Waickman AT, Jalkanen S, Niemela J, Airas L, Bynoe MS. CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9325–9330. doi: 10.1073/pnas.0711175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Day YJ, Marshall MA, Huang L, McDuffie MJ, Okusa MD, Linden J. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: inhibition of chemokine induction. American journal of physiology. 2004;286:G285–293. doi: 10.1152/ajpgi.00348.2003. [DOI] [PubMed] [Google Scholar]
- 26.Lau CL, Zhao Y, Kron IL, Stoler MH, Laubach VE, Ailawadi G, Linden J. The role of adenosine A2A receptor signaling in bronchiolitis obliterans. The Annals of thoracic surgery. 2009;88:1071–1078. doi: 10.1016/j.athoracsur.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alam MS, Kurtz CC, Wilson JM, Burnette BR, Wiznerowicz EB, Ross WG, Rieger JM, Figler RA, Linden J, Crowe SE, Ernst PB. A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4+ helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal immunology. 2009;2:232–242. doi: 10.1038/mi.2009.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. The Journal of clinical investigation. 1990;85:1150–1157. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose FR, Hirschhorn R, Weissmann G, Cronstein BN. Adenosine promotes neutrophil chemotaxis. The Journal of experimental medicine. 1988;167:1186–1194. doi: 10.1084/jem.167.3.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panther E, Idzko M, Herouy Y, Rheinen H, Gebicke-Haerter PJ, Mrowietz U, Dichmann S, Norgauer J. Expression and function of adenosine receptors in human dendritic cells. Faseb J. 2001;15:1963–1970. doi: 10.1096/fj.01-0169com. [DOI] [PubMed] [Google Scholar]
- 31.Allen-Gipson DS, Spurzem K, Kolm N, Spurzem JR, Wyatt TA. Adenosine promotion of cellular migration in bronchial epithelial cells is mediated by the activation of cyclic adenosine monophosphate-dependent protein kinase A. J Investig Med. 2007;55:378–385. doi: 10.2310/6650.2007.00019. [DOI] [PubMed] [Google Scholar]
- 32.Tsutsui S, Schnermann J, Noorbakhsh F, Henry S, Yong VW, Winston BW, Warren K, Power C. A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J Neurosci. 2004;24:1521–1529. doi: 10.1523/JNEUROSCI.4271-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen GQ, Chen YY, Wang XS, Wu SZ, Yang HM, Xu HQ, He JC, Wang XT, Chen JF, Zheng RY. Chronic caffeine treatment attenuates experimental autoimmune encephalomyelitis induced by guinea pig spinal cord homogenates in Wistar rats. Brain research. 2010;1309:116–125. doi: 10.1016/j.brainres.2009.10.054. [DOI] [PubMed] [Google Scholar]
- 34.Brown DA, Sawchenko PE. Time course and distribution of inflammatory and neurodegenerative events suggest structural bases for the pathogenesis of experimental autoimmune encephalomyelitis. The Journal of comparative neurology. 2007;502:236–260. doi: 10.1002/cne.21307. [DOI] [PubMed] [Google Scholar]
- 35.Engelhardt B, Wolburg-Buchholz K, Wolburg H. Involvement of the choroid plexus in central nervous system inflammation. Microscopy research and technique. 2001;52:112–129. doi: 10.1002/1097-0029(20010101)52:1<112::AID-JEMT13>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 36.Steffen BJ, Breier G, Butcher EC, Schulz M, Engelhardt B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. The American journal of pathology. 1996;148:1819–1838. [PMC free article] [PubMed] [Google Scholar]
- 37.Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. The Journal of experimental medicine. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bynoe MS, Evans JT, Viret C, Janeway CA., Jr Epicutaneous immunization with autoantigenic peptides induces T suppressor cells that prevent experimental allergic encephalomyelitis. Immunity. 2003;19:317–328. doi: 10.1016/s1074-7613(03)00239-5. [DOI] [PubMed] [Google Scholar]
- 40.Carman AJ, Mills JH, Krenz A, Kim DG, Bynoe MS. Adenosine receptor signaling modulates permeability of the blood-brain barrier. J Neurosci. 2011;31:13272–13280. doi: 10.1523/JNEUROSCI.3337-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spangrude GJ. Assessment of lymphocyte development in radiation bone marrow chimeras. In: Coligan John E, et al., editors. Current protocols in immunology. Unit 4. Chapter 4. 2008. p. 6. [DOI] [PubMed] [Google Scholar]
- 42.Spangrude GJ, Brooks DM, Tumas DB. Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood. 1995;85:1006–1016. [PubMed] [Google Scholar]
- 43.Staples JE, Fiore NC, Frazier DE, Jr, Gasiewicz TA, Silverstone AE. Overexpression of the anti-apoptotic oncogene, bcl-2, in the thymus does not preventthymic atrophy induced by estradiol or 2,3,7, 8-tetrachlorodibenzo-p-dioxin. Toxicology and applied pharmacology. 1998;151:200–210. doi: 10.1006/taap.1998.8446. [DOI] [PubMed] [Google Scholar]
- 44.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clinical and experimental immunology. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhasin M, Wu M, Tsirka SE. Modulation of microglial/macrophage activation by macrophage inhibitory factor (TKP) or tuftsin (TKPR) attenuates the disease course of experimental autoimmune encephalomyelitis. BMC immunology. 2007;8:10. doi: 10.1186/1471-2172-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lassmann H, Schmied M, Vass K, Hickey WF. Bone marrow derived elements and resident microglia in brain inflammation. Glia. 1993;7:19–24. doi: 10.1002/glia.440070106. [DOI] [PubMed] [Google Scholar]
- 47.Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 48.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiological reviews. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 49.El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:189–197. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. 1997;90:1600–1610. [PubMed] [Google Scholar]
- 51.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T Cells: Mechanisms of Differentiation and Function. Annual review of immunology. 2012 doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar V, Stellrecht K, Sercarz E. Inactivation of T cell receptor peptide-specific CD4 regulatory T cells induces chronic experimental autoimmune encephalomyelitis (EAE) The Journal of experimental medicine. 1996;184:1609–1617. doi: 10.1084/jem.184.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–259. doi: 10.1182/blood-2007-03-081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moreau JL, Huber G. Central adenosine A(2A) receptors: an overview. Brain Res Brain Res Rev. 1999;31:65–82. doi: 10.1016/s0165-0173(99)00059-4. [DOI] [PubMed] [Google Scholar]
- 55.Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC. Tissue distribution of adenosine receptor mRNAs in the rat. British journal ofpharmacology. 1996;118:1461–1468. doi: 10.1111/j.1476-5381.1996.tb15561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elliott JI, Douek DC, Altmann DM. Mice lacking alpha beta + T cells are resistant to the induction of experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 1996;70:139–144. doi: 10.1016/s0165-5728(96)00106-3. [DOI] [PubMed] [Google Scholar]
- 57.Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol. 2007;179:1254–1263. doi: 10.4049/jimmunol.179.2.1254. [DOI] [PubMed] [Google Scholar]
- 58.Li Y, Oskouian RJ, Day YJ, Rieger JM, Liu L, Kern JA, Linden J. Mouse spinal cord compression injury is reduced by either activation of the adenosine A2A receptor on bone marrow-derived cells or deletion of the A2A receptor on non-bone marrow-derived cells. Neuroscience. 2006;141:2029–2039. doi: 10.1016/j.neuroscience.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 59.Dai SS, Zhou YG. Adenosine 2A receptor: a crucial neuromodulator with bidirectional effect in neuroinflammation and brain injury. Reviews in the neurosciences. 2011;22:231–239. doi: 10.1515/RNS.2011.020. [DOI] [PubMed] [Google Scholar]
- 60.Rivo J, Zeira E, Galun E, Einav S, Linden J, Matot I. Attenuation of reperfusion lung injury and apoptosis by A2A adenosine receptor activation is associated with modulation of Bcl-2 and Bax expression and activation of extracellular signal-regulated kinases. Shock (Augusta, Ga. 2007;27:266–273. doi: 10.1097/01.shk.0000235137.13152.44. [DOI] [PubMed] [Google Scholar]
- 61.Hasko G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, Marton A, Szabo C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. Faseb J. 2000;14:2065–2074. doi: 10.1096/fj.99-0508com. [DOI] [PubMed] [Google Scholar]
- 62.Kreckler LM, Gizewski E, Wan TC, Auchampach JA. Adenosine suppresses lipopolysaccharide-induced tumor necrosis factor-alpha production by murine macrophages through a protein kinase A- and exchange protein activated by cAMP-independent signaling pathway. The Journal of pharmacology and experimental therapeutics. 2009;331:1051–1061. doi: 10.1124/jpet.109.157651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meininger CJ, Schelling ME, Granger HJ. Adenosine and hypoxia stimulate proliferation and migration of endothelial cells. The American journal of physiology. 1988;255:H554–562. doi: 10.1152/ajpheart.1988.255.3.H554. [DOI] [PubMed] [Google Scholar]
- 64.Lutty GA, Mathews MK, Merges C, McLeod DS. Adenosine stimulates canine retinal microvascular endothelial cell migration and tube formation. Current eye research. 1998;17:594–607. [PubMed] [Google Scholar]
- 65.Othman T, Yan H, Rivkees SA. Oligodendrocytes express functional A1 adenosine receptors that stimulate cellular migration. Glia. 2003;44:166–172. doi: 10.1002/glia.10281. [DOI] [PubMed] [Google Scholar]
- 66.Knight D, Zheng X, Rocchini C, Jacobson M, Bai T, Walker B. Adenosine A3 receptor stimulation inhibits migration of human eosinophils. Journal of leukocyte biology. 1997;62:465–468. doi: 10.1002/jlb.62.4.465. [DOI] [PubMed] [Google Scholar]
- 67.Duffy SM, Cruse G, Brightling CE, Bradding P. Adenosine closes the K+ channel KCa3.1 in human lung mast cells and inhibits their migration via the adenosine A2A receptor. European journal of immunology. 2007;37:1653–1662. doi: 10.1002/eji.200637024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi IY, Lee JC, Ju C, Hwang S, Cho GS, Lee HW, Choi WJ, Jeong LS, Kim WK. A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. The American journal of pathology. 2011;179:2042–2052. doi: 10.1016/j.ajpath.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Save S, Mohlin C, Vumma R, Persson K. Activation of adenosine A2A receptors inhibits neutrophil transuroepithelial migration. Infection and immunity. 2011;79:3431–3437. doi: 10.1128/IAI.05005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacology & therapeutics. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 71.Grenz A, Homann D, Eltzschig HK. Extracellular adenosine: a safety signal that dampens hypoxia-induced inflammation during ischemia. Antioxidants & redox signaling. 2011;15:2221–2234. doi: 10.1089/ars.2010.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal”. Science signaling. 2009;2:pe6. doi: 10.1126/scisignal.256pe6. [DOI] [PubMed] [Google Scholar]
- 73.Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunological reviews. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. The New England journal of medicine. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 76.Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008;111:5571–5580. doi: 10.1182/blood-2007-11-126763. [DOI] [PubMed] [Google Scholar]
- 77.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nature immunology. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 78.Moon C, Ahn M, Jeong C, Kim H, Shin T. Immunohistochemical study of netrin-1 in the spinal cord with rat experimental autoimmune encephalomyelitis. Immunological investigations. 2011;40:160–171. doi: 10.3109/08820139.2010.525570. [DOI] [PubMed] [Google Scholar]
- 79.Moreno B, Fernandez-Diez B, Di Penta A, Villoslada P. Preclinical studies of methylthioadenosine for the treatment of multiple sclerosis. Multiple sclerosis (Houndmills, Basingstoke, England) 2010;16:1102–1108. doi: 10.1177/1352458510375968. [DOI] [PubMed] [Google Scholar]
- 80.Moreno B, Hevia H, Santamaria M, Sepulcre J, Munoz J, Garcia-Trevijano ER, Berasain C, Corrales FJ, Avila MA, Villoslada P. Methylthioadenosine reverses brain autoimmune disease. Annals of neurology. 2006;60:323–334. doi: 10.1002/ana.20895. [DOI] [PubMed] [Google Scholar]
- 81.Munshi R, Clanachan AS, Baer HP. 5′-Deoxy-5′-methylthioadenosine: a nucleoside which differentiates between adenosine receptor types. Biochemical pharmacology. 1988;37:2085–2089. doi: 10.1016/0006-2952(88)90560-6. [DOI] [PubMed] [Google Scholar]