

Abstract
While nanoparticles maximize the amount of chemotherapeutic drug in tumors relative to normal tissues, nanoparticle-based drugs are not accessible to the majority of cancer cells because nanoparticles display patchy, near-perivascular accumulation in tumors. To overcome the limitations of current drugs in their molecular or nanoparticle form, we developed a nanoparticle based on multi-component nanochains to deliver drug to the majority of cancer cells throughout a tumor while reducing off-target delivery. The nanoparticle is composed of three magnetic nanospheres and one doxorubicin-loaded liposome assembled in a 100-nm-long chain. These nanoparticles display prolonged blood circulation and significant intratumoral deposition in tumor models in rodents. Furthermore, the magnetic particles of the chains serve as a mechanical transducer to transfer radiofrequency energy to the drug-loaded liposome. The defects on the liposomal walls trigger the release of free drug capable of spreading throughout the entire tumor, which results in a wide-spread anticancer effect.
Keywords: Multi-component nanoparticle, magnetic nanoparticles, radiofrequency-triggered drug release, doxorubicin, nanochain, chemotherapy
Although potent chemotherapeutic drugs are available to oncologists, the clinical utility of these agents is limited by their toxicity, leading to use of suboptimal doses that are often lower than necessary for cancer eradication in vivo. For example, doxorubicin (DOX) exemplifies a potent chemotherapeutic agent with highly problematic systemic toxicity.1 While chemotherapeutic agents are distributed within tumors and healthy tissues in a non-specific manner, nanoparticle-based drugs have been developed to exploit a feature of the tumor microenvironment, the so-called ‘Enhanced Permeability and Retention’ (EPR) effect.2 Nanoparticles function as carriers for selective entry into tumors through their altered vasculature while reducing off-target delivery.3
While nanoparticles maximize the amount of drug in the tumor relative to normal tissues,3 the drug is not available to the majority of cancer cells as nanoparticles display patchy, near-perivascular accumulation in tumors.4-6 Nanoparticles en route to their target face numerous biobarriers created by the abnormal tumor physiology including erratic blood flow, low convective transport due to high interstitial pressure, abnormal extracellular matrix (ECM), large diffusion distances in some tumor regions, and binding to tumor and stroma cells and the ECM. These biobarriers limit the extravasation and interstitial transport of nanoparticles into tumors.5 Nanoparticles typically display a patchy distribution within the well-vascularized regions of tumors, while they are absent in the avascular regions of tumors.7-9 Even after successful extravasation, nanoparticles remain proximal to the vessel wall.4 Due to negligible convection in the tumor interstitium, nanoscale agents rely solely on passive diffusion to be transported through the ECM. Unfortunately, diffusion of nanoparticles is very inefficient as it is much slower than diffusion of small molecules. Additionally, tissue penetration of nanoparticles is further limited by cellular obstacles and ECM components.10-12
While free drug in its molecular form quickly spreads in the tumor interstitium,13-15 nanoparticles release their content slowly, once they deposit at the target site.15, 16 This slow release generates a low temporal and spatial concentration gradient of the drug, resulting in non-cytotoxic levels of the drug distal from the particle.15 While the slow release of drug from nanoparticles does not favor cytotoxic effects, it improves the drug's safety profile during the particle's circulation in the blood.3, 17, 18
Here, we show that we can integrate the advantages of the molecular and nanoparticle mode of chemotherapeutics into a single agent based on the nanochain technology and show how this combined mode can be used to significantly improve the outcome of chemotherapy. The nanoparticle is composed of three iron oxide (IO) nanospheres and one DOX-loaded liposome assembled together in a 100-nm-long chain (abbreviated as DOX-NC). Animal studies indicated that the DOX-NC nanoparticle displayed prolonged blood residence time and enhanced deposition into tumors. Furthermore, animals bearing mammary cancer xenografts showed an improved response, when the DOX-NC treatment was followed by the application of a radiofrequency (RF) field as measured by decreased tumor growth and prolonged survival. We show that the IO tail of the DOX-NC particle composed of magnetic nanoparticles can serve as a mechanical transducer to transfer RF energy to the liposome membrane. Thus, once DOX-NC has extravasated to the tumor site, RF-induced disruption of the liposomal membrane integrity liberates drug molecules into their free form that can efficiently diffuse into the tumor interstitium (Fig. 1a). This results in a wide-spread anticancer effect as confirmed with histological analysis of apoptosis.
Fig. 1.
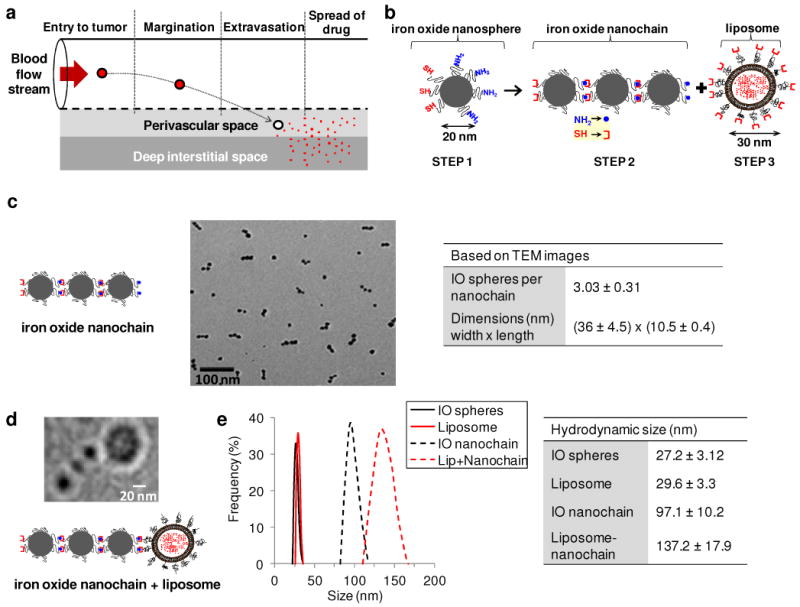
Characterization of the DOX-NC nanoparticle. (a) Illustration of the required steps for the successful delivery of nanoparticle-based drug to tumors. (b) Diagram of the DOX-NC nanoparticle and its constituent components including a nanochain composed of three iron oxide (IO) spheres and one liposome. (c) TEM image of magnetic nanochains composed of three IO spheres. The table summarizes the main characteristics of the magnetic nanochains obtained from visual analysis of TEM images (minimum count was 200 particles; data presented as mean ± s.d). (d) TEM image of a nanochain particle composed of three IO spheres and one DOX-loaded liposome. (e) Size distribution of the parent nanoparticles and DOX-loaded nanochains obtained by DLS measurements (data presented as mean ± s.d.)
Results
Fabrication and characterization of multi-component nanochains
The nanochain technology19 is based on a two-step approach to fabricate nanochains using solid-phase chemistry. In the first step, amine-functionalized IO nanospheres were attached on a solid support via a crosslinker containing a disulfide bridge. Liberation of the nanosphere using thiolytic cleavage created thiols on the portion of the particle's surface that interacted with the solid support resulting in a particle with two faces, one displaying only amines and the other only thiols. Therefore, we were able to topologically control the conversion of amines on the surface of the IO nanospheres into thiols, resulting in a particle with asymmetric surface chemistry (ASC). In the second step, employing solid-phase chemistry and step-by-step addition of particles, the two unique faces on the same IO nanosphere served as fittings to assemble them into IO nanochains (Fig. 1b).
The magnetic nanochains were analyzed via visual inspection of multiple TEM images. As shown in Figure 1c, the magnetic nanochains were synthesized in a highly controlled manner. Most of the nanochains are linear and consist of 3 IO spheres. To evaluate the robustness of the nanochain synthesis, the number of IO nanospheres per nanochain was measured in multiple TEM images (minimum count was 200 particles). While 16% of the total particles in the suspension were the parent (unbound) IO spheres, the majority of the particles (73%) comprised of nanochains with 3 IO spheres (8 and 4% were nanochains with 2 or 4 IO spheres, respectively). Importantly, our methodology offers exceptional flexibility in synthesizing nanochains consisting of various types of constituent members with different functions. Specifically, in the last step of synthesis, we attached one drug-loaded liposome per magnetic nanochain (Fig. 1d). The final nanoparticle consisted of three IO spheres and one DOX-loaded liposome with the overall geometrical dimensions of the DOX-NC particle being about 100 × 30 nm (length × width), which was essentially the summation of the lengths of its constituent IO spheres and liposome. A design criterion was to use a liposome with a size that is comparable to that of the IO spheres of the magnetic nanochain. We therefore used DOX-loaded liposomes with a hydrodynamic diameter of about 30 nm, which were fabricated using a combination of extrusion and sonication. As shown in Fig. 1e, the hydrodynamic size of the final DOX-NC particle and each component separately, as measured by dynamic light scattering (DLS), verified the TEM findings. It should be noted that DLS measured the effective hydrodynamic diameter based on the diffusion of the particles. Due to the high intraliposomal space available for drug encapsulation and the efficient remote loading technique,20 the DOX cargo of DOX-NC was high (i.e. 6.8 × 10−5 ng/DOX-NC particle).
In vitro on-command triggered drug release using RF
Through their interaction with magnetic fields, the magnetic component (IO spheres) of the DOX-NC particle efficiently converts magnetic energy to mechanical energy, which is dependent on the strength and frequency of the magnetic field, as well as the configuration of the IO spheres in the nanochain.21 Thus, drug release can be remotely triggered due to defects of the liposomal membrane caused by the oscillation of the magnetic ‘tail’ of the DOX-NC particle in the presence of an RF field (Fig. 2a). Magnetic field generation was accomplished using an RF source and a solenoidal coil that was size-matched to the sample, which was placed inside the coil. Fig. 2b shows that the release of DOX can be triggered in a controlled manner under the RF field (10 kHz frequency, 1-50 W dissipated power) at a very low concentration of DOX-NC particles expected to deposit in tumor tissues during in vivo applications. Notably, the release rate could be modulated by adjusting the operating parameters of the RF field. We should emphasize that no temperature increase occurred in the DOX-NC suspension under the ‘mild’ RF field (experiment was performed at room temperature).
Fig. 2.
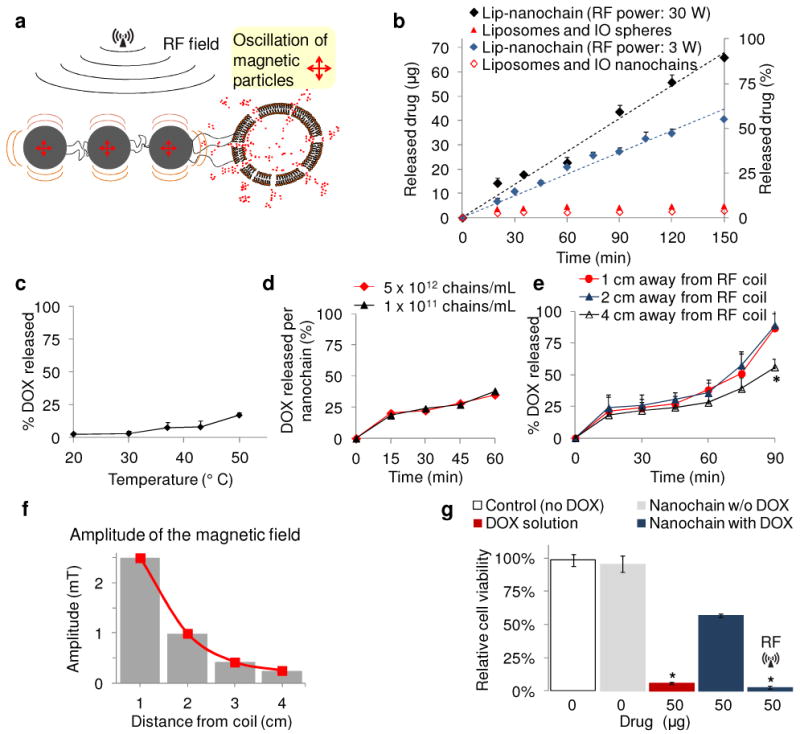
In vitro evaluation of the RF-triggered release profile of DOX from DOX-NC particles. (a) Illustration of the defects on the liposome caused by ‘vibration’ of the IO spheres under an RF field. (b) Triggered release from DOX-NC particles using an RF field at 10 kHz and different energy outputs (the sample was located 1 cm away from the RF coil). The samples were exposed to the RF field for the entire duration of the experiment. Besides DOX-NC particles, the RF field (30 W) was applied to mixtures of liposomes with IO nanospheres or IO nanochains at a ratio of 1:3 (liposome: IO spheres). (c) Effect of temperature on the drug release from DOX-NC particles (incubation time was 60 min). (d) Drug release from DOX-NC particles at different particle concentration under an RF field at 10 kHz/30W (the sample was located 1 cm away from the RF coil). (e) Drug release from DOX-NC particles at different distance from the RF source (RF field: 10 kHz/30W). (f) Amplitude of the magnetic field at different distances from the RF source (RF field: 10 kHz/30W). (g) Cytotoxicity of DOX-NC (with or without RF) on 13762 MAT B III cells. Control treatments included black nanochains, free DOX, and liposomal DOX. The two data points marked with asterisks are statistically different compared to the other conditions (P<0.01).
The DOX release profile from DOX-NC particles in the absence of RF is shown in Fig. 2c. To investigate the effect of temperature on the release rate, the DOX-NC suspension was incubated at different temperatures for 60 min. A 5% release of the DOX cargo was released at 37 °C, which is consistent with the behavior of the parent liposome. The liposome component of the DOX-NC particle is composed of the phospholipid DPPC, which does not result in thermosensitive liposomes. While DPPC membranes have a transition temperature of 41 °C, the addition of cholesterol to the liposomal membrane has a significant stabilizing effect.22, 23 Due to the increased transition temperature of the DPPC/cholesterol membrane (Tm>50 °C)24 and the stable entrapment of the precipitated DOX in the liposome,17 a minor increase in release of DOX is expected at elevated temperature due to increase of DOX solubility.23 Thus, the time course of the release profiles showed an initial burst in the first 5 min followed by a plateau (data not shown). A temperature of 50 °C was required to cause a significant release in 60 min, which is significantly higher than the observed temperature of the release experiment under the RF field.
To further investigate whether mechanical vibration is the release mechanism, we measured the release from suspensions of dramatically different concentrations of DOX-NC under the same RF field. As shown in Fig. 2d, the same release rate per DOX-NC particle is achieved from low concentrations as well as very high concentration of the particles. So far the in vitro studies excluded bulk heating of the DOX-NC suspensions under the RF field. However, significant local heating can be generated around nanoparticles.25, 26 To explore the possibility of local heating, a fluorophore linked on the surface of the DOX-NC particle was used as a thermometer based on an established method.26 As shown in Fig. S2 in the Supporting Information, there is no significant heat generation around the DOX-NC particles. Thus, contrary to heat-induced drug release, we can conclude that the triggered release mechanism of DOX-NC is concentration-independent and is probably based on mechanical forces that occur on the single particle scale. We then investigated the dependence of the release rate on the distance of the DOX-NC suspension from the RF source. Fig. 2e shows that the release rate is significantly lower at 4 cm away from the RF coil after a 90 min exposure (P<0.01), which is consistent with the relationship of the magnetic field strength to distance (Fig. 2f). Furthermore, Fig. 2g shows the cytotoxic effect on mammary adenocarcinoma cells (13762 MAT BIII) of released DOX from DOX-NC, non-released DOX-NC, empty nanochains (no DOX), and free DOX. The empty nanochain had no effect on cancer cells. While DOX-NC had moderate cytotoxicity, the RF-triggered release of DOX from DOX-NC had significantly higher cytotoxic effects (P<0.01) due to release of free DOX. Notably, the released drug is 100% bioavailable.
Blood circulation and tumor deposition of nanochains
Plasma clearance studies were performed on animals without tumors in order to evaluate only the effects of phagocytic clearance. Following an intravenous injection of DOX-NC at a dose of 0.5 mg DOX per kg body weight, Fig. 3a shows that the 100-nm-long DOX-NC particle exhibited prolonged blood residence time (blood t1/2∼26 hours). For comparisons, we used a long-circulating 100-nm liposome (blood t1/2∼18 hours) due to its long and successful history of encapsulating DOX for clinical use. In addition, the concentration profiles of DOX-NC in the blood measuring either the DOX levels (liposome component) or the fluorescently tagged IO spheres (IO tail component) matched suggesting that the structure of the DOX-NC particle remains intact during circulation in blood.
Fig. 3.

Blood circulation and organ distribution of the DOX-NC particles in rats. (a) Plasma clearance of DOX-loaded liposomes (100 nm in diameter) and DOX-NC in rats at a dose of 0.5 mg/kg DOX (n=5). Besides DOX, fluorescence spectroscopy was used to measure Alexa 488 on the iron oxide particles (*P<0.05). (b) Organ and tumor distribution 24 h after administration of the DOX-loaded liposomes and DOX-NC at a dose of 0.5 mg DOX/kg in the rat 13762 MAT B III tumor model (n=6; *P<0.05).
Using a dose of 0.5 mg/kg DOX, we evaluated the organ and tumor distribution of DOX-NC in the 13762 MAT B III tumor model, which is a rat-syngenic aggressive mammary adenocarcinoma. The animals were euthanized 24 hours after intravenous administration of DOX-NC or 100-nm liposomes, and the organs and tumors were extracted and analyzed for DOX content. The accumulation of DOX-NC in the heart, lungs and kidney was about 5% or less of the injected dose (Fig. 3b), which was comparable to the behavior of the 100-nm liposomes. More importantly, the uptake of DOX-NC by the liver was significantly lower than that of liposomes (P<0.01). Taking under consideration that nanoparticles are primarily cleared by the reticuloendothelial system, the low uptake of DOX-NC by the liver correlates to its prolonged blood residence. DOX-NC outperformed the 100-nm liposomes as indicated by their higher intratumoral accumulation (Fig. 3b, P<0.01). This is likely due to the fact that nanoparticle extravasation into tumors is directly proportional to their blood residence time.27
Therapeutic effectiveness in a rat breast tumor model
The MAT B III tumor-bearing animals were intravenously injected with DOX-NC at a low dose (0.5 mg DOX per kg body weight). Typical dosage of liposomal DOX is 10-20 times higher and ranges from 5-10 mg/kg in animal studies.28, 29 Since a significant amount of DOX-NC is deposited into tumors by 24 hours after injection, the RF field was utilized at this time point. As shown in Fig. 4a, 24 hours after injection, the RF field (10 kHz/3-5 W) was applied for 60 min using the RF coil positioned 1 cm from the animal and oriented such that the magnetic field was directed toward the tumor. Iron staining of histological sections using Prussian blue showed that DOX-NC particles were well-distributed within the tumor interstitium at 24 hours post-injection (Fig. 4b). In contrast, no Prussian blue staining was observed in tumors treated with liposomal DOX (images not shown). Without the application of RF, direct fluorescence (red) imaging of the histological sections failed to detect free DOX (images not shown). Hence, the drug is still incorporated into the nanochain with the fluorescence signal of intra-liposomal DOX being quenched. Following the application of the RF field, free DOX was widely spread in the tumor extravascular space and localized in the nuclei of the tumor cells (Fig. 4c).
Fig. 4.
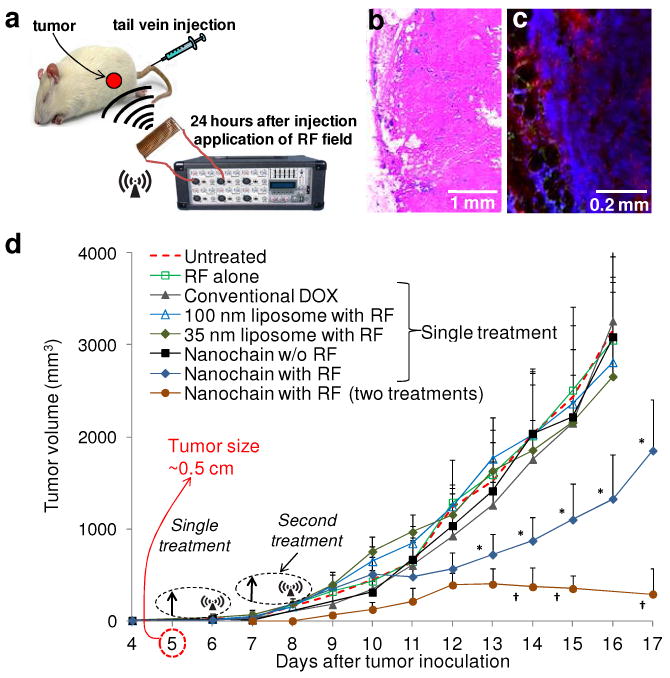
In vivo treatment of breast tumor-bearing rats using DOX-NC particles. (a) Schematic of the therapeutic protocol. (b) Histological evaluation of the distribution of systemically administered DOX-NC particles (blue: Prussian blue stain) in a tumor. (c) Application of an RF field released DOX molecules (red) that localized in the nuclei of cancer cells (blue: DAPI). (d) Measurement of the tumor growth of 13763 MAT B III tumors in rats after systemic administration of DOX-NC at a dose of 0.5 mg/kg DOX (arrow; day 5) followed by application of the RF field (day 6). Control treatments included saline (untreated), RF alone, free DOX, 100-nm liposomal DOX (with RF), 35-nm liposomal DOX (with RF) and DOX-NC (without RF). Another group of animals received a second injection of DOX-NC (arrow; days 7) followed by RF application (day 8). Data points marked with asterisks are statistically significant relative to all the other single-treated groups. Data points marked with crosses are statistically significant relative to all groups (n=6; * and † P<0.05).
After we recognized that DOX-NC can efficiently deposit into tumors, the tumor response to DOX-NC was evaluated by quantitatively following the tumor size for several days after injection. Based on the fact that DOX-NC displayed significant accumulation in the tumor at 24 hours post-injection, the application of the RF field 24 hours after injection of DOX-NC significantly suppressed tumor growth as shown in Fig. 4d (P<0.01). As expected, application of the RF alone had no effect on the tumor growth rate. Animals treated with the same low dose of DOX of clinically used free DOX, 35-nm or 100-nm liposomal DOX followed by RF failed to produce any therapeutic benefits. While a single treatment of liposomal DOX has generated therapeutic benefits in animal tumor models, the administrated dose was 10-20 times higher than the dose we used.28-32 Even though DOX-NC achieves higher accumulation in the tumor than the 100-nm liposomal DOX (as shown in Fig. 3b), there was no substantial effect on the tumor growth rate (without the application of RF). In conjunction to the slow release of DOX from DOX-NC, we speculate that this is related to the very low dose of DOX. Based on the same dose per treatment, we also employed a two-cycle treatment using DOX-NC (at days 5 and 7 after tumor inoculation) followed by RF application (at days 6 and 8). As shown in Fig. 4d, the multiple treatments accomplished greater tumor shrinkage than a single treatment.
Furthermore, the therapeutic efficacy of the DOX-NC treatment followed by RF was determined by comparing the survival times of treated animals to untreated animals (Fig. S1 in Supporting Information). The group treated with DOX-NC followed by RF exhibited a statistically significant increase in survival time (25.1±3.8 days) compared to the untreated group and the other groups that received single treatment (survival ∼15.2±2.4 days). Notably, the two-cycle treatment using DOX-NC followed by RF prolonged the survival to a greater extent than the single treatment (46±8.1 days). These in vivo studies using systemic administration of DOX-NC at a dose of 0.5 mg/kg DOX demonstrate that 1) nanochains effectively extravasate into tumors and 2) RF application to DOX-NC-treated tumors enhanced the therapeutic outcome.
Histological evaluation of the anticancer efficacy
In addition to the improved therapeutic outcome as measured by lower tumor growth and prolonged survival, we sought to verify that the anticancer activity of DOX-NC is based on improved distribution of free drug after application of the RF field. Since DOX is a weak fluorophore, direct fluorescence imaging of DOX provided a qualitative demonstration of the RF-triggered drug release. We then performed histological analysis of apoptosis using the more sensitive TUNEL assay to quantitatively evaluate the extent and topology of the apoptotic cells in response to DOX-NC. It should be noted that red color indicates apoptotic cells in Fig. 5 and not molecules of DOX. Following a single injection of a DOX treatment, animals were euthanized 24 hours post-injection and tumors were excised. Visual inspection of histological images of tumors treated with free DOX at the regular dose (i.e. 5 mg/kg) showed a substantial number of apoptotic cells (Fig. 5a). Tumors treated with 100-nm liposomes (Fig. 5b) or DOX-NC (Fig. 5c) at the low dose of 0.5 mg/kg DOX displayed a small number of apoptotic cells primarily in the well-vascularized rim. Importantly, negligible apoptosis was observed after systemic administration of empty nanochains (no DOX cargo) followed by RF (Fig. 5d), suggesting that limited or no RF heating of the magnetic nanospheres takes place. However, RF application on DOX-NC-treated animals resulted in massive apoptosis in both the well-vascularized rim and the less vascularized inner core (Fig. 5e).
Fig. 5.
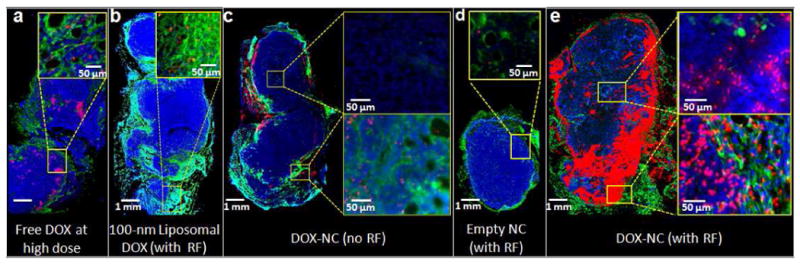
Histological evaluation of the apoptotic effect of DOX-NC in the rat MAT B III model. (a) Fluorescence image of a histological section of a tumor 48 h after IV injection of free DOX at 5 mg/kg. The specific endothelial antigen CD31 was stained (green). Nuclei (blue) were stained with DAPI. Apoptotic cell nuclei were stained with TUNEL (red). (b) No significant apoptosis was observed in a tumor 48 h after systemic administration of 100-nm liposomal DOX at 0.5 mg/kg (RF was applied 24 h after injection). (c) Few apoptotic cells were found in a tumor 48 h after systemic administration of DOX-NC at 0.5 mg/kg. (d) Negligible apoptosis was found in a tumor 48 h after systemic administration of an empty nanochain (RF was applied 24 h after injection). (e) A significant number of apoptotic cells was found in a tumor 48 h after systemic administration of liposomal DOX at 0.5 mg/kg (RF was applied 24 h after injection).
To obtain a quantitative evaluation, the total number of cells was counted based on the nuclear stain (DAPI) in multiple histological sections (minimum 20) per tumor, whereas apoptotic cells were quantified based on TUNEL-stained nuclei. The percent of apoptotic cells relative to the total number of cancer cells was used as a measure of the anticancer efficacy. Using the CD31 staining of the microvasculature, we drew ROIs to distinguish apoptosis in the rim from the core of the tumor. Fig. 6a shows that 2.3 and 13.8 % of the cancer cells were apoptotic in the case of tumors treated with the 100-nm liposome and DOX-NC, respectively. In good agreement with the previous in vivo studies (e.g. tumor distribution and survival), DOX-NC exhibited greater cytotoxicity than the 100-nm liposomes. As expected for both the liposomes and DOX-NC, the anticancer effect was more profound in the rim of the tumors, where about 8 times more apoptotic cells were observed compared to the less vascularized core (Fig. 6b). On the other hand, the combination of DOX-NC and RF resulted in 34.2% of the cancer cells being apoptotic, which was a significant increase of apoptosis compared to any other treatment (P<0.01). This is also in good agreement with our previous observation that released DOX was found in the cancer cell nuclei. In fact, previous studies have shown that nuclear DNA functions as a sink for DOX.13-15 More importantly, the apoptotic results were equally elevated in the core and the rim of the tumors. Application of RF on DOX-NC-treated tumors exhibited 5 and 18 times greater apoptosis in the core of tumors compared to DOX-NC (no RF) and liposomes (with RF), respectively. Taking under consideration this rat breast tumor model is highly aggressive, having a core with little or no vascularization, the apoptotic index indicates that RF liberates bioavailable drug into non-vascularized regions resulting in a wide-spread anticancer effect throughout the entire tumor.
Fig. 6.
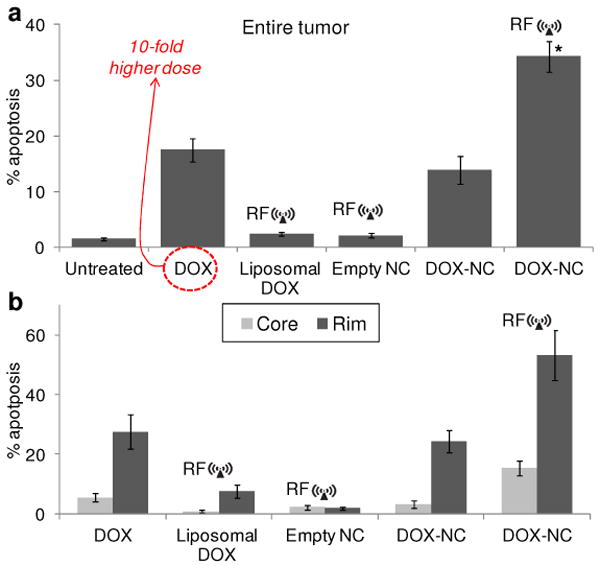
Quantitative histological evaluation of apoptosis in the rat MAT B III model. (a) A quantitative analysis of the fluorescence images was performed by comparing the total number of cancer and apoptotic cells of an entire tumor as measured in at least 20 histological sections per tumor (about 10,000 cells per section). The apoptotic effect on tumors treated with DOX-NC followed by RF was compared to the other DOX-based treatments (n=3 rats per group; * P<0.01). (b) Regional apoptosis in the tumor was measured based on the degree of vascularization. Using the endothelial cells staining (CD31), the well-vascularized rim of the tumor was distinguished from its core.
Since the intratumoral penetration of therapeutic molecules and nanoparticles is highly variable between different types of cancer and different species, we tested DOX-NC in an orthotopic 4T1 mammary tumor model in mice. As shown in Fig.7a and b, tumors treated with the 35-nm liposomal DOX exhibited more apoptotic cells than the 100-nm liposomal DOX. Similarly to the MAT B III model, the 4T1 tumors treated with DOX-NC (Fig. 7c) displayed higher levels of apoptosis than both liposomal treatments. As shown in Fig. 7d, tumors treated with DOX-NC followed by RF substantially increased the number of apoptotic cells. Fig. 7e summarizes the quantification of the apoptotic index of the various treatments, which is highly consistent with the data obtained from the MAT B III model. For example, the percent of apoptotic cells was elevated ∼2 fold in tumors treated with the combination of DOX-NC and RF (37% apoptotic cells in the tumor mass; P<0.01) compared to DOX-NC without RF (20%). We should note the 4T1 tumors do not present a clear hypervascularized rim and a less vascularized/avascular core resulting in a more consistent rate of apoptosis throughout the tumor.
Fig. 7.
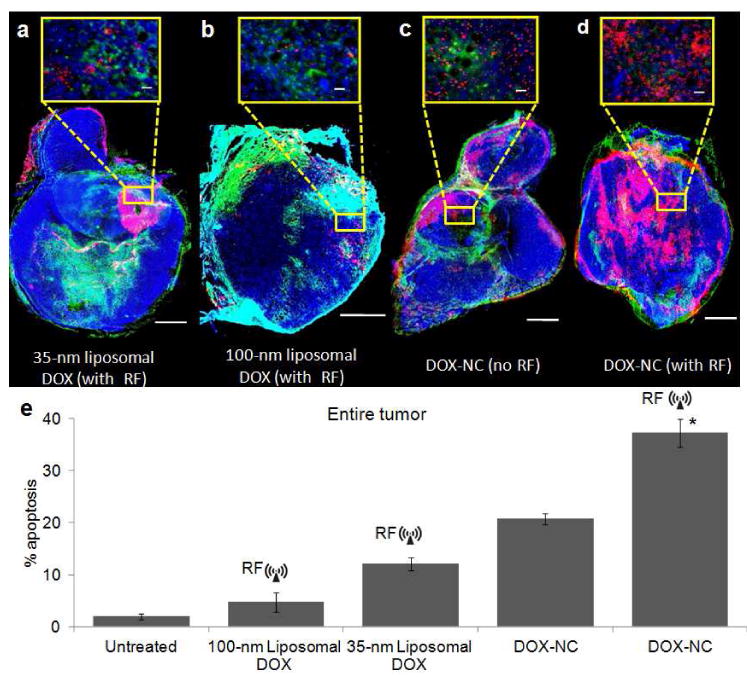
Histological evaluation of apoptosis in the mouse 4T1 model. (a) Fluorescence image of a histological section of a tumor 48 h after IV injection of 35-nm liposomal DOX at 0.5 mg/kg (CD31: green, DAPI: blue, TUNEL: red). RF was applied 24 h after injection. The scale bar is 1 mm (scale bar of the inset is 50 μm) (b) No significant apoptosis was observed in a tumor 48 h after systemic administration of 100-nm liposomal DOX at 0.5 mg/kg (RF was applied 24 h after injection). (c) More apoptotic cells were found in a tumor 48 h after systemic administration of DOX-NC at 0.5 mg/kg. (d) A significant number of apoptotic cells was found in a tumor 48 h after systemic administration of DOX-NC at 0.5 mg/kg followed by RF application 24 h after injection. (e) A quantitative analysis of apoptosis was performed by comparing the total number of cancer and apoptotic cells of an entire tumor (minimum 20 histological sections per tumor; n=3 mice per group; * P<0.01).
The histological studies evaluated the anticancer efficacy of DOX-NC using a single administration of the agent at a low dose of 0.5 mg/kg DOX. Overall, the apoptotic index in both animal models indicates that the RF-triggered release from DOX-NC substantially improved the interstitial transport and spatial distribution of the drug compared to the control treatments.
Discussion
We tested the therapeutic efficacy of the DOX-NC nanoparticles in two tumor models of triple-negative breast cancer. Triple-negative breast tumors lack estrogen, progesterone, and HER2 receptors and exhibit a highly aggressive phenotype. Since neither hormone therapy nor drugs that target HER2 are likely to be effective against these tumors, the standard treatment for triple-negative breast cancer is chemotherapy comprising predominantly of anthracyclines (e.g. DOX) in its free33-35 or nanoparticle forms.33, 36 Our study utilized two models of triple negative tumors: the rat syngenic MAT B III and mouse syngenic 4T1 mammary adenocarcinoma models. Both transplantation models form highly aggressive, invasive and metastatic tumors in the context of an intact immune system, and in the 4T1 model, tumors were evaluated in the context of the mammary microenvironment. These well-established breast tumor models have been widely used to study tumor angiogenesis,37 primary tumor growth,7, 9, 32 invasion, and metastases.38 Importantly, we showed that treatment of these highly aggressive tumors with the multi-component DOX-NC particles followed by an RF field facilitated rapid release of free DOX molecules capable of spreading to deep interstitial and avascular regions, which are otherwise inaccessible.
Various triggered release mechanisms have been applied in the design of nanoparticle systems to address the drug delivery limitations to tumors. Such systems include temperature or pH sensitive liposomes or polymeric nanoparticles.23, 39-42 However, the release mechanism of these particles relies on changes in environmental factors (e.g. pH, temperature), which may be non-uniform throughout the tumor volume. For example, pH sensitive nanoparticles require either mildly acidic tissue or intracellular uptake and exposure to the acidic conditions of endosomes.39 Since nanoparticles deposit in the near-perivascular space, these regions typically exhibit near-physiological pH especially in the well-vascularized rim of tumors. On the other hand, release of drug from nanoparticles due to intracellular uptake occurs in the perivascular regions involving a small number of cancer cells relative to the bulk of the tumor. Alternatively, thermal energy can be used as a stimulus for drug release. Temperature sensitive nanoparticles typically require a metal nanoparticle (e.g. gold or iron) to mediate heat generation through the absorption of an external field (e.g. NIR for gold, RF for iron) to accomplish a temperature elevation to about 42-45 °C in surrounding tissue.40, 43 However, to overcome heat dissipation, these strategies require the spatial concentration of the metal particles to be sufficiently high to elevate the tissue temperature, which is feasible in vascularized tumors. However, due to insufficient concentration of the metal particles, hyperthermia cannot be achieved in avascular (or not well-vascularized) tumors or tumor regions, resulting in failure to trigger the drug release from nanoparticles present in these locations. In fact, these are precisely the tumor regions that are difficult to treat, since they are inaccessible by current drug delivery systems. In the case of DOX-NC however, the release mechanism is not based on environmental factors, because mechanical forces induced by an RF field can trigger the release from a single DOX-NC particle. In this study, we demonstrate that drug release can take place even in these ‘difficult to treat’, less vascularized locations, where the concentration of nanoparticles is typically low. Of course, there are reasons other than tumor dosing and drug transport in the tumor interstitium that govern tumor responsiveness to chemotherapy. Effectiveness also depends on cellular uptake and potential multidrug resistance (MDR) mechanisms (e.g. drug efflux proteins, drug entrapment in cytoplasmic vesicles). However, by improving the temporal and spatial intratumoral accumulation of chemotherapeutic drugs, substantially improved cancer therapies can be achieved with all the benefits of nanoparticle-based chemotherapy.
When magnetic nanoparticles are subjected to an external, oscillating magnetic field, there are two relaxation mechanisms (Brownian and Néel relaxation) that govern their magnetization response in an effort to align with the applied field.21 Brownian relaxation, the physical rotation of the entire nanoparticle, is typically the dominant relaxation mechanism for nanoparticles larger than about 25 nm. In the case of the DOX-NC nanoparticles, Brownian relaxation is restricted by the bonds between the constituent nanospheres, such that Brownian motion may be observed as a mechanical “vibration” of the chain, rather than true rotational motion. Néel relaxation is dominant for nanoparticles smaller than 15 nm, and is highly dependent on a nanoparticle's crystal structure. To reorient its magnetic moment with an applied field, the nanoparticle must overcome an energy barrier, which results in the dissipation of excess heat. This phenomenon has been exploited for hyperthermia, but for efficient application, requires high spatial concentrations of nanoparticles. The size of the constituent nanospheres in the DOX-NC particles places them in between the Brownian and Néel regimes, and it is possible that their response to the 10 kHz field could be a mixture of mechanical oscillations and local heating. However, our in vitro studies show that no significant increase of temperature occurs locally at the particle scale and no significant drug release occurs from the DOX-NC particle. Thus these data suggest that a local temperature increase is not the dominant mechanism and that mechanical vibration at the selected frequency is enough to cause significant drug release. Future work includes optimizing the DOX-NC particle and the parameters of the magnetic field parameters (e.g. frequency, magnitude) for more efficient and rapid release.
To address the limitations of using environmental factors to stimulate drug release, other investigators have used ultrasound to release drug from particles showing promising results.44, 45 For example, upon sonication of DOX-loaded polymeric nanobubbles in the presence of perfluoropentane, drug release resulted in tumor regression in animal studies.46 Similar to the RF-triggered release of drug from DOX-NC, drug-loaded liposomes or nanoparticles coupled to the surface of microbubbles release their cargo due to ultrasound-induced rupture of the particles.47, 48
Notably, one of the advantages of the nanochain technology is the control of the shape and size of nanoparticles. By defining the topology of two different functional groups on the surface of the parent nanospheres, we were able to assemble them in a linear orientation with a high degree of uniformity. While other strategies have resulted in well-defined structures at the nanoscale,49-53 they are typically appropriate to only one type of material. In addition to the well-defined geometry, the multi-component nature of DOX-NC resulted in a combination of features: 1) on-command triggered drug release using an external RF field and 2) prolonged blood circulation and enhanced deposition into tumors. Interestingly, we observed that the 100-nm-long DOX-NC displayed longer blood residence times than the 100-nm liposomes. This is in good agreement with recent studies54-57 indicating that the oblate shape of particles favors their circulation in the blood due to lower uptake by macrophages. Future work will focus on further evaluating the effect of size and shape of the nanochains on blood circulation and extravasation into tumors. In addition, further in vivo studies need to be performed to identify the time point that the intratumoral accumulation of the particles peaks, which is the optimal point to apply the RF field.
While the deposition of DOX-NC into tumors was the necessary first step, our primary objective was to effectively increase the bioavailability of the drug cargo of the nanoparticle by delivering the active compound to cancer cell nuclei. To study the cytotoxic effects of DOX-NC, the in situ apoptosis in tumor tissue in the two animal models was used as a quantitative measure. DOX is a weak fluorophore and hence its release was readily detected. Increased release of DOX was associated with an elevation of apoptosis as shown by TUNEL. Being an intercalating agent, the cytostatic effect of DOX primarily leads to apoptosis, since it involves intercalation into chromosomal DNA,58 and DNA damage via inhibition of topoisomerase II and RNA polymerase II,59, 60 resulting in double-stranded DNA breaks.61 It should be noted that literature indicates that different DOX doses activate different regulatory mechanisms to induce either apoptosis or cell death through mitotic catastrophe.62 However, mitotic catastrophe has been observed when cancer cells are exposed to low DOX levels (e.g. 50 ng/mL) for at least 6 days. In our animal studies, the histological evaluation of apoptosis took place two days after administration of DOX-NC (or the control treatments). In addition to apoptosis, we hypothesize that a portion of the cancer cells in the tumors will eventually undergo cell death through mitotic catastrophe due to chronic exposure to low DOX concentrations.
Conclusion
In conclusion, we demonstrated that a nanochain-based chemotherapeutic enabled the delivery of the cytotoxic drug, doxorubicin to the majority of cancer cells throughout a tumor. Successful drug delivery to tumors requires that a long-circulating nanoparticle 1) enters the tumor microcirculation, 2) navigates through the tumor leaky vasculature into the tumor interstitium, and 3) releases the drug close to cancer cells. The nanoparticle system shown here meets all of these requirements. Due to their prolonged blood circulation, the nanochains capitalized on the EPR effect, and showed a high concentration in the tumor. Furthermore, once these multi-component nanoparticles extravasated into the tumor site, RF-triggered drug release resulted in a wide-spread cytotoxic effect throughout the entire tumor. Taking under consideration that RF can penetrate deep into tissues, we envision that this platform technology could effectively deliver drugs to primary and metastatic tumors with all the benefits of reduced side effects and substantial impact on cancer treatment.
Methods
Synthesis and characterization of multi-component nanochains
The nanochains were synthesized following our previously published method.19 Briefly, solid-phase chemistry was used to partially modify the surface functionality of nanospheres. CLEAR resin (Peptides International Inc, Louisville, KY) functionalized with amines was modified with a homobifunctional cleavable cross-linker reactive towards amines (DTSSP). Amine-functionalized IO nanospheres were introduced, allowed to bind to the solid support and then cleaved off using a reducing agent (TCEP). The same type of resin was used and the modified spheres with surface asymmetry were introduced in a step-by-step manner. As a final component, an amine functionalized DOX-loaded liposome was added before recovering the chain via a reducing agent. The chains were characterized in terms of their size (DLS), structure (TEM), and magnetic relaxivity (Bruker minispec relaxometer). Details of the synthesis and the analytical characterization of the DOX-NC nanoparticles are shown in the Supporting Information.
In vitro RF-triggered drug release
The DOX-NC suspension was exposed to an RF field using a custom-made solenoid (10 kHz frequency at a power of 2-30 Watts into the samples, solenoid's resistance ∼5 Ohms). Triggered release from the DOX-NC particles was measured using the fluorescence properties of DOX (λex/em=485/590 nm). The cytotoxicity of released drug from the DOX-NC particles was compared to the liposomal drug and free drug. Briefly, cytotoxicity studies were performed by seeding 13762 MAT BIII cells at a density of 105 cells/well in a 6-well plate 24 h before incubation with the formulations. Prior to incubation, cells were washed three times with fresh medium and then incubated with the treatment for 180 minutes at a concentration of 150 μM doxorubicin per well. The cells were washed three times with fresh medium and incubated for 48 h at 37°C and 5% CO2 in a humidified environment. The number of viable cells was determined using a formazan-based cell counting assay (CCK-8). Untreated cells were served as live controls for normalization of the data.
Pharmacokinetic studies
All animal procedures were conducted under a protocol approved by the CWRU IACUC. Female Fisher rats were given an IV injection of DOX-NC at a dose of 0.5 mg/kg DOX. Blood was collected from the orbital sinus at various time points before and after injection. Plasma was isolated by centrifugation (2200g, 15 min), and DOX was extracted after lysis in 30% MeOH and heating at 60 °C for 20 min. The solution was then vortexed and centrifuged. Fluorescent readings of the samples were obtained to detect DOX (λex/λem = 485/590) and Alexa-350 on the chain (λex/λem = 346/442). Plasma samples obtained immediately prior to injection were used to correct for background fluorescence. Further details are shown in Supporting Information.
Animal tumor models
The rat tumor model was established by a subcutaneous injection of 1 × 106 13762 MAT B III cells into the right flank of female Fisher rats. Mouse tumors were generated orthotopically in female BALB/c mice by injection of 0.5 × 106 4T1 cells into the inguinalmammary fat pad. Once the appropriate tumor size was established (diameter ∼0.5 cm), the animals were used in the in vivo studies. Each cell line required different lag times to produce a tumor lesion of about 0.5 cm in size (5 and 8 days for the MAT B III and 4T1 model, respectively). Based on our prior experience,7, 32, 63 we chose this tumor size as the starting point of the animal studies, since the primary tumor mass is sufficiently large to present angiogenic, necrotic and invasive areas, and therefore may be more informative and relevant to human disease.
Organ distribution
Twenty four hours after intravenous (IV) injection of the DOX-NC particles or liposomal DOX at a dose of 0.5 mg/kg DOX to the tails of the rat tumor model, the animals were anesthesized and transcardially perfused with heparinized PBS followed by 4% paraformaldehyde in PBS. The organs and tumors were then retrieved, washed with PBS, blotted dry, weighed and DOX was measured following an established protocol.9 Organ and tumor samples from an animal treated with a saline injection were used to correct for background fluorescence.
Survival study
Once the appropriate tumor sizes were established (diameter ∼0.5 cm), the rat tumor model was IV injected with DOX-NC at a dose of 0.5 mg/kg DOX. After 24 hours from injection, animals were exposed to the RF field operated as described previously. Following the same dose and schedule, control groups included animals treated with DOX-NC (but no RF), only RF, liposomal DOX, liposomal DOX with RF, free DOX, and saline. In addition to the single treatments, another group followed two cycles of treatment. The subsequent treatments were 2 days apart at the same DOX dose followed by exposure to RF following an identical protocol to the first cycle. The tumor growth was monitored every day using caliper measurements. The tumor growth was allowed to progress until the animals showed abnormal symptoms, at which point the animal were euthanized in a CO2 chamber. Time of death was determined to be the following day.
Histological evaluation
The rat MAT B III and the mouse 4T1 models were used in the histological studies. Animals treated with DOX-NC were exposed to the RF field 24 hours post-injection. After 24 hours from the application of the RF field, the animals were anesthetized with an IP injection of ketamine/xylazine and transcardially perfused with heparinized PBS followed by 4% paraformaldehyde in PBS. Controls included animals treated with DOX-NC (no RF), 35-nm liposomal DOX (with RF), 100-nm liposomal DOX (with RF), free DOX, RF alone and saline. The tumors were explanted and post-fixed overnight in 4% paraformaldehyde in PBS. The fixed tumors were soaked in 30% sucrose (w/v) in PBS at 4 °C for cryosectioning. Serial sections of 12 μm thickness were collected using a cryostat (Leica CM 300).
To visualize the tumor microvasculature, the tissue slices were immunohistochemically stained for the specific endothelial antigen CD31 (BD Biosciences, Pharmingen). The tissues were also stained with the nuclear stain DAPI. To evaluate the spread of the released DOX in relation to location of DOX-NC particles, Prussian blue stain was used to detect iron. Direct fluorescence (red) imaging of tumor sections were performed for imaging DOX. Apoptosis was detected using a TUNEL assay (Promega). The tissue sections were imaged at 20× on the Zeiss Axio Observer Z1 motorized FL inverted microscope. To obtain an image of the entire tumor section, a montage of each section was made using the automated tiling function of the microscope. The total number of cells was counted based on the nuclear stain (DAPI) in multiple histological sections per tumor (minimum 20), whereas apoptotic cells were quantified based on TUNEL-stained nuclei.
Statistical analysis
Means were determined for each variable in this study and the resulting values from each experiment were subjected to one-way analysis of variance with post hoc Bonferroni test. A P value of less than 0.01 was used to confirm significant differences. Normality of each data set was confirmed using the Anderson-Darling test.
Supplementary Material
Acknowledgments
We thank Dr. James Basilion for discussions, Michael Calabrese and Richard Pham for help with the fabrication and characterization of nanochains, and Kristen Lozada for help with the 4T1 tumor model. This work was partially supported by a grant from the American Cancer Society IRG-91-022-18 (EK) and a pilot grant from the Case Comprehensive Cancer Center P30 CA043703 (EK). L.B and R.T. were supported by a fellowship from the NIH Interdisciplinary Biomedical Imaging Training Program (5T32EB007509).
References
- 1.Von Hoff DD, Layard MW, Basa P, Davis HL, Jr, Von Hoff AL, Rozencweig M, Muggia FM. Risk Factors for Doxorubicin-Induced Congestive Heart Failure. Ann Intern Med. 1979;91:710–717. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 2.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 3.Lasic DD. Doxorubicin in Sterically Stabilized Liposomes. Nature. 1996;380:561–562. doi: 10.1038/380561a0. [DOI] [PubMed] [Google Scholar]
- 4.Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor Vascular Permeability, Accumulation, and Penetration of Macromolecular Drug Carriers. J Natl Cancer Inst. 2006;98:335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 5.Yuan F. Transvascular Drug Delivery in Solid Tumors. Semin Radiat Oncol. 1998;8:164–175. doi: 10.1016/s1053-4296(98)80042-8. [DOI] [PubMed] [Google Scholar]
- 6.Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, Brown EB, Izumi Y, Campbell RB, Berk DA, et al. Role of Tumor-Host Interactions in Interstitial Diffusion of Macromolecules: Cranial Vs. Subcutaneous Tumors. Proc Natl Acad Sci USA. 2001;98:4628–4633. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karathanasis E, Chan L, Karumbaiah L, McNeeley K, D'Orsi CJ, Annapragada AV, Sechopoulos I, Bellamkonda RV. Tumor Vascular Permeability to a Nanoprobe Correlates to Tumor-Specific Expression Levels of Angiogenic Markers. PLoS ONE. 2009;4:e5843. doi: 10.1371/journal.pone.0005843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Microvascular Permeability and Interstitial Penetration of Sterically Stabilized (Stealth) Liposomes in a Human Tumor Xenograft. Cancer Res. 1994;54:3352–3356. [PubMed] [Google Scholar]
- 9.Karathanasis E, Chan L, Balusu SR, D'Orsi CJ, Annapragada AV, Sechopoulos I, Bellamkonda RV. Multifunctional Nanocarriers for Mammographic Quantification of Tumor Dosing and Prognosis of Breast Cancer Therapy. Biomaterials. 2008;29:4815–4822. doi: 10.1016/j.biomaterials.2008.08.036. [DOI] [PubMed] [Google Scholar]
- 10.Grantab R, Sivananthan S, Tannock IF. The Penetration of Anticancer Drugs through Tumor Tissue as a Function of Cellular Adhesion and Packing Density of Tumor Cells. Cancer Res. 2006;66:1033–1039. doi: 10.1158/0008-5472.CAN-05-3077. [DOI] [PubMed] [Google Scholar]
- 11.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of Extracellular Matrix Assembly in Interstitial Transport in Solid Tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 12.Kostarelos K, Emfietzoglou D, Papakostas A, Yang WH, Ballangrud A, Sgouros G. Binding and Interstitial Penetration of Liposomes within Avascular Tumor Spheroids. Int J Cancer. 2004;112:713–721. doi: 10.1002/ijc.20457. [DOI] [PubMed] [Google Scholar]
- 13.Terasaki T, Iga T, Sugiyama Y, Sawada Y, Hanano M. Nuclear Binding as a Determinant of Tissue Distribution of Adriamycin, Daunomycin, Adriamycinol, Daunorubicinol and Actinomycin D. J Pharmacobiodyn. 1984;7:269–277. doi: 10.1248/bpb1978.7.269. [DOI] [PubMed] [Google Scholar]
- 14.Marafino BJ, Jr, Giri SN, Siegel DM. Pharmacokinetics, Covalent Binding and Subcellular Distribution of [3H]Doxorubicin after Intravenous Administration in the Mouse. J Pharmacol Exp Ther. 1981;216:55–61. [PubMed] [Google Scholar]
- 15.Laginha KM, Verwoert S, Charrois GJ, Allen TM. Determination of Doxorubicin Levels in Whole Tumor and Tumor Nuclei in Murine Breast Cancer Tumors. Clin Cancer Res. 2005;11:6944–6949. doi: 10.1158/1078-0432.CCR-05-0343. [DOI] [PubMed] [Google Scholar]
- 16.Nagayasu A, Uchiyama K, Kiwada H. The Size of Liposomes: A Factor Which Affects Their Targeting Efficiency to Tumors and Therapeutic Activity of Liposomal Antitumor Drugs. Adv Drug Deliv Rev. 1999;40:75–87. doi: 10.1016/s0169-409x(99)00041-1. [DOI] [PubMed] [Google Scholar]
- 17.Lasic DD, Frederik PM, Stuart MC, Barenholz Y, McIntosh TJ. Gelation of Liposome Interior. A Novel Method for Drug Encapsulation. FEBS Lett. 1992;312:255–258. doi: 10.1016/0014-5793(92)80947-f. [DOI] [PubMed] [Google Scholar]
- 18.Papahadjopoulos D, Allen TM, Gabizon A, Mayhew E, Matthay K, Huang SK, Lee KD, Woodle MC, Lasic DD, Redemann C, et al. Sterically Stabilized Liposomes: Improvements in Pharmacokinetics and Antitumor Therapeutic Efficacy. Proc Natl Acad Sci U S A. 1991;88:11460–11464. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peiris PM, Schmidt E, Calabrese M, Karathanasis E. Assembly of Linear Nano-Chains from Iron Oxide Nanospheres with Asymmetric Surface Chemistry. PLoS One. 2011;6:e15927. doi: 10.1371/journal.pone.0015927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolotin E, Cohen R, Bar L, Emanuel N, Ninio S, Lasic D, Barenholz Y. Ammonium Sulfate Gradients for Efficient and Stable Remote Loading of Amphipathic Weak Bases into Liposomes and Ligandoliposomes. Journal of Liposome Research. 1994;4:455–479. [Google Scholar]
- 21.Rauwerdink AM, Weaver JB. Concurrent Quantification of Multiple Nanoparticle Bound States. Med Phys. 2011;38:1136–1140. doi: 10.1118/1.3549762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunt CA. Liposomes Disposition in Vivo. V. Liposome Stability in Plasma and Implications for Drug Carrier Function. Biochim Biophys Acta. 1982;719:450–463. doi: 10.1016/0304-4165(82)90233-1. [DOI] [PubMed] [Google Scholar]
- 23.Agarwal A, Mackey MA, El-Sayed MA, Bellamkonda RV. Remote Triggered Release of Doxorubicin in Tumors by Synergistic Application of Thermosensitive Liposomes and Gold Nanorods. ACS Nano. 2011;5:4919–4926. doi: 10.1021/nn201010q. [DOI] [PubMed] [Google Scholar]
- 24.Lasic D, Martin F. Stealth Liposomes. CRC Press Inc; 1995. [Google Scholar]
- 25.Govorov AO, Richardson HH. Generating Heat with Metal Nanoparticles. Nano Today. 2007;2:30–38. [Google Scholar]
- 26.Huang H, Delikanli S, Zeng H, Ferkey DM, Pralle A. Remote Control of Ion Channels and Neurons through Magnetic-Field Heating of Nanoparticles. Nat Nanotechnol. 2010;5:602–606. doi: 10.1038/nnano.2010.125. [DOI] [PubMed] [Google Scholar]
- 27.Gabizon A, Papahadjopoulos D. Liposome Formulations with Prolonged Circulation Time in Blood and Enhanced Uptake by Tumors. Proc Natl Acad Sci USA. 1988;85:6949–6953. doi: 10.1073/pnas.85.18.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gabizon A, Goren D, Horowitz AT, Tzemach D, Lossos A, Siegal T. Long-Circulating Liposomes for Drug Delivery in Cancer Therapy: A Review of Biodistribution Studies in Tumor-Bearing Animals. Adv Drug Deliv Rev. 1997;24:337–344. [Google Scholar]
- 29.Rose PG. Pegylated Liposomal Doxorubicin: Optimizing the Dosing Schedule in Ovarian Cancer. Oncologist. 2005;10:205–214. doi: 10.1634/theoncologist.10-3-205. [DOI] [PubMed] [Google Scholar]
- 30.McNeeley KM, Karathanasis E, Annapragada AV, Bellamkonda RV. Masking and Triggered Unmasking of Targeting Ligands on Nanocarriers to Improve Drug Delivery to Brain Tumors. Biomaterials. 2009;30:3986–3995. doi: 10.1016/j.biomaterials.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Agarwal A, Jaye DL, Giegerman CM, Bellamkonda RV. Rational Identification of a Novel Peptide for Targeting Nanocarriers to 9L Glioma. J Biomed Mater Res A. 2008 doi: 10.1002/jbm.a.31762. [DOI] [PubMed] [Google Scholar]
- 32.Karathanasis E, Suryanarayanan S, Balusu SR, McNeeley K, Sechopoulos I, Karellas A, Annapragada AV, Bellamkonda RV. Imaging Nanoprobe for Prediction of Outcome of Nanoparticle Chemotherapy by Using Mammography. Radiology. 2009;250:398–406. doi: 10.1148/radiol.2502080801. [DOI] [PubMed] [Google Scholar]
- 33.Campos S. Liposomal Anthracyclines: Adjuvant and Neoadjuvant Therapy for Breast Cancer. Oncologist. 2003;8(2):10–16. doi: 10.1634/theoncologist.8-suppl_2-10. [DOI] [PubMed] [Google Scholar]
- 34.O'Shaughnessy J. Liposomal Anthracyclines for Breast Cancer: Overview. Oncologist. 2003;8(2):1–2. doi: 10.1634/theoncologist.8-suppl_2-1. [DOI] [PubMed] [Google Scholar]
- 35.Rivera E. Liposomal Anthracyclines in Metastatic Breast Cancer: Clinical Update. Oncologist. 2003;8(2):3–9. doi: 10.1634/theoncologist.8-suppl_2-3. [DOI] [PubMed] [Google Scholar]
- 36.Clinicaltrials.gov sponsored by NIH. [Accessed 11/11/2011]; URL: http://clinicaltrials.gov/
- 37.Parish CR, Freeman C, Brown KJ, Francis DJ, Cowden WB. Identification of Sulfated Oligosaccharide-Based Inhibitors of Tumor Growth and Metastasis Using Novel in Vitro Assays for Angiogenesis and Heparanase Activity. Cancer Res. 1999;59:3433–3441. [PubMed] [Google Scholar]
- 38.Xing RH, Mazar A, Henkin J, Rabbani SA. Prevention of Breast Cancer Growth, Invasion, and Metastasis by Antiestrogen Tamoxifen Alone or in Combination with Urokinase Inhibitor B-428. Cancer Res. 1997;57:3585–3593. [PubMed] [Google Scholar]
- 39.Simoes S, Moreira JN, Fonseca C, Duzgunes N, de Lima MC. On the Formulation of pH-Sensitive Liposomes with Long Circulation Times. Adv Drug Deliv Rev. 2004;56:947–965. doi: 10.1016/j.addr.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 40.Brazel CS. Magnetothermally-Responsive Nanomaterials: Combining Magnetic Nanostructures and Thermally-Sensitive Polymers for Triggered Drug Release. Pharm Res. 2009;26:644–656. doi: 10.1007/s11095-008-9773-2. [DOI] [PubMed] [Google Scholar]
- 41.Tai LA, Tsai PJ, Wang YC, Wang YJ, Lo LW, Yang CS. Thermosensitive Liposomes Entrapping Iron Oxide Nanoparticles for Controllable Drug Release. Nanotechnology. 2009;20:135101. doi: 10.1088/0957-4484/20/13/135101. [DOI] [PubMed] [Google Scholar]
- 42.Lee JH, Jang JT, Choi JS, Moon SH, Noh SH, Kim JW, Kim JG, Kim IS, Park KI, Cheon J. Exchange-Coupled Magnetic Nanoparticles for Efficient Heat Induction. Nat Nanotechnol. 6:418–422. doi: 10.1038/nnano.2011.95. [DOI] [PubMed] [Google Scholar]
- 43.Yavuz MS, Cheng Y, Chen J, Cobley CM, Zhang Q, Rycenga M, Xie J, Kim C, Song KH, Schwartz AG, et al. Gold Nanocages Covered by Smart Polymers for Controlled Release with Near-Infrared Light. Nat Mater. 2009;8:935–939. doi: 10.1038/nmat2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, Cai X, Kim C, Sun G, Zhang Y, Deng R, Yang M, Chen J, Achilefu S, Wang LV, et al. Gold Nanocages Covered with Thermally-Responsive Polymers for Controlled Release by High-Intensity Focused Ultrasound. Nanoscale. 2011;3:1724–1730. doi: 10.1039/c0nr00932f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moon GD, Choi SW, Cai X, Li W, Cho EC, Jeong U, Wang LV, Xia Y. A New Theranostic System Based on Gold Nanocages and Phase-Change Materials with Unique Features for Photoacoustic Imaging and Controlled Release. J Am Chem Soc. 2011;133:4762–4765. doi: 10.1021/ja200894u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rapoport N, Gao Z, Kennedy A. Multifunctional Nanoparticles for Combining Ultrasonic Tumor Imaging and Targeted Chemotherapy. J Natl Cancer Inst. 2007;99:1095–1106. doi: 10.1093/jnci/djm043. [DOI] [PubMed] [Google Scholar]
- 47.Lum AF, Borden MA, Dayton PA, Kruse DE, Simon SI, Ferrara KW. Ultrasound Radiation Force Enables Targeted Deposition of Model Drug Carriers Loaded on Microbubbles. J Control Release. 2006;111:128–134. doi: 10.1016/j.jconrel.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kheirolomoom A, Dayton PA, Lum AF, Little E, Paoli EE, Zheng H, Ferrara KW. Acoustically-Active Microbubbles Conjugated to Liposomes: Characterization of a Proposed Drug Delivery Vehicle. J Control Release. 2007;118:275–284. doi: 10.1016/j.jconrel.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang X, El-Sayed IH, Qian W, El-Sayed MA. Cancer Cell Imaging and Photothermal Therapy in the Near-Infrared Region by Using Gold Nanorods. JACS. 2006;128:2215–2120. doi: 10.1021/ja057254a. [DOI] [PubMed] [Google Scholar]
- 50.Sardar R, Shumaker-Parry JS. Asymmetrically Functionalized Gold Nanoparticles Organized in One-Dimensional Chains. Nano Letters. 2008;8:731–736. doi: 10.1021/nl073154m. [DOI] [PubMed] [Google Scholar]
- 51.Park JH, von Maltzahn G, Zhang L, Derfus AM, Simberg D, Harris TJ, Ruoslahti E, Bhatia SN, Sailor MJ. Systematic Surface Engineering of Magnetic Nanoworms for in Vivo Tumor Targeting. Small. 2009;5:694–700. doi: 10.1002/smll.200801789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park JH, von Maltzahn G, Zhang LL, Schwartz MP, Ruoslahti E, Bhatia SN, Sailor MJ. Magnetic Iron Oxide Nanoworms for Tumor Targeting and Imaging. Advanced Materials. 2008;20:1630–1635. doi: 10.1002/adma.200800004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai Q, Worden JG, Trullinger J, Huo Q. A “Nanonecklace” Synthesized from Monofunctionalized Gold Nanoparticles. J Am Chem Soc. 2005;127:8008–8009. doi: 10.1021/ja042610v. [DOI] [PubMed] [Google Scholar]
- 54.Chithrani BD, Ghazani AA, Chan WC. Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 55.Sharma G, Valenta DT, Altman Y, Harvey S, Xie H, Mitragotri S, Smith JW. Polymer Particle Shape Independently Influences Binding and Internalization by Macrophages. J Control Release. 147:408–412. doi: 10.1016/j.jconrel.2010.07.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Shape Effects of Filaments Versus Spherical Particles in Flow and Drug Delivery. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Champion JA, Mitragotri S. Role of Target Geometry in Phagocytosis. Proc Natl Acad Sci USA. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muller I, Niethammer D, Bruchelt G. Anthracycline-Derived Chemotherapeutics in Apoptosis and Free Radical Cytotoxicity (Review) Int J Mol Med. 1998;1:491–494. doi: 10.3892/ijmm.1.2.491. [DOI] [PubMed] [Google Scholar]
- 59.Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-Induced DNA Damage Mediated by Mammalian DNA Topoisomerase Ii. Science. 1984;226:466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- 60.Levin M, Silber R, Israel M, Goldfeder A, Khetarpal VK, Potmesil M. Protein-Associated DNA Breaks and DNA-Protein Cross-Links Caused by DNA Nonbinding Derivatives of Adriamycin in L1210 Cells. Cancer Res. 1981;41:1006–1010. [PubMed] [Google Scholar]
- 61.Kellner U, Sehested M, Jensen PB, Gieseler F, Rudolph P. Culprit and Victim -- DNA Topoisomerase II. Lancet Oncol. 2002;3:235–243. doi: 10.1016/s1470-2045(02)00715-5. [DOI] [PubMed] [Google Scholar]
- 62.Eom YW, Kim MA, Park SS, Goo MJ, Kwon HJ, Sohn S, Kim WH, Yoon G, Choi KS. Two Distinct Modes of Cell Death Induced by Doxorubicin: Apoptosis and Cell Death through Mitotic Catastrophe Accompanied by Senescence-Like Phenotype. Oncogene. 2005;24:4765–4777. doi: 10.1038/sj.onc.1208627. [DOI] [PubMed] [Google Scholar]
- 63.Yori JL, Seachrist DD, Johnson E, Lozada KL, Abdul-Karim FW, Chodosh LA, Schiemann WP, Keri RA. Kruppel-Like Factor 4 Inhibits Tumorigenic Progression and Metastasis in a Mouse Model of Breast Cancer. Neoplasia. 2011;13:601–610. doi: 10.1593/neo.11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.