

Arrhythmogenic cardiomyopathy is the most arrhythmogenic form of human heart disease and a major cause of sudden death in the young.1,2 It was first described as a right ventricular disease (arrhythmogenic right ventricular cardiomyopathy or ARVC), but is now recognized to include biventricular and left dominant forms which may be misdiagnosed as dilated cardiomyopathy or myocarditis. Arrhythmias occur early in the natural history of arrhythmogenic cardiomyopathy, often preceding structural remodeling of the myocardium.1, 2 This so-called “concealed phase” of the disease is unique among the primary myocardial disorders. In hypertrophic cardiomyopathy, for example, arrhythmic risk is related to the underlying substrate of myocyte disarray, hypertrophy, fibrosis and small-vessel disease. And in dilated cardiomyopathy, arrhythmias generally arise in the context of significant ventricular dilatation and contractile dysfunction accompanied by changes in the expression, activity and spatial distribution of ion channel proteins and currents. In contrast, there is something fundamentally arrhythmogenic about arrhythmogenic cardiomyopathy particularly in its early stage in which frequent arrhythmias arise in otherwise apparently normal hearts. In this sense, arrhythmogenic cardiomyopathy is more reminiscent of the ion channelopathies than other forms of non-ischemic cardiomyopathy.
Arrhythmogenic cardiomyopathy has been linked to mutations in genes encoding desmosomal proteins (PKP2, DSG2, DSC2, DSP and JUP).1, 2 Desmosomes are cell-cell adhesion organelles. They are particularly abundant in heart and skin, tissues that normally experience mechanical stress. Not surprisingly therefore, clinical phenotypes in patients with desmosomal mutations take the form of myocardial and cutaneous diseases. In fact, patients with arrhythmogenic cardiomyopathy often exhibit disease flares in response to stress or exercise, emphasizing the importance of biomechanical determinants of disease. However, the relationship between changes in biomechanical properties of the myocardium produced by desmosomal mutations and heart rhythm abnormalities in arrhythmogenic cardiomyopathy is poorly characterized. Advances in understanding this relationship could reveal important basic mechanisms of arrhythmogenesis.
Dependence of Intercellular Electrical Coupling on Intercellular Mechanical Coupling
As a general rule, defects in intercellular mechanical coupling are associated with an inability to establish and/or maintain normal cell-cell communication via gap junctions.3 This dependency appears to be especially critical in myocardium in which large gap junctions, required for safe impulse propagation, are typically surrounded by extensive points of cell-cell adhesion within intercalated disks. The high density of protein packing in gap junctions makes them stiff and susceptible to rupture in response to shear. The intimate juxtaposition of gap junctions and adhesion junctions at intercalated disks may, therefore, have evolved to protect gap junctions from mechanical stress associated with contraction.3
Multiple lines of evidence support the idea that normal electrical coupling at gap junctions depends on normal mechanical coupling at intercalated disks. For example, when adult cardiac myocytes are disaggregated and allowed to re-associate, the first connections formed are adherens junctions and desmosomes. 4 Gap junctions appear only after stable mechanical coupling has been reestablished. Genetic interventions that reduce expression of gap junction proteins have no apparent effect on the number or size of mechanical junctions, 5 whereas genetic ablation of adhesion junction proteins causes a significant reduction in gap junction number and size and increases risk of arrhythmias.6 If mutations in desmosomal genes compromise cell-cell mechanical coupling in arrhythmogenic cardiomyopathy, then it follows that gap junction remodeling might be a fundamental feature of the myocardium in this disease. Surprisingly little is known about whether and how disease-causing desmosomal mutations affect the biomechanical properties of myocardium, but as detailed below, diffuse gap junction remodeling, demonstrable even during the concealed phase, appears to be a frequent feature of arrhythmogenic cardiomyopathy.
Gap Junction Remodeling in Arrhythmogenic Cardiomyopathy
Gap junction remodeling in arrhythmogenic cardiomyopathy was first described in 4 patients with Naxos disease, a rare cardiocutaneous syndrome with the clinical triad of woolly hair, palmoplantar keratoderma and ARVC caused by a recessive deletion mutation in the gene encoding the mechanical junction protein plakoglobin (also known as γ-catenin).7 The amount of immunoreactive signal for the major ventricular gap junction protein, Cx43, was found to be markedly reduced at intercalated disks in right and left ventricular myocardium. One of these patients was known to carry the recessive mutation and exhibited the characteristic hair and skin features of the disease, but died at age 7 of acute non-lymphocytic leukemia. At autopsy, the heart appeared normal (Figure 1). However, Holter monitoring had documented frequent ventricular extrasystoles mainly of RV origin, and progressive depolarization abnormalities including QRS prolongation and epsilon waves (Figure 1).7 Immunofluorescent analysis showed marked reduction in the amount of Cx43 signal in the heart, and immunoblotting showed apparent loss of highly phosphorylated Cx43 (Figure 1), which is known to be selectively located within gap junctions as opposed to intracellular sites. Electron microscopy showed fewer and smaller gap junctions at intercalated disks, thus confirming the immunohistochemical findings.7 Although demonstrated in only a single example of concealed disease, these observations suggest that gap junction remodeling occurs early in the natural history of arrhythmogenic cardiomyopathy before the onset of myocardial degeneration and fibrofatty tissue accumulation.
Figure 1.

A. 12-lead electrocardiogram at rest from a 7-year-old patient with Naxos disease in the “concealed phase” showing QRS prolongation (to 110 ms) and epsilon waves in leads V1 through V3, and deeply inverted T waves in leads V1 through V3. B. Low magnification sections of left and right ventricles at autopsy showing no evidence of myocardial degeneration or fibrofatty tissue accumulation (trichrome Heidenhain stain). C. Representative confocal images showing loss of immunoreactive Cx43 signal at intercalated disks in the left ventricle of this patient compared with a normal control. D. Immunoblot of Cx43 in left and right ventricular myocardium of this patient showing apparent loss of highly phosphorylated Cx43 (arrows) compared with a normal control. Figure modified from Kaplan et al.7
Gap junction remodeling has also been reported in the heart in Carvajal syndrome, another cardiocutaneous syndrome characterized by woolly hair, palmoplantar keratoderma and a biventricular form of arrhythmogenic cardiomyopathy caused by a recessive deletion mutation in the desmosomal protein desmoplakin.8 These initial reports were followed by now extensive evidence that gap junction remodeling occurs not only in severe, highly penetrant cardiocutaneous syndromes caused by recessive mutations, but also in classical ARVC associated with autosomal dominant mutations in all known desmosomal genes and in patients with clinically documented disease but no known mutation. 9 In the great majority of cases, gap junction remodeling occurred diffusely and involved not only the right ventricular free wall but also the left ventricle and interventricular septum in which myocardial degeneration and fibrofatty tissue accumulation do not typically occur.9 Similar findings have been reported in a spontaneous canine model of ARVC.10
Additional lines of evidence support the notion that gap junction remodeling may be linked to basic disease mechanisms in arrhythmogenic cardiomyopathy. For example, reduced immunoreactive signal for Cx43 at cell junctions has been demonstrated in neonatal rat ventricular myocytes transfected to express mutant isoforms of plakophilin-2 (R79X, 179fs) known to cause ARVC in humans.11 Moreover, siRNA-mediated plakophilin-2 silencing in neonatal rat ventricular myocyte cultures leads to a decrease in total Cx43 content, a significant redistribution of Cx43 to the intracellular space, and a decrease in dye coupling between cells.12 However, apparently normal Cx43 signal at cell-cell junctions has been reported in 3 patients from Newfoundland with a highly penetrant and lethal form of arrhythmogenic cardiomyopathy associated with a mutation in TMEM43, a transmembrane protein of uncertain function.13 Taken together, these observations indicate that while remodeling of gap junctions may not occur in all forms of the disease spectrum, it does seem to be a frequent feature of arrhythmogenic cardiomyopathy. That it arises early in the course of the disease in most cases suggests a mechanistic link to the fundamental disease pathway in ARVC.
Future Directions
To a large extent, demonstration of gap junction remodeling in arrhythmogenic cardiomyopathy has been based on immunohistochemistry. In fact, there is little if any direct evidence that gap junction remodeling is responsible for conduction abnormalities or contributes to the highly arrhythmogenic phenotype in arrhythmogenic cardiomyopathy, although it is certainly plausible to propose such a role. Moreover, little is known about the underlying mechanisms linking expression of mutant forms of desmosomal proteins to gap junction remodeling. If gap junction remodeling is, in fact, related mainly to abnormal biomechanical properties caused by mutant desmosomal proteins, then it might be expected to develop first in areas of greatest mechanical stress, especially in response to exercise. However, there have been no studies to determine whether gap junction remodeling occurs initially in such areas, including those destined to become sites of fibrofatty scar formation.
We have developed an overarching hypothesis to explain the pathogenesis of arrhythmogenic cardiomyopathy. It includes several potential mechanisms by which mutant desmosomal proteins may lead to gap junction remodeling. As shown in Figure 2, we propose that abnormal responses to mechanical load caused by mutant desmosomal proteins induce cytokine expression by cardiac myocytes and destabilize junctional plakoglobin (γ-catenin). Increasing evidence shows that redistribution of plakoglobin from junctional to intracellular pools occurs in arrhythmogenic cardiomyopathy9 and this can perturb canonical and non-canonical Wnt signaling pathways potentially altering gene expression and Ca2+ homeostasis.14 Engagement of Frizzled class receptors by Wnt ligands inhibits glycogen synthase kinase 3β, resulting in stabilization and accumulation of β-catenin and γ-catenin which translocate to the cell nucleus and interact with Tcf transcription factors to modulate expression of target genes. One such gene encodes Cx43,15 thus implicating a possible mechanism by which altered Wnt signaling mediated by pathological accumulation of plakoglobin could affect Cx43 expression.
Figure 2.
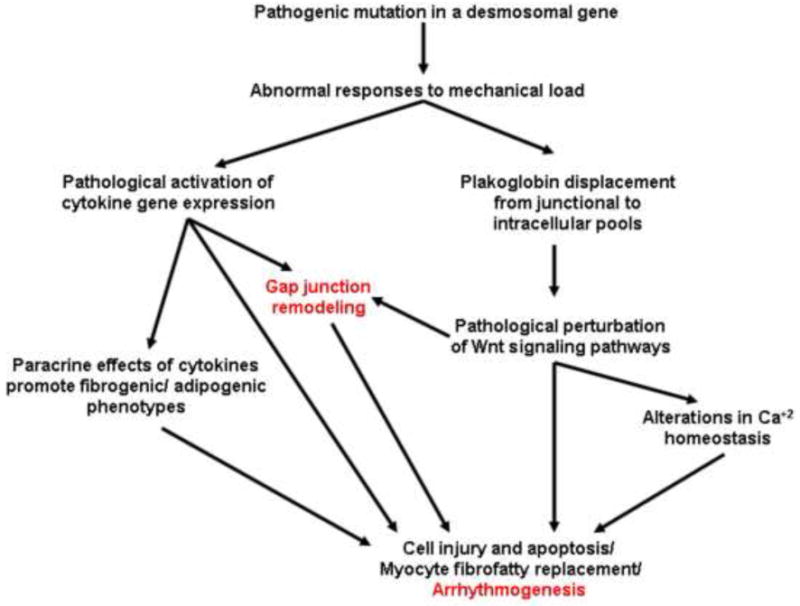
A proposed disease pathway in arrhythmogenic cardiomyopathy showing potential mechanisms of gap junction remodeling
We have recently reported elevated circulating levels of multiple pro-inflammatory cytokines and provided evidence of TNFα and IL-17 expression by the myocardium in patients with arrhythmogenic cardiomyopathy.16 We have also shown that selected cytokines including TNFα and IL-17 can cause redistribution of plakoglobin from junctions to intracellular sites in normal cardiac myocytes.16 Cytokines expressed by injured cardiac myocytes in arrhythmogenic cardiomyopathy could also affect gap junctions in the heart. For example, Cx43 mRNA levels are reduced in the rat heart following an inflammatory state induced by administration of bacterial lipolysaccharide (LPS) or after hepatic ischemia and reperfusion.17 Reduced Cx43 mRNA levels also occurred in sham-operated animals suggesting that downregulation of this gene was part of a generalized inflammatory response. Serum analysis showed high levels of TNFα following administration of LPS. Cx43 promoter activity was also reduced significantly in H9c2 cells incubated with TNFα in concentrations as low as 1ng/ml of medium.17 Collectively, these results suggest that inflammatory cytokines implicated in the pathogenesis of arrhythmogenic cardiomyopathy may promote gap junction remodeling. If cytokines do play an important role, then anti-inflammatory therapy could be beneficial. However, we know of no studies on this question and, based on previous experience with broad-spectrum anti-inflammatory therapy in heart failure in which adverse effects related to immunosuppression were observed,18 we would recommend a cautious approach. Targeted therapy could be of value if a specific mediator were shown to play a critical role in arrhythmogenesis.
In conclusion, the characteristic electrocardiographic abnormalities in ARVC strongly suggest a conduction defect. The presence of epsilon waves and QRS prolongation presumably reflect slow, heterogeneous electrical conduction that could be related to gap junction remodeling. However, it remains to be determined to what extent changes in electrical coupling contribute to the highly arrhythmogenic phenotype in arrhythmogenic cardiomyopathy, and how gap junction remodeling interacts with other potential arrhythmogenic mechanisms in this deadly disease. Moreover, the triggers of arrhythmias during the concealed phase of disease remain undefined. One promising avenue for future investigation is more detailed elucidation of the manifold mechanisms implicated in dysregulated Wnt signaling pathways. Future advances could lead to fundamental insights into arrhythmogenesis, not only in arrhythmogenic cardiomyopathy but in other more common heart diseases, and suggest new therapeutic targets to treat or prevent sudden death.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 2.Saffitz JE. The pathobiology of arrhythmogenic cardiomyopathy. Annu Rev Pathol. 2011;28:299–321. doi: 10.1146/annurev-pathol-011110-130151. [DOI] [PubMed] [Google Scholar]
- 3.Saffitz JE. Dependence of electrical coupling on mechanical coupling in cardiac myocytes – insights gained from cardiomyopathies caused by defects in cell-cell communication. Ann NY Acad Sci. 2005;1047:336–344. doi: 10.1196/annals.1341.030. [DOI] [PubMed] [Google Scholar]
- 4.Kostin S, Hein S, Bauer EP, Schaper J. Spatiotemporal development and distribution of intercellular junctions in adult rat cardiomyocytes in culture. Circ Res. 1999;85:154–167. doi: 10.1161/01.res.85.2.154. [DOI] [PubMed] [Google Scholar]
- 5.Saffitz JE, Green KG, Kraft WJ, Schechtman KB, Yamada KA. Effects of diminished expression of connexin43 on gap junction number and size in ventricular myocardium. Am J Physiol (Heart Circ Physio) 2000;278:H1662–H1670. doi: 10.1152/ajpheart.2000.278.5.H1662. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Levin MD, Xiong Y, Petrenko N, Patel VV, Radice GL. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J Mol Cell Cardiol. 2008;44:597–606. doi: 10.1016/j.yjmcc.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaplan SR, Gard JJ, Protonotarios N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 2004;13:26–32. doi: 10.1016/S1054-8807(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 9.Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 10.Oxford EM, Everitt M, Coombs W, et al. Molecular composition of the intercalated disc in a spontaneous canine animal model of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2007;4:1196–1205. doi: 10.1016/j.hrthm.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joshi-Mukherjee R, Coombs W, Musa H, Oxford E, Taffet S, Delmar M. Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (ARVC)- related plakophilin-2 (PKP2) mutations. Heart Rhythm. 2008;5:1715–1723. doi: 10.1016/j.hrthm.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 2007;101:703–711. doi: 10.1161/CIRCRESAHA.107.154252. [DOI] [PubMed] [Google Scholar]
- 13.Christensen AH, Andersen CB, Tybjaerd-Hansen A, Haunso S, Svendsen JH. Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clin Genet. 2001;80:256–264. doi: 10.1111/j.1399-0004.2011.01623.x. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Gras E, Lombardi R, Giocondo MJ, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ai Z, Fischer A, Spray DC, Brown AMC, Fishman GI. Wnt-1 regulation of connexin43 in cardiac myocytes. J Clin Invest. 2000;105:161–171. doi: 10.1172/JCI7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asimaki A, Tandri H, Duffy ER, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circulation: Arrhythm Electrophysiol. 2011 Aug 22; doi: 10.1161/CIRCEP.111.964890. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Cobo M, Gingalewski C, Drujan D, De Maio A. Downregulation of connexin 43 gene expression in rat heart during inflammation; the role of tumour necrosis factor. Cytokine. 1999;11:216–224. doi: 10.1006/cyto.1998.0422. [DOI] [PubMed] [Google Scholar]
- 18.Gullestad L, Kjekshus J, Damås JK, Ueland T, Yndestad A, Aukrust P. Agents targeting inflammation in heart failure. Expert Opin Investig Drugs. 2005;14:557–66. doi: 10.1517/13543784.14.5.557. [DOI] [PubMed] [Google Scholar]