

Summary
RIG-I is a cytosolic pathogen recognition receptor that initiates immune responses against RNA viruses. Upon viral RNA recognition, anti-viral signalling requires RIG-I redistribution from the cytosol to membranes where it binds the adaptor protein, MAVS. Here we identify the mitochondrial targeting chaperone protein, 14-3-3ε, as a RIG-I-binding partner and essential component of a translocation complex or “translocon” containing RIG-I, 14-3-3ε, and the TRIM25 ubiquitin ligase. The RIG-I translocon directs RIG-I redistribution from the cytosol to membranes where it mediates MAVS-dependent innate immune signalling during acute RNA virus infection. 14-3-3ε is essential for the stable interaction of RIG-I with TRIM25, which facilitates RIG-I ubiquitination and initiation of innate immunity against hepatitis C virus and other pathogenic RNA viruses. Our results define 14-3-3ε as a key component of a RIG-I translocon required for innate antiviral immunity.
Introduction
Retinoic acid inducible gene-I (RIG-I) plays a major role in pathogen recognition and intracellular signaling of innate immunity that initiates the immune response to RNA virus infection. In particular, RIG-I is essential for resistance to hepatitis C virus (HCV) infection (Gale and Foy, 2005). During acute HCV infection RIG-I recognizes and binds to the poly-uridine/cytosine (poly-U/UC) motif of the HCV RNA, thus signaling an innate immune response that restricts hepatocyte permissiveness for infection (Loo et al., 2006). Viral control of these processes supports chronic HCV infection in nearly 200 million people, thus marking RIG-I regulation of hepatic defenses as a critical determinant impacting HCV infection and immunity (Liu and Gale, 2010; Sumpter et al., 2005).
RIG-I is an RNA helicase encoding tandem amino-terminal caspase activation and recruitment domains (CARDs). RIG-I functions as a cytosolic pathogen recognition receptor (PRR) (Loo and Gale Jr, 2011) and mediates immune signaling after binding to pathogen associated molecular pattern (PAMP) motifs within viral RNA that accumulate during acute infection. RIG-I recognizes 5′ triphosphate (5′-ppp), double-stranded RNA and poly-U/UC motifs as non-self PAMPs through the actions of its repressor domain (RD) (Cui et al., 2008; Saito et al., 2007), thus releasing it from autorepression and resulting in its signaling activation (Jiang et al., 2011). Active RIG-I then mediates downstream signaling by binding to the adaptor protein, MAVS, located on the outer membrane of the mitochondria, peroxisomes, and on mitochondria-associated membrane (MAM), the last being a subdomain of the ER and the main site of intracellular innate immune and inflammatory signaling (Dixit et al., 2010; Horner et al., 2011; Zhou et al., 2011). RIG-I binds to MAVS through CARD interactions. This process results in establishment of a MAVS signalosome that drives the activation of IRF-3 and NF-κB, resulting in the expression of IFN-β and a variety of antiviral and immunomodulatory genes that confer antiviral defense and that regulate immunity to infection (Loo and Gale Jr, 2011). To initiate immune signaling RIG-I must traverse the cytoplasm and relocalize to the MAM compartment for MAVS binding (Horner et al., 2011). This process is dependent upon K63-linked ubiquitination of RIG-I on K172R by the E3 ubiquitin ligase Tripartite motif-containing protein 25 (TRIM25). RIG-I interaction with TRIM25 and redistribution to membrane compartments represents a major step in immune signaling but how these processes are directed to trigger innate immunity has not been defined (Gack et al., 2007). Here we identify 14-3-3ε as a RIG-I binding protein and essential chaperone of a translocation complex or “RIG-I translocon” containing RIG-I, 14-3-3ε, and TRIM25 (Dougherty and Morrison, 2004; Gack et al., 2008), that directs RIG-I redistribution from the cytosol to the membrane for MAVS interaction and immune signaling during acute RNA virus infection. We show that in human hepatocytes translocon assembly is controlled by the RIG-I RD and induced within minutes of HCV PAMP RNA-ligand binding by RIG-I. Thus, approaches to modulate RIG-I translocon activity may offer therapeutic strategies to suppress virus infection.
Results
RIG-I undergoes cytosol-to-membrane translocation during acute virus infection
We conducted membrane flotation by sucrose gradient centrifugation of human hepatoma (Huh7) cell extracts to separate membrane and cytosolic cell compartments for analysis of protein localization. The distribution of endogenous RIG-I, tubulin (a cytoplasmic marker protein), and protein markers of ER (calnexin), MAM (MFN2), outer- (VDAC) and inner-(Cox-1) mitochondrial membrane were then examined in the resulting cellular fractions. To assess RIG-I distribution during acute virus infection, Huh7 cells were infected with Sendai virus (SenV), an RNA virus that accurately models HCV activation of RIG-I signaling (Saito et al., 2008). In mock-infected cells calnexin and MFN2 were distributed within fractions 2 and 3, reflecting their membrane-association while tubulin was present only in fractions 5-7, demonstrating an effective separation of total membrane from the cytosolic compartments (Fig. 1a). We noticed that compared to resting cells, mitochondrial markers in SenV-infected cells, such as Cox-1 and VDAC, shifted from fractions 2-5 to fraction 2. This differencemight be due to alteredcontext of mitochondria during viral infection, as mitochondria elongate during infection (Castanier, 2009; Horner, 2011), resulting in additional membrane surface and altered density. RIG-I was exclusively present within the cytosolic fractions of mock-infected cells. However, within 4 hrs of SenV infection RIG-I appeared in the membrane fractions and was present there through the 24 hr analysis. RIG-I similarly redistributed to membrane during acute SenV infection of 293 cells (Supplementary Fig. 1a). We further evaluated the distribution of YFP-RIG-I expressed in Huh7 cells. Fluorescence microscopy analysis showed YFP-RIG-I distributed throughout the cytosol in mock-infected cells but was rapidly redistributed to a perinuclear pattern during an acute SenV infection time-course (Fig. 1b). The redistribution of RIG-I coincided with induction of the innate immune response as marked by the phosphorylation/activation of IRF-3 (see Supplementary Fig. 2b, lanes 1-3). We examined the extent of YFP-RIG-I redistribution in mock-infected Huh7 cells or after 24 hrs of infection with SenV, corresponding to the peak of innate immune induction(Horner et al., 2011). In mock-infected cells YFP-RIG-I was distributed throughout the cytoplasm and bounded by the plasma membrane (Fig. 1c, vectors I and II). In contrast, YFP-RIG-I redistributed to concentrate within a perinuclear membranous pattern during acute SenV infection (Fig. 1c, vectors III and IV) wherein a significant majority of infected cells exhibited this pattern of YFP-RIG-I redistribution (Fig. 1c.). We observed a similar cytosol-to-membrane redistribution of YFP-RIG-I during acute SenV-infection of PH5CH8 hepatocytes that corresponded to the temporal activation of IRF-3 and induction of IFIT1 expression (Supplementary Fig. 1b) (Ikeda et al., 1998). Taken together, these results demonstrate that that innate immune signaling is coupled to RIG-I cytosol-to-membrane redistribution during acute RNA virus infection.
Figure 1.
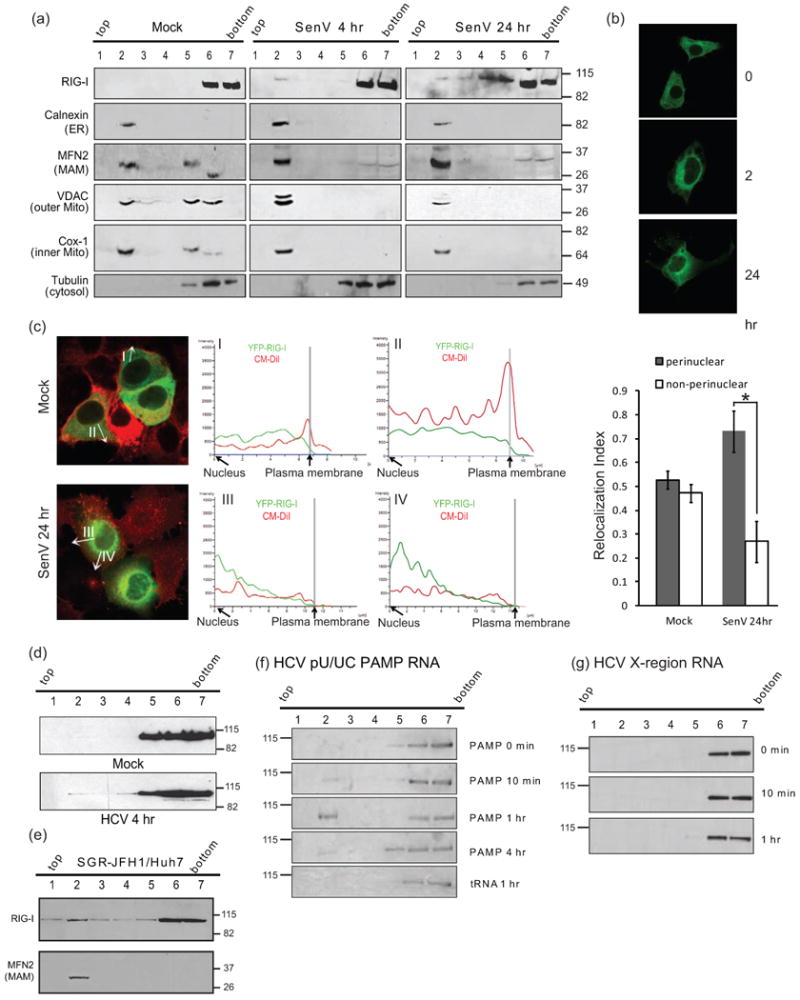
Translocation of RIG-I during acute virus infection. (a) Acute SenV infection. Membrane markers were monitored with RIG-I after 4 and 24 hr of infection of Huh7 cells. Fraction numbers and their gradient positions are noted at the top. Fractions 2 and 3 contain membranes. Fractions 5-7 contain cytosol. (b) Redistribution of YFP-RIG-I over an acute timecourse of SenV infection. (c) Intracellular distribution of YFP-RIG-I (green) in mock-infected cells and in cells 4 hrs after SenV infection. Graphs show the levels of YFP-RIG-I present along the vector marked in each fluorescent micrograph (white arrow) spanning from nucleus to plasma membrane. Relocalization index desmonstrates a significant RIG-I redistribution during acute virus infection. Error bars represent +/- standard deviation.*, p value <0.05. (d) Gradient position of Flag-RIG-I in Huh7 cells after mock infection or 4 hr HCV infection. (e) RIG-I distribution in gradients from Huh7-HCV replicon (SGR-JFH1) cells. (f) Gradient position of Flag-RIG-I in Huh7 cells 10 min to4 hr after transfection of cells with HCV (PAMP) RNA and (g) X-region motif RNA.
We next assessed RIG-I distribution during acute HCV infection in Huh7 cells expressing Flag-RIG-I. Immunoblot analysis of membrane flotation fractions revealed that a portion of RIG-I redistributed to membrane fractions within 4 hrs of HCV infection (Fig. 1d). RIG-I membrane association was also observed in Huh7 cells harbouring a subgenomic HCV replicon RNA (Fig. 1e). We therefore evaluated whether or not HCV PAMP RNA is sufficient to stimulate the intracellular redistribution of RIG-I. Huh7 cells expressing Flag-RIG-I were transfected with in vitro transcribed RNA containing 5′ppp and encoding the poly-U/UC PAMP or the non-stimulatory X-region RNA motif of the HCV RNA genome (Saito et al., 2008). RIG-I distribution among gradient fractions was then evaluated by immunoblot analysis. Remarkably, Flag-RIG-I exhibited redistribution to membrane fractions within 10 min, peaking at 1 hr, and re-accumulating in the cytosol fractions by 4 hrs after cells were transfected with poly-U/UC PAMP RNA but not X-region RNA (Fig. 1, f and g). The redistribution of RIG-I corresponded with an accumulation of poly-U/UC PAMP but not X-region RNA within the membrane fraction of transfected cells (Supplementary Fig. 1c), reflecting the formation of PAMP RNA/RIG-I complex and further demonstrating RIG-I ligand-binding specificity (Saito et al., 2008). These results reveal that ligand binding by RIG-I triggers its cytosol-to-membrane redistribution during acute HCV infection, and demonstrate that RIG-I redistribution is triggered upon binding to PAMP RNA.
Requirements for RIG-I membrane association
Recent studies show that RIG-I is recruited to the MAM where it binds MAVS to initiate innate immune signaling (Horner et al., 2011). To determine if RIG-I redistribution was dependent on MAVS we evaluated the subcellular distribution of RIG-I within Huh7 cells stably expressing a non-targeting vector shRNA (NT-Huh7) or shRNA targeting MAVS. In both cell lines RIG-I was relocalized to the membrane fractions within 4 hrs and through 24 hrs post-infection with SenV (Fig. 2a), indicating that MAVS is not required for RIG-I relocalization and membrane association. This finding is consistent with the presence of membrane-associated RIG-I in HCV replicon cells (Fig. 1e), in which MAVS is cleaved from the MAM by the HCV protease NS3/4A during chronic HCV replication (Horner et al., 2011; Loo et al., 2006). It is therefore likely that RIG-I is recruited to a membrane domain prior to interacting with MAVS during viral infection.
Figure 2.
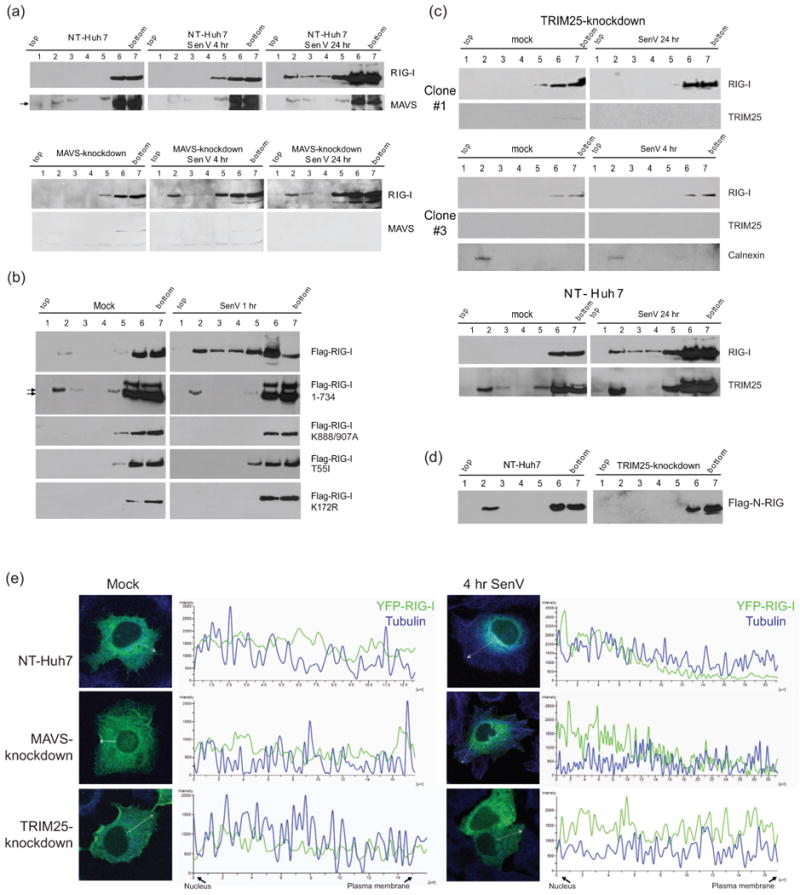
Requirements for RIG-I translocation. (a-d) Protein distribution across membrane flotation gradients was monitored by immunoblot assay. (a) NT-Huh7 and MAVS-knockdown Huh7 cells after 4 or 24 hr SenV infection or mock infection. (b) Distribution of wtor mutant FLAG-RIG-I in Huh7 cells after 1 hr of mock-infection (left) or SenV-infection (right). Arrows denote positions of FLAG-RIG-I 1-734. (c) Distribtion of endogenous RIG-I and TRIM25 from within TRIM25-knockdown (upper) and NT-Huh7 cells (lower). Cells were mock-infected (left) or infected with SenV for 4 or 24 hr prior to harvest. The same blots were stripped and re-probed to detect RIG-I and TRIM25. (d) Distribution of constitutively active FLAG N-RIG mutants expressed in NT control or TRIM25-knockdown Huh7 cells. (e) Distribution of YFP-RIG-I (green) and tubulin (blue) in NT control, MAVS-knockdown, and TRIM25-knockdown Huh7 cells. Mock-infected SenV-infected cells were imaged by confocal microscopy and analyzed for the fluorescence intensity across the sections indicated by white vectors.
To define the requirements for RIG-I redistribution, we assessed the subcellular distribution of Flag-tagged wild-type (wt) and mutant RIG-I constructs expressed in Huh7 cells. Because RIG-I is held in an inactive conformation in the absence of bound RNA ligand (Jiang et al., 2011; Saito et al., 2007), ectopically expressed wt Flag-RIG-I does not mediate significant signaling in the absence of virus infection and thus maintains its cytosolic location (Fig. 2b). These observations also imply that the membrane relocalization of RIG-I during acute viral infection is not merely driven by an increase in RIG-I abundance that results from innate immune signaling. However, wt Flag-RIG-I redistributed to a membrane association within 1 hr of SenV infection (Fig. 2b). Moreover, Flag-RIG-I 1-734, which is truncated to lack the RD and is constitutively active in signaling (Saito et al., 2007), was membrane-associated when expressed in Huh7 cells regardless of virus infection (Fig. 2b). In contrast, the RNA binding defective mutant, Flag-RIG-I K888/907A(Takahasi et al., 2008), failed to redistribute to membrane fractions after SenV infection. Importantly, RIG-I T55I and K172R mutants, which are essential for TRIM25 binding and ubiquitination (Gack et al., 2007), also failed to redistribute upon SenV infection (Fig. 2b). Thus, inactive or “signaling-off” RIG-I resides in the cytosol but upon activation during acute virus infection RIG-I translocates to a membrane compartment in a manner dependent upon RNA binding and controlled by the RD, TRIM25 interaction, and K172 ubiquitination.
We found that TRIM25 was typically distributed within both cytosol and membrane fractions in either Huh7 cells that encode wt RIG-I or Huh7.5 cells that encode the T55I RIG-I mutant regardless of virus infection, suggesting that TRIM25 localization itself is not regulated through interaction with RIG-I (Supplementary Fig. 2a), though membrane-associated TRIM25 may be involved in other processes which operate independently of RIG-I signaling. We conducted membrane flotation analysis of NT-Huh7 cells and TRIM25-knockdown stable Huh7 cells to assess whether TRIM25 is required for RIG-I relocalization during viral infection. As shown in Fig. 2c, loss of TRIM25 expression resulted in abrogation of virus-induced cytosol-to-membrane redistribution of RIG-I. Furthermore, in TRIM25-knockdown Huh7 cells, constitutively-active Flag-N-RIG (see Fig. 3b; Sumpter et al., 2005) remained localized in the cytosol (Fig. 2d). The absence of TRIM25 but not MAVS expression resulted in an inability of YFP-RIG-I to redistribute from cytosol to membrane upon SenV infection (Fig. 2e). In mock-infected NT-Huh7 cells, RIG-I distributed to the cytosol, with TRIM25 distributed in both cytosol and membrane compartments (Fig. 2c). We found that virus-induced RIG-I redistribution was followed by induction of IRF-3 phosphorylation in NT-Huh7 cells but not in TRIM25-knockdown Huh7 cells (Supplementary Fig. 2b). Thus, TRIM25 is necessary for virus-induced RIG-I translocation and innate immune signaling. Since the constitutive membrane distribution of TRIM25 itself is not sufficient to confer cytosol-to-membrane redistribution of RIG-I (see Fig. 2c; Supplemental Fig. 4), the translocation activity of the RIG-I/TRIM25 complex is likely mediated by additional components of a RIG-I translocation complex or “RIG-I translocon”.
Figure 3.

14-3-3ε is a RIG-I binding partner in a RIG-I translocon containing TRIM25. (a) Complex formation between endogenous RIG-I, 14-3-3ε, and TRIM25. The recovered products from each immunoprecipitation reaction (IP) or input extract (lysate) were analyzed by immunoblot assay for RIG-I, TRIM25 and 14-3-3ε. (b) RIG-I mutants used in (c) and (e). Domain structure of RIG-I is shown with sites of the specific mutations. The region encoding the N-RIG construct is in brackets. (c) Impaired 14-3-3ε interaction of RIG-I point-mutants. HEK293 cells expressing Flag-tagged wt or mutant RIG-I were co-expressed with Myc-14-3-3ε and cells were infected with 0 to 200 HAU SenV for 16 hr follwed by anti-Flag immunoprecipitation of cell extracts. The products were immunoblotted with anti-TRIM25 or anti-Myc antibody. (d) Anti-Flag immunoprecipitates of RIG-I(wt) from (c) were eluted and subjected to anti-Myc immunoprecepitation. (e) Mapping of the 14-3-3ε binding domain in RIG-I. Flag-tagged wt RIG-I or RIG-I mutants were co-expressed with Myc-14-3-3ε in 293 cells. Cells were mock-infected (-) or infected with SenV (+). Cell extracts were subjected to immunoprecipitation with anti-FLAG antibody and products were analyzed by immunoblot with anti-Myc antibody for 14-3-3ε. (f) and (g), Distribution across membrane floatation gradients was monitored by immunoblot assay. Cells were mock-infected or infected with SenV for 4 or 16 hr prior to harvest. (f) Distribution of endognous 14-3-3σ and 14-3-3ε from Huh7 cells. (g) Distribution of RIG-I, 14-3-3ε, and TRIM25 in NT- or 14-3-3ε-knockdown Huh7 cells.
14-3-3ε is an essential component of the RIG-I translocon
To identify additional components that might participate in the cytosol-to-membrane translocation of a RIG-I translocon we conducted mass spectrometry analysis to define proteins recovered within anti-FLAG immunoprecipitation of extracts from Huh7 cells expressing FLAG-N-RIG or irrelevant FLAG-tagged protein. A known mitochondrial targeting chaperone protein, 14-3-3ε (Dougherty and Morrison, 2004), was identified by proteomics analysis of recovered proteins that specifically associated with FLAG-N-RIG but not with irrelevant FLAG-control (Supplementary Table 1). In co-immunoprecipitation experiments using anti-RIG-I antibody we confirmed that 14-3-3ε could form a complex with RIG-I that was further enhanced by SenV infection (Fig. 3a). Remarkably, we also found that this complex contained increasing amounts of TRIM25 during the course of infection. RIG-I and 14-3-3ε could bind to TRIM25 in a similar virus-induced manner, as revealed by immunoblot analysis of anti-TRIM25 immunoprecipitation products (Fig. 3a). TRIM25 is known to bind to 14-3-3σ, an ubiquitin and ISG15 ligase (Zhang and Zhang, 2011), suggesting it may bind to a variety of 14-3-3 isoforms. To evaluate TRIM25 interaction specificity with 14-3-3ε we conducted anti-TRIM25 co-immunoprecipitation and immunoblot analysis of extracts from Huh7 cells co-expressing Myc-TRIM25 and Flag-14-3-3ε, 14-3-3η, or 14-3-3σ (binding control). As shown in Supplementary Fig. 3a, 14-3-3ε formed a stable virus-induced complex with TRIM25 whereas 14-3-3η and 14-3-3σ exhibited constitutive TRIM25 binding. These results demonstrate that RIG-I, 14-4-3ε, and TRIM25 interact and form a complex during acute virus infection.
To define the site(s) of 14-3-3ε binding within RIG-I, and determine if RIG-I/14-4-3ε/TRIM25 form a virus-inducible ternary complex, we assessed Myc-14-3-3ε binding to wt Flag-RIG-I and various FLAG-RIG-I mutants in mock-infected or SenV-infected cells. Neither 14-3-3ε nor TRIM25 could bind to RIG-I K888/907A, indicating that PAMP RNA binding and resulting conformational change of RIG-I promote formation of the translocon (Fig. 3c). Importantly, while 14-3-3ε bound efficiently to RIG-I S8A phosphorylation mutant (Fig. 3e) (Nistal-Villan et al., 2010), the interactions of 14-3-3ε with RIG-I T55I or RIG-I K172R were impaired (Fig. 3c). As the latter two RIG-I mutants lack the ability to bind or to be ubquitinated by TRIM25, respectively (Gack et al., 2008; Gack et al., 2007), TRIM25 binding and modification of RIG-I are thereby essential to stabilize the interaction of RIG-I with 14-3-3ε, suggesting that the proteins can form a ternary complex. To confirm that 14-3-3ε, RIG-I, and TRIM25 form a virus-inducible ternary complex, Myc-14-3-3ε and Flag-RIG-I were co-expressed and co-immunoprecipitated by anti-Flag antibody. The immunoprecipitates were then eluted by Flag peptide competition and then further subjected to a second round of immunoprecipitation with anti-Myc antibody. After two rounds of immunoprecipitation 14-3-3ε, RIG-I, and TRIM25 were recovered in a stable complex (Fig. 3d), indicating ternary complex formation in response to acute virus infection. Furthermore, 14-3-3ε bound only to wt Flag-RIG-I and N-RIG, and binding required the RIG-I CARDs but neither the helicase domain nor the RD (Fig. 3e).
To determine if 14-3-3ε plays a specific role in RIG-I translocation during virus infection, we monitored distribution of14-3-3ε and 14-3-3σ (as a control) from membrane flotation analysis. 14-3-3σ was only found in the cytosolic fractions of cells; in contrast, 14-3-3ε was localized to both membrane and cytosolic fractions regardless of infection (Fig. 3f). We note that a preliminary proteomics analysis of cell fractions showed that 14-3-3ε and TRIM25 but not 14-3-3σ are components of MAM and ER in Huh7 cells (data not shown). These results are consistent with previous studies of 14-3-3ε defining it as a MAM/mitochondrial-targeting chaperone protein, and indicate that 14-3-3ε is a component of the RIG-I translocon (Aslan et al., 2009). Indeed SenV infection could induce IRF-3 phosphorylationin 14-3-3σ-knockdown Huh7 cells but not in 14-3-3ε-knockdown Huh7 cells (Supplementary Fig. 3b), demonstrating the specificity of 14-3-3ε function in RIG-I-dependent signaling. We therefore assessed membrane association of RIG-I during SenV infection of NT-Huh7 cells and in 14-3-3ε knockdown cells. In NT-Huh7 cells the relocalization of RIG-I from cytosol to membrane occurred within 24 hr of infection but RIG-I translocation failed to occur in 14-3-3ε-knockdown cells (Fig. 3g). Moreover, when expressed in 14-3-3ε-knockdown cells YFP-RIG-I failed to redistribute during SenV infection (Supplementary Fig. 3c). These results indicate that 14-3-3ε forms an essential complex with RIG-I and TRIM25 to facilitate the cytosol-to-membrane translocation of RIG-I during acute virus infection. 14-3-3ε, TRIM25, and RIG-I therefore comprise the RIG-I translocon.
The RIG-I translocon is essential for antiviral innate immunity
To assess the role of the RIG-I translocon in controlling virus infection we first evaluated RIG-I signaling actions in Huh7 and HEK293 cells that ectopically co-express 14-3-3ε and TRIM25. The co-expression of 14-3-3ε and TRIM25 significantly enhanced SenV signaling to the IFN-β promoter activity during acute infection whereas expression of either protein alone did not enhance signaling (Fig. 4a-b). 14-3-3ε/TRIM25 co-expression also enhanced ISG56 mRNA induction by SenV (Fig. 4a). Thus, 14-3-3ε and TRIM25 cooperatively serve as accessory proteins of RIG-I signaling. We also assessed the IFN-β promoter activity during SenV infection of 14-3-3ε knockdown cells. As shown in Fig. 4c, innate immune signaling was attenuated in both 14-3-3ε- and TRIM25-knockdown cells but remained intact in the NT control HEK293 cells. Additionally, in HEK293 and Huh7 cells expressing either NT shRNA or shRNA targeting 14-3-3σ, N-RIG expression induced robust signaling to the IFN-β promoter but signaling was specifically blunted in 14-3-3ε- or TRIM25-knockdown Huh7 cells (Supplementary Fig. 4a-b). This decrease in IFN-β promoter activity correlated with increased susceptibility to infection with vesicular stomatitus virus expressing GFP (VSV-GFP) of both 14-3-3ε and TRIM25-knockdown cells as compared to NT control cells (Fig. 4d). Importantly, when infected with HCV, 14-3-3ε-knockdown or TRIM25-knockdown Huh7 cells both exhibited significantly enhanced permissiveness for infection as compared to NT-Huh7 cells (Fig. 4e). Thus, the RIG-I translocon is essential for innate immune signaling and host defence against RNA virus infection. To define the level within the RIG-I pathway that 14-3-3ε and TRIM25 operate, we evaluated IFN-β promoter signaling in 14-3-3ε- or TRIM25-knockdown Huh7 cells upon ectopic expression of N-RIG, MAVS or constitutively active IRF-3 (IRF-3-5D). In contrast to N-RIG, and despite that MAVS signaling was decreased in14-3-3ε-knockdown cells, both MAVS and IRF-3-5D could signal to the IFN-β promoter in the absence of 14-3-3ε or TRIM25 expression (Supplementary Fig.4 b-d). We note that the reduction in signaling by ectopic MAVS could reflect an additional yet uncharacterized role for 14-3-3ε in MAVS-specific signaling processes that are independent of RIG-I. Together these results imply that like TRIM25, 14-3-3ε is essential for RIG-I signaling of innate immunity, and that 14-3-3ε and TRIM25 both operate within the RIG-I pathway at a point between RIG-I and MAVS to direct downstream signaling from the level of RIG-I itself.
Figure 4.
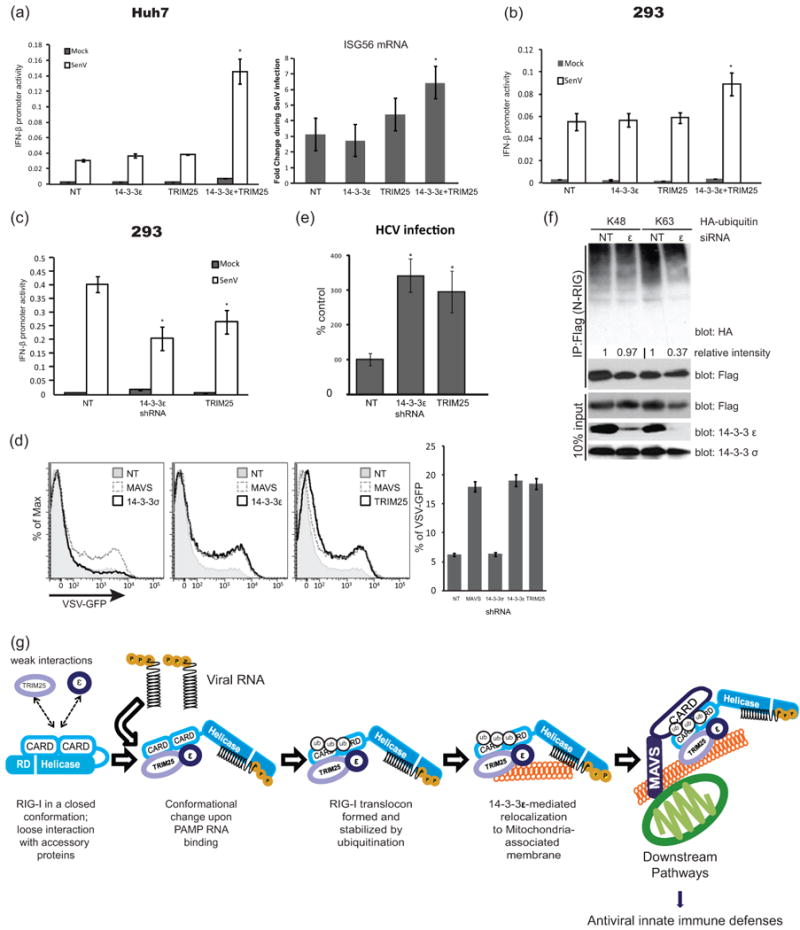
14-3-3ε is essential for antiviral signaling activity of the RIG-I translocon. Ectopic expression of both 14-3-3ε and TRIM25 in (a) Huh7 and (b) HEK293 cells enhanced IFN-β promoter activity and ISG56 mRNA expression during SenV infection. (c) IFN-β promoter activity in NT-control, 14-3-3ε-knockdown, or TRIM25-knockdown 293 cells 24 hr after SenV infection. (d) VSV-GFP positive cells of 14-3-3σ-knockdown, 14-3-3ε-knockdown or TRIM25-knockdown Huh7 cells were anlyzed by flow cytometryafter 12 hr of VSV-GFP infection at MOI 0.01. (e) Levels of HCV-infected cells in cultures of NT-, 14-3-3ε-knockdown or TRIM25-knockdown-Huh7 cells were determined by focus forming assay after 48 hr HCV infection. (f) 14-3-3ε is required for K63 ubquitination of the RIG-I CARDs. Flag-N-RIG was co-expressed with HA-tagged K48 or K63 ubiquitin in Huh7 cells transfected with NT-control or 14-3-3ε-specific siRNA. Cell extracts were subject to immunoprecipitation with anti-Flag antibody. Ubiquitination level of N-RIG products was evaluated by anti-HA immunoblot, quantified using Image J software, and expressed as a value relative to the corresponding NT-control. Error bars represent +/- standard deviation. *, p value <0.05; **, p value <0.01. (g) Model of RIG-I translocon assembly and function.
14-3-3ε is known to modulate ubiquitin ligase activity (Nagaki et al., 2006). Since TRIM25 functions as the E3 ubiquitin ligase of K63-linked ubiquitination of the RIG-I CARDs for signaling activation (Gack et al., 2007), we evaluated the role of 14-3-3ε in regulating the ubiquitination of Flag-N-RIG when co-expressed with HA-tagged K48 or K63 ubiquitin in NT-Huh7 control cells or 14-3-3ε-knockdown cells. Immunoblot analysis of anti-Flag immunoprecipitation products recovered from the cell extracts revealed differential levels of N-RIG-ubiquitin products. Whereas N-RIG-K63 ubiquitin products were overall reduced in 14-3-3ε-knockdown cells, N-RIG -K48 ubiquitin products remained unchanged between NT-Huh7 and 14-3-3ε-knockdown cells (Fig. 4f). Notably, the reduction of N-RIG-K63 ubiquitin products corresponds with the loss of immune signaling and enhanced permissiveness to virus infection imposed by loss of 14-3-3ε expression in knockdown cells (compare Figs. 4a-e). These results indicate that 14-3-3ε interaction with RIG-I serves to stimulate or stabilize TRIM25 modification of the RIG-I translocon by promoting RIG-I-K63 ubiquitination, translocon interactions and innate immune signaling.
Discussion
RIG-I translocation to the MAM/mitochondrial interface is essential for MAVS interaction, downstream activation of IRF-3 (Horner et al., 2011), and the onset of innate antiviral immunity. Our results reveal that this process is mediated through a RIG-I translocon involving 14-3-3ε, TRIM25, and ubiquitination of RIG-I. 14-3-3 proteins are involved in a wide range of cellular processes by serving as scaffold or chaperone proteins (Muslin et al., 1996; Yang et al., 2006). In particular, 14-3-3ε facilitates the cytoplasm-to-mitochondrion membrane shuttling of BAD and BAX (Nomura et al., 2003). Our results now define 14-3-3ε as an essential cofactor of the membrane distribution of RIG-I for immune signaling against RNA viruses. RIG-I activation is initiated upon binding of PAMP RNA to release RD autorepression of CARD signaling (Saito et al., 2007). Our results infer that RD control of CARD exposure also promotes interaction with 14-3-3ε and TRIM25 for translocon formation. In the case of HCV, this process occurs minutes after cell expression of PAMP RNA (Fig. 1). We propose that RIG-I modification by TRIM25 serves to sustain its “signaling-on” conformation initiated by ligand-binding to the RD, and that 14-3-3ε then facilitates stable RIG-I-TRIM25 interaction/ubiquitination of RIG-I (Gack et al., 2007) and translocon activity to bind MAVS for downstream innate immune signaling (Fig. 4g), consistent with both biochemical and structural models of RIG-I activation (Jiang et al., 2011; Saito et al., 2007). Governance of RIG-I translocon activity by PAMP RNA binding, K63 ubiquitination, and 14-3-3ε chaperone function offers multiple checkpoints for control of the immune response.
RIG-I is the prototypical member of the RLR family, which also includes melanoma differentiation- associated gene 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2). While MDA5 has been shown to act as an essential PRR for recognition of specific viruses both independently of and cooperatively with RIG-I, LGP2 serves as a regulator of RLR signaling (Komuro and Horvath, 2006; Saito et al., 2007; Satoh et al., 2010). Similar to RIG-I, MDA5 and LGP2 are cytosolic proteins and effect innate immune signaling control through MAVS interaction (Loo and Gale Jr, 2011), which is expected to involve their cytosol-to-membrane redistribution. One scenario for this redistribution is that the RLRs share a common process of virus-induced translocon assembly and function that is triggered by ligand binding and regulated by signaling-adapter protein interaction. As 14-3-3 proteins themselves can function as signaling-adaptor proteins whose actions are modified by their phosphorylation state, it is possible that RIG-I translocon assembly is further governed by 14-3-3ε phosphorylation or other modifications (Dougherty and Morrison, 2004). In this sense translocon assembly could be responsive to a variety of virus-induced signals that serve to trigger or enhance innate immunity. In addition to altering protein localization, the 14-3-3 proteins are also involved in modulating enzymatic activity, and regulating protein modification (Dougherty and Morrison, 2004), consistent with the function of 14-3-3ε in promoting RIG-I ubiquitination by TRIM25 and supporting translocon activity. Moreover, 14-3-3 proteins have been implicated in pathogen resistance of both mammalian and plant cells (Brandt et al., 1992; Ohman et al., 2010). Together with our current observations, these studies implicate a broad role for 14-3-3 family proteins in eukaryotic innate immunity.
Regulation of innate immunity and RIG-I signaling controls the outcome of RNA virus infection and immunity (Loo and Gale Jr, 2011). RIG-I signaling is induced during acute HCV infection as the virus spreads from cell to cell in the liver wherein it drives a hepatic innate immune response aimed at suppressing HCV infection (Liu and Gale, 2010). To evade hepatic immunity, HCV directs the proteolysis of MAVS by the viral NS3/4A protease to release it from the MAM and ablate RIG-I signaling, thus abrogating the innate immune actions of RIG-I (Horner et al., 2011; Loo et al., 2006; Saito et al., 2008). Other studies show that HCV core protein can interact with 14-3-3ε to induce Raf-1 kinase activity and to regulate the release of BAX from mitochondria, thereby imposing control of host cell signaling and apoptosis (Aoki et al., 2000; Lee et al., 2007). These observations underscore an important role for 14-3-3ε in HCV infection and pathogenesis. Other pathogenic RNA viruses also target RIG-I or its signaling partners for regulation and immune evasion (Loo and Gale Jr, 2011). A notable example is influenza A virus of which the NS1 protein of certain strains can bind to RIG-I to disrupt complex formation with TRIM25 and block innate immune signaling (Gack et al., 2009), suggesting that it may disrupt the actions of the RIG-I translocon. Targeting 14-3-3ε function and virus-responsive signaling networks of RIG-I translocon regulation may therefore offer attractive therapeutic strategies to modulate immune signaling for the control of RNA virus infection.
Experimental Procedures
Cells
Huh7 and Huh7.5 (human hepatoma), and HEK293 cells have been described (Saito et al., 2008). MAVS, TRIM25, 14-3-3ε, and 14-3-3σ-knock-down cells were produced through transduction and selection of cells with gene-specific pLKO.1-puro vector-based shRNA expression cassettes purchased from Sigma.
Mass-spectrometry and protein identification
Products recovered from immunoprecipitation reactions of cell extracts were processed for peptide analysis and peptides were separated by nano-HPLC using a 5cm Magic C18 Column (Michrom Bioresources, Auburn, CA) using a buffer system of (A) water + 0.1% formate and (B) acetonitrile + 0.1% formate (Avantor Performance Materials, Phillipsburg, NJ) in 60 minute gradient from 98% A / 2% B to 5% A / 95% B. Peptides were detected using a linear ion trap mass spectrometer (LTQ, Thermo Fisher Scientific). An initial mass scan from 400-2000 m/z was followed by 4 data dependent collision induced dissociation MS/MS scans for peptide identifications. Normalized collision energy of 35% was used for CID. MS/MS data was searched against the human IPI v3.28 database using SEQUEST. The results of these searches were analyzed using Peptideprophet and Proteinprophet to determine the likelihood of a confident protein identification. Only proteins with a probability greater than 0.95 were considered.
Membrane floatation assay
Membrane preparation was performed as previously described (Liu et al., 2009). Cell lysates in 0.5 ml of hypotonic buffer were mixed with 3 ml of 72% sucrose in low-salt buffer (LSB) and overlaid with 4 ml of 55% sucrose in LSB, followed by addition of 1.5 ml of 10% sucrose in LSB and centrifugation at 38,000 rpm in a Beckman SW41Ti rotor for 14 h at 4°C. Fractions were collected from the top of the gradient, diluted with PBS, and further concentrated by an Amicon YM-30 filter unit (Millipore).
Virus infection
Huh7 cells were cultured infected with SenV, HCV (strain HCV 2a/JFH1) (Wakita and Kato, 2006) or VSV-GFP (Loo et al., 2006) in serum-free media at 37° C. One hour later, the cells were rinsed in PBS and incubated in media containing 10% FBS. At the time of harvest, cells were washed twice with PBS and fixed in 3% Paraformadehyde:PBS solution. Cells were immunostained with anti-HCV serum or visualized for GFP as described (Loo et al., 2006).
Supplementary Material
Highlights.
RIG-I undergoes cytosol-to-membrane translocation during acute virus infection.
RNA ligand binding and CARD domains of RIG-I are required for RIG-I translocation.
14-3-3ε is an essential chaperone protein component of the RIG-I translocon.
RIG-I/14-3-3ε/TRIM25 translocon is essential for antiviral innate immunity.
Acknowledgments
We thank our colleagues for reagents, and members of the Gale laboratory for discussion. Supported by NIH grants AI060389, AI88778, DA024563.
Footnotes
Additional methods are found in Supplementary Materials.
Author Contributions. H.M.L. conducted molecular studies of RIG-I membrane-association, protein-interaction, and signaling analyses. Y.M.L., G.A.Z., and M.G.K. conducted mass-spectrometry. S.M.H generated MAVS-knockdown cells and provided insightful discussions. M.G. directed the research. H.M.L. and M.G. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki H, Hayashi J, Moriyama M, Arakawa Y, Hino O. Hepatitis C virus core protein interacts with 14-3-3 protein and activates the kinase Raf-1. J Virol. 2000;74:1736–1741. doi: 10.1128/jvi.74.4.1736-1741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan JE, You H, Williamson DM, Endig J, Youker RT, Thomas L, Shu H, Du Y, Milewski RL, Brush MH, et al. Akt and 14-3-3 control a PACS-2 homeostatic switch that integrates membrane traffic with TRAIL-induced apoptosis. Mol Cell. 2009;34:497–509. doi: 10.1016/j.molcel.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt J, Thordal-Christensen H, Vad K, Gregersen PL, Collinge DB. A pathogen-induced gene of barley encodes a protein showing high similarity to a protein kinase regulator. Plant J. 1992;2:815–820. [PubMed] [Google Scholar]
- Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2009;11:133–138. doi: 10.1038/embor.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Morrison DK. Unlocking the code of 14-3-3. J Cell Sci. 2004;117:1875–1884. doi: 10.1242/jcs.01171. [DOI] [PubMed] [Google Scholar]
- Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci U S A. 2008;105:16743–16748. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Sugiyama K, Mizutani T, Tanaka T, Tanaka K, Sekihara H, Shimotohno K, Kato N. Human hepatocyte clonal cell lines that support persistent replication of hepatitis C virus. Virus Res. 1998;56:157–167. doi: 10.1016/s0168-1702(98)00063-x. [DOI] [PubMed] [Google Scholar]
- Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale M, Patel SS, Marcotrigiano J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro A, Horvath CM. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol. 2006;80:12332–12342. doi: 10.1128/JVI.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Park SO, Joe CO, Kim YS. Interaction of HCV core protein with 14-3-3epsilon protein releases Bax to activate apoptosis. Biochem Biophys Res Commun. 2007;352:756–762. doi: 10.1016/j.bbrc.2006.11.098. [DOI] [PubMed] [Google Scholar]
- Liu HM, Gale M. Hepatitis C Virus Evasion from RIG-I-Dependent Hepatic Innate Immunity. Gastroenterol Res Pract. 2010;2010:548390. doi: 10.1155/2010/548390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HM, Aizaki H, Choi KS, Machida K, Ou JJ, Lai MM. SYNCRIP (synaptotagmin-binding, cytoplasmic RNA-interacting protein) is a host factor involved in hepatitis C virus RNA replication. Virology. 2009;386:249–256. doi: 10.1016/j.virol.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Gale M., Jr Immune Signaling by RIG-I-like Receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Nagaki K, Yamamura H, Shimada S, Saito T, Hisanaga S, Taoka M, Isobe T, Ichimura T. 14-3-3 Mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase Nedd4-2 and epithelial Na+ channels. Biochemistry. 2006;45:6733–6740. doi: 10.1021/bi052640q. [DOI] [PubMed] [Google Scholar]
- Nistal-Villan E, Gack MU, Martinez-Delgado G, Maharaj NP, Inn KS, Yang H, Wang R, Aggarwal AK, Jung JU, Garcia-Sastre A. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-beta production. J Biol Chem. 2010;285:20252–20261. doi: 10.1074/jbc.M109.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H, Tsujimoto Y. 14-3-3 Interacts directly with and negatively regulates pro-apoptotic Bax. J Biol Chem. 2003;278:2058–2065. doi: 10.1074/jbc.M207880200. [DOI] [PubMed] [Google Scholar]
- Ohman T, Lietzen N, Valimaki E, Melchjorsen J, Matikainen S, Nyman TA. Cytosolic RNA recognition pathway activates 14-3-3 protein mediated signaling and caspase-dependent disruption of cytokeratin network in human keratinocytes. J Proteome Res. 2010;9:1549–1564. doi: 10.1021/pr901040u. [DOI] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M., Jr Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A. 2010;107:1512–1517. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M, Jr, Inagaki F, Fujita T. Nonself RNA-Sensing Mechanism of RIG-I Helicase and Activation of Antiviral Immune Responses. Molecular Cell. 2008;29:428–440. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Yang X, Lee WH, Sobott F, Papagrigoriou E, Robinson CV, Grossmann JG, Sundstrom M, Doyle DA, Elkins JM. Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc Natl Acad Sci U S A. 2006;103:17237–17242. doi: 10.1073/pnas.0605779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. J Interferon Cytokine Res. 2011;31:119–130. doi: 10.1089/jir.2010.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.