

Abstract
(S)-5-Fluoro-2-(2,2,6,6-tetramethylpiperidin-1-yloxymethyl)-1-tosylindoline, a 2-methyleneoxy-substituted chiral indoline, was synthesized on multigram scale using an efficient copper-catalyzed enantioselective intramolecular alkene aminooxygenation. The synthesis is accomplished in four steps and the indoline is obtained in 89% ee (>98% after one recrystallization). Other highlights include efficient gram-scale synthesis of the (4R,5S)-di-Ph-box ligand and efficient separation of a monoallylaniline from its bis(allyl)aniline by-product by distillation under reduced pressure.
Keywords: indoline, aminooxygenation, copper-catalyzed, alkene, TEMPO
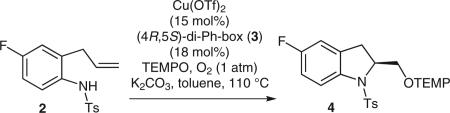
Chiral indolines are found in a number of bioactive compounds.1 While a number of chiral indoline syntheses exist, many involve resolution of racemates or use of stoichiometric chiral substrates and auxiliaries.1 We have recently reported that N-sulfonyl-2-allylanilines react under copper catalysis to provide a range of functionalized chiral indolines where the specific functionalization depends upon the reaction components.2 In 2008, we reported the first catalytic enantioselective intramolecular alkene aminooxygenation using (2,2,6,6-tetramethylpiperdin-1-yl)oxyl (TEMPO) radical as the oxygen source.2a Herein is reported a four step, five gram synthesis of (S)-5-fluoro-2-(2,2,6,6-tetramethylpiperidin-1-yloxymethyl)-1-tosylindoline (4) (Scheme 1). This method makes use of ambient pressure O2 as a green oxidant and the procedure has been optimized beyond our initial report2a for reaction scale, catalyst loading, and reaction time. We have previously shown that the N–O bond of the TEMPO adducts can be reduced with Zn (MeOH, aq NH4Cl, 70 °C) to liberate the free alcohol without diminishing enantiopurity (Scheme 2).2a
Scheme 1.

Copper-catalyzed enantioselective indoline synthesis
Scheme 2.

Reduction of the N–O bond gives the alcohol
We were particularly interested in demonstrating the method starting with a substituted aniline, as the unsubstituted (S)-(+)-2-indolinemethanol is commercially available. Our synthesis of N-allyl-4-fluoroaniline (1) required implementation of a simple vacuum distillation procedure for its separation from 4-fluoroaniline and its N-bis(allyl)-4-fluoroaniline side product. The separation of mono-, di-, and trialkylamines can be a challenge3 and we demonstrate that vacuum distillation can address this challenge in this instance.
A larger-scale preparation of the (4R,5S)-di-Ph-box (box = bisoxazoline) ligand 3 via a simple dialkylation of commercially available 2,2′-methylenebis[(4R,5S)-4,5-diphenyl-2-oxazoline] is also described here.2c,4
Our originally reported catalytic enantioselective intra-molecular aminooxygenation of N-sulfonyl-2-allylani-lines provided for the synthesis of a number of substituted chiral indolines (e.g., 4) with good to excellent yield and moderate to excellent enantioselectivities.2aN-Tosyl substrates provided higher reactivity, yields, and enantioselectivities than their N-nosyl and N-mesyl counterparts, and aniline substituents such as F, Cl, OMe, and CN were all tolerated.2a In that report, the reaction scales were limited to 40–50 mg (ca. 0.13 mmol) of sulfonamide (e.g., 2) and TEMPO radical (3 equiv) served as both the oxygen atom source and the oxidant (Table 1, entry 1).2a It was subsequently found that upon scale-up to 0.33 mmol the reactions did not go to completion (Table 1, entry 2). A similar reactivity problem with less reactive γ-pentenylsulfonamide substrates was solved by introducing an O2 atmosphere (1 atm, balloon) and this was found to be a general solution to the indoline scale-up problem (Table 1, entry 3).2a,5 The reaction time was reduced from 24 to 6 hours and the catalyst loading was lowered from 20 mol% Cu(OTf)2 to 15 mol% Cu(OTf)2 without diminishing efficiency or selectivity (Table 1, entries 3–7). The presence of K2CO3 is not needed (Table 1, entries 5–9). Both PhCF3 and PhMe are good solvents for the reaction, so PhMe was used in the scale-up due to its more common industrial usage (Table 1, entries 6 and 7). The use of lower O2 atmosphere was probed by using 10% O2 in N2 and it was found that at the 15 mol% Cu(OTf)2 loading, enantioselectivity was reduced (Table 1, entry 8). At 20 mol% Cu(OTf)2 loading, the level of enantioselectivity was restored (Table 1, entry 9). For the purposes of this reaction scale up, minimization of catalyst loading was deemed more important, so the largest scale (13.9 mmol) reaction was performed with 100% O2 atmosphere (1 atm, balloon, Table 1, entry 7).6
Table 1.
Optimization of the Aminooxygenationa
 | ||||
|---|---|---|---|---|
| Entry | 2 (mmol) | Time (h) | Yield (%) | ee (%) |
| 1bcd | 0.131 | 24 | 83 | 89 |
| 2bcd | 0.33 | 24 | 45 | nde |
| 3b | 0.33 | 6 | 85 | 91 |
| 4 | 0.33 | 6 | 87 | 91 |
| 5f | 0.33 | 6 | 87 | 91 |
| 6f | 3.27 | 6 | 83 | 91 |
| 7f | 13.9 | 6 | 89 | 89 |
| 8fg | 0.33 | 24 | 88 | 73 |
| 9cfg | 0.33 | 24 | 85 | 93 |
Reaction conditions: Cu(OTf)2 (15 mol%) and (4R,5S)-di-Ph-box 3 (18 mol%) were dissolved in PhMe and heated at 70 °C for 2.5 h. To the cooled mixture was added sulfonamide 2 (1 equiv), K2CO3 (1 equiv), TEMPO (3 equiv), and additional PhMe (0.08 M final concentration with respect to 2). The reaction was heated with stirring under O2 (1 atm, balloon). Yields refer to product isolated after flash chromatography on silica gel. Enantiomeric excess was determined by chiral HPLC.
Reaction was run in PhCF3.
20 mol% Cu(OTf)2 and 25 mol% ligand 3 was used.
The reaction was run under argon instead of O2.
nd = not detected.
No K2CO3 was used in this reaction.
10% O2 in N2 (1 atm, balloon) was used.
The scaled-up reaction sequence shown in Scheme 1 first involved N-allylation of 4-fluoroaniline. A 2.5:1 stoichiometry of 4-fluoroaniline to allyl bromide was used to minimize formation of the N,N-diallyl-4-fluoroaniline side product, which was harder to separate from N-allyl-4-fluoroaniline than the parent 4-fluoroaniline. The ratio of mono to diallylation product is also a function of the aniline substituent, so in some cases, for example, aniline, a stoichiometry closer to 1:1 is sufficient.7 While the crude mixture generated from this allylation of 4-fluoroaniline can be separated chromatographically, the separation is difficult and less practical on a larger scale. Distillation under vacuum (0.5 mmHg), however, did adequately separate N-allyl-4-fluoroaniline (1) and proved a very efficient method for isolating nearly pure sample (4.50 g, 75% yield). Subsequent BF3?OEt2 promoted aza-Claisen8 and N-tosylation were uneventful and provided crystalline sulfonamide 2 (6.49 g) in 75% yield over two steps.
We have observed that the (4R,5S)-diphenylbis(oxazo-line) ligand 3 provides superior reactivity and enantioselectivity, compared to the commercially available (4R)-Ph-box ligand, in challenging Cu(II)-catalyzed alkene aminofunctionalization reactions.2 This ligand was key in enabling the catalyst loading to be reduced from 20 to 15 mol%. The synthesis of this ligand has been reported previously.4a While the published route was sufficient to produce the ligand, we found that employing a dialkylation protocol initially introduced by Denmark4b can provide the ligand directly from commercially available material.2c In the event, we obtained 1.37 grams (86% yield) of ligand 3 using this protocol, which is of sufficient quantity to allow for the subsequent large scale aminooxygenation reaction. Thus, enantioselective aminooxygenation of 2 under the conditions described in Table 1, entry 7 provided 5.47–5.69 grams (86–89%) of indoline 4 in 89% yield. One recrystallization from hexanes provided 4.28 grams of indoline 4 in >98% ee as determined by chiral HPLC.
In conclusion, the very promising copper-catalyzed enantioselective intramolecular alkene aminooxygenation reaction2a has been demonstrated on a multigram scale and chiral indoline 4 was produced in high enantiomeric excess. The reaction scale-up study included optimization of solvent, time, and catalyst loading, and the scale-up problem (lack of conversion at larger scale) was solved by introduction of O2 as an environmentally benign oxidant (1 atm, balloon). Implementation of this method for the synthesis of chiral indolines is now a practical choice for synthetic organic chemists.
All reagents were used out of the bottle as purchased from the supplier without further purification, unless otherwise noted. 1H NMR spectra were recorded in CDCl3 (using 7.26 ppm for reference of residual CHCl3) at 300, 400, or 500 MHz unless otherwise noted. 13C NMR spectra were recorded in CDCl3 (using 77.0 ppm as internal reference) at 75 MHz, unless otherwise noted. IR spectra were taken neat using a Nicolet-Impact 420 FTIR. Wave numbers in cm–1 are reported for characteristic peaks. High-resolution mass spectra were obtained at SUNY Buffalo's mass spectrometry facility on a ThermoFinnigan MAT XL spectrometer. Optical rotations were obtained using a Rudolph Autopol 1 fitted with a microcell with a 100 mm path length. Melting points are reported as uncorrected.
N-Allyl-4-fluoroaniline (1)
A 250 mL round-bottomed flask equipped with a magnetic stir bar was charged with K2CO3 (12.7 g, 91.6 mmol, 1.90 equiv), sealed with a rubber septum, and purged with argon. The flask was charged with anhyd DMF (40.0 mL) and 4-fluoroaniline (11.6 mL, 121 mmol, 2.50 equiv) was added via syringe, and the mixture was stirred for 10 min. Allyl bromide (4.19 mL, 48.2 mmol, 1.0 equiv) was added via syringe over a period of 5 min. The reaction vessel was then equipped with a reflux condenser and sealed with a rubber septum, with a needle outlet exiting through a bubbler to relieve the pressure. The reaction mixture was then heated for 24 h at 70 °C. The reaction vessel was allowed to cool to r.t. before H2O (75 mL) was added and the mixture was stirred for 10 min. The mixture was extracted with EtOAc (3 × 50 mL) and the combined organic phases were washed with brine (3 × 50 mL), dried (Na2SO4, 65 g), filtered, and concentrated by rotary evaporation (20 mmHg, 60 °C) to afford a dark red oil. Purification of the crude oil by fractional distillation under vacuum afforded the 1 as a colorless oil, isolated as a 28:1 mixture of 1 and the diallylaniline; yield: 4.50 g (75%); bp 50–58 °C/0.5 mmHg.
IR (film): 3426, 1613, 1523, 1313, 1220 cm–1.
1H NMR (400 MHz, CDCl3): δ = 6.92–6.87 (m, 2 H), 6.58–6.54 (m, 2 H), 5.92 (m, 1 H), 5.31–5.16 (m, 2 H), 3.74 (dt, J = 1.6, 5.2 Hz, 2 H), 3.65 (br s, 1 H).
13C NMR (75 MHz, CDCl3): δ = 155.6 (d, J = 234.9 Hz), 144.0 (d, J = 2.3 Hz), 135.3, 116.0 (d, J = 44.9 Hz), 115.4, 113.7 (d, J = 8.1 Hz), 47.0.
HRMS-ESI: m/z [M + H]+ calcd for C9H11FN: 152.0870; found: 152.0865.
N-(2-Allyl-4-fluorophenyl)-4-methylbenzenesulfonamide (2)
An oven-dried 250 mL pressure tube (82.5 mm O.D. × 142 mm) was equipped with a stir bar, sealed with a rubber septum, and flushed with argon. The tube was charged with N-allyl-4-fluoroaniline (1; 4.50 g, 29.5 mmol, 1.0 equiv) and xylenes (70.0 mL), both added via syringe. The pressure tube was cooled to –78 °C and the solution was stirred for 10 min. BF3?OEt2 (4.08 mL, 35.4 mmol, 1.2 equiv) was added via syringe and the solution was stirred for 10 min at –78 °C, then brought to r.t. The reaction vessel was sealed with a screw cap, placed in an oil bath, and heated at 180 °C for 10 h. Upon cooling to r.t., the reaction was quenched with aq 2 M NaOH (52 mL). The mixture was extracted with EtOAc (3 × 60 mL) and the combined organic phases were washed with brine (2 × 60 mL), fil tered, dried (Na2SO4, 65 g), filtered, and concentrated by rotary evaporation (20 mmHg, 50 °C) to afford a dark red oil. Residual solvents were removed under vacuum (1 mmHg at 22 °C). The crude 2-allyl-4-fluoroaniline (4.53 g) was obtained as an oil in sufficient purity for use in the subsequent step. A 250 mL round-bottomed flask containing the crude 2-allyl-4-fluoroaniline (2; 4.50 g, 29.5 mmol, 1 equiv) was equipped with a magnetic stir bar, sealed with a septum, and purged with argon. The oil was dissolved in CH2Cl2 (29.0 mL) and treated with pyridine (7.23 mL, 88.7 mmol, 3.0 equiv), both added via syringe. This solution was stirred 10 min and then treated with TsCl (6.67 g, 34.9 mmol, 1.2 equiv). After stirring for 16 h at r.t., the reaction mixture was washed with aq 1 M HCl (3 × 60 mL). The combined aqueous layer washes were extracted with CH2Cl2 (1 × 30 mL). All organic phases were then combined and washed with brine (2 × 60 mL), dried (Na2SO4, 50 g), filtered, and concentrated in vacuo (20 mmHg, 35 °C) to afford a dark red oil. Purification by flash chromatography on silica gel (using a gradient of 0–15% EtOAc in hexanes) afforded the sulfon-amide 2 as a pale yellow solid; yield: 6.49 g (75% over two steps); mp 78 °C.
IR (film): 3263, 1590, 1497, 1167, 1093 cm–1.
1H NMR (500 MHz, CDCl3): δ = 7.50–7.48 (dd, J = 8.0, 2.0 Hz, 2 H), 7.20–7.14 (m, 3 H), 6.79 (m, 1 H), 6.73 (dd, J = 2.0, 9.0 Hz, 1 H), 6.56 (br s, 1 H), 5.63 (m, 1 H), 5.02 (dd, J = 1.5, 8.5 Hz, 1 H), 5.85 (dd, 1.5, 17.0 Hz, 1 H), 3.00 (d, J = 6.0 Hz, 2 H), 2.93 (s, 3 H).
13C NMR (75 MHz, CDCl3): δ = 161.0 (d, J = 246.0 Hz), 143.9, 136.5 (d, J = 6.9 Hz), 134.8, 130.4 (d, J = 3.4 Hz), 129.6, 128.2, 127.8 (d, J = 9.2 Hz), 127.0, 117.4, 117.0 (d, J = 23.0 Hz), 114.1 (d, J = 21.8 Hz), 35.7, 21.5.
HRMS (ESI): m/z calcd for C16H16FNO2S + Na [M + Na]+ : 328.0778; found: 328.0772.
2,2-Bis[2-(4R,5S-diphenyl-1,3-oxazolinyl)]propane (3)
An oven-dried 250 mL round-bottomed flask equipped with a stir bar was charged with 2,2′-methylenebis[(4R,5S)-4,5-diphenyl-2-oxazoline] (1.50 g, 3.27 mmol, 1 equiv) and the flask was sealed with a rubber septum, and purged with argon. The compound was dissolved in anhyd THF (100 mL) and treated with i-Pr2NEt (0.47 mL, 3.33 mmol, 1 equiv) and TMEDA (1.00 mL, 6.67 mmol, 2 equiv), added via syringe. The flask was placed in a –75 °C bath and the mixture was stirred for 5 min and then treated with aq 1.6 M n-BuLi in hexanes (4.10 mL, 6.56 mmol, 2 equiv), added dropwise via syringe. The reaction was then warmed to –20 °C over 15 min. Once at –20 °C, this temperature was maintained for 30 min, after which the temperature was lowered to –75 °C and MeI (0.42 mL, 6.73 mmol, 2 equiv) was added via syringe. Upon completion of the addition, the cold bath was removed and the reaction mixture was stirred at 22 °C under argon for 16 h. The reaction was quenched with sat. aq NH4Cl (50 mL) and diluted with H2O (25 mL) to dissolve any salts that may form. The mixture was extracted with Et2O (3 × 100 mL) and the combined organics were washed with brine (100 mL), dried (MgSO4, 1 g), filtered, and concentrated in vacuo (20 mmHg, 40 °C). The crude white solid was purified by flash chromatography on silica gel (50% EtOAc in hexanes) to afford ligand 3 as a fluffy white solid; yield: 1.37 g (86%); mp 160 °C; [α]D25 +362.0 (c = 1.0, CH2Cl2 {Lit.4a [α]D25 +367.0 (c = 1.05, CH2Cl2)}.
IR (film): 3030, 2931, 1656, 1454, 1143, 1114, 975 cm–1.
1H NMR (400 MHz, CDCl3): δ = 7.02 (s, 10 H), 6.96 (s, 10 H), 5.97 (d, J = 10 Hz, 2 H), 5.59 (d, J = 10.4 Hz, 2 H), 1.92 (s, 6 H).
13C NMR (75 MHz, CDCl3): δ = 170.4, 137.5, 136.2, 127.9, 127.6, 127.4, 126.9, 126.6, 86.3, 73.8, 39.6, 24.8.
HRMS-ESI: m/z [M + H]+ calcd for C33H31N2O2: 487.2374; found: 487.2380.
(S)-5-Fluoro-2-(2,2,6,6-tetramethylpiperidin-1-yloxymethyl)-1-tosylindoline (4)
An oven-dried, 500 mL, 14/20 neck size, round-bottomed flask equipped with a magnetic stirring bar was brought into a glove box under an argon environment, where the flask was charged with Cu(OTf)2 (0.76 g, 2.1 mmol, 0.15 equiv) and (4R,5S)-diphenylbis(oxazoline) 3 (1.22 g, 2.5 mmol, 0.18 equiv); then the flask was sealed with a rubber septum. The flask was removed from the glove box and placed under argon. Freshly dried toluene (78 mL) was added via syringe and the mixture was heated at 70 °C for 2.5 h. This catalyst solution was allowed to cool to r.t., then TEMPO (6.51 g, 41.7 mmol, 3.00 equiv) was added as a solid and sulfonamide 2 (4.25 g, 13.9 mmol, 1 equiv) was added via syringe as a solution in toluene (99 mL). The resulting solution was kept under an O2 atmosphere (1 atm, balloon using a glass 14/20 side arm adapter tapered to connect to a short rubber vacuum hose connected to the O2 filled balloon) and was heated at 110 °C (oil bath) for 6 h. The reaction mixture was cooled to r.t., the stir bar was removed, and the mixture was concentrated by rotary evaporation (20 mmHg, 50 °C) to afford a brown oil. The crude oil was purified by flash chromatography on silica gel (0–30% EtOAc in hexanes gradient), affording indoline 4 as an off-white solid in 89% ee (enantiomeric excess determined by chiral HPLC on Chiralpak AD-RH); yield: 5.47–5.69 g (86–89%); mp 87–89 °C; [α]D22 +92.6 (c = 1.0, CHCl3); ee = 89%, determined by Varian Prostar High Performance Liquid Chromatography using the Chiralpak AD-RH: 10% i-PrOH–hexane; 0.4 mL/min; λ = 254 nm; tR (major) = 9.32 min, tR (minor) = 8.14 min. The optical purity of indoline 4 was further enriched to >98% ee by recrystallization from hexanes (4.28 g of optically pure 4).
IR (film): 2936, 1600, 1480, 1356, 1263, 1163 cm–1.
1H NMR (500 MHz, CDCl3): δ = 7.58 (dd, J = 5.0, 3.5 Hz, 1 H), 7.51–7.49 (d, J = 8.0, 2.0 Hz, 2 H), 7.16 (d, J = 8.0 Hz, 2 H), 6.87 (dt, J = 9.0 Hz, 1 H), 6.74 (dd, J = 2.5, 5.5 Hz, 1 H), 4.34 (m, 1 H), 3.98–3.93 (m, 2 H), 2.76 (d, J = 16.5 Hz, 1 H), 2.66 (dd, J = 9.5, 16.0 Hz, 1 H), 2.36 (s, 3 H), 1.44–1.38 (m, 5 H), 1.23 (d, J = 10.0 Hz, 1 H), 1.15 (d, J = 7.5 Hz, 6 H), 0.95 (s, 3 H), 0.83 (s, 3 H).
13C NMR (75 Hz, CDCl3): δ = 160.3 (d, J = 243.0 Hz), 143.9, 138.0 (d, J = 2.6 Hz), 135.1 (d, J = 9.2 Hz), 134.8, 129.6, 127.0, 118.2 (d, J = 9.2 Hz), 113.9 (d, J = 23.1 Hz), 111.8 (d, J = 27.5 Hz), 78.7, 61.4, 59.9, 39.5, 33.1, 31.7, 21.5, 20.0, 19.8, 16.9.
HRMS-ESI: m/z [M + 1]+ calcd for C25H34FN2O3S: 461.2275; found: 461.2274.
Anal. Calcd for C25H33FN2O3S: C, 65.19; H, 7.22; N, 6.09. Found: C, 65.28, H, 7.09; N, 6.09.
Supplementary Material
Acknowledgment
We thank the National Institutes of Health (GM078383) for support of this research.
Footnotes
Supporting Information for this article is available online at http://www.thieme-connect.com/ejournals/toc/synthesis.
References
- 1.Anas S, Kagan HB. Tetrahedron: Asymmetry. 2009;20:2193. [Google Scholar]
- 2.a Fuller PH, Kim J-W, Chemler SR. J. Am. Chem. Soc. 2008;130:17638. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liwosz TW, Chemler SR. J. Am. Chem. Soc. 2012;134:2020. doi: 10.1021/ja211272v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bovino MT, Chemler SR. Angew. Chem. Int. Ed. 2012;51:3923. doi: 10.1002/anie.201109044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a Seaman W, Norton AR, Woods JT, Bank HN. J. Am. Chem. Soc. 1945;67:1571. [Google Scholar]; b Goetz-Luthy N. J. Chem. Educ. 1949;26:271. [Google Scholar]; c Bannister WW, Pennace JR, Smith DW, Chelton CF, Wareing JA, Curby WA. J. Chem. Educ. 1973;50:578. [Google Scholar]
- 4.Original synthesis of ligand 3: Masamune S, Lowenthal RE. Chem. Abstr. ??? 1994 US Patent 5298623 A 19940329, The method used in the present work: Denmark SE, Stiff CM. J. Org. Chem. 2000;65:5875. doi: 10.1021/jo0007175.
- 5.a We believe loss of conversion upon scale-up is due to self-quenching of the TEMPO radical (disproportionation), which O2 might prevent. TEMPO reactivity: Vogler T, Studer A. Synthesis. 2008:1979.
- 6.a These reactions, as performed (1 atm O2, balloon), are not expected to pose a fire/explosion hazard, but use of 100% O2 atmosphere with organic solvents can be an industrial concern. There is precedent for turning a batch process using O2, balloon, into a continuous flow process that uses 8% O2 in N2: Ye X, Johnson MD, Diao T, Yates MH, Stahl SS. Green Chem. 2010;12:1180. doi: 10.1039/c0gc00106f. and references cited therein. b After these studies were completed, we found that under O2 atmosphere the amount of TEMPO can be reduced to 1.5 equiv.
- 7.Muniz K, Hovelmann CH, Streuff J. J. Am. Chem. Soc. 2008;130:763. doi: 10.1021/ja075041a. [DOI] [PubMed] [Google Scholar]
- 8.Yip K-T, Yang M, Law K-L, Zhu N-Y, Yang D. J. Am. Chem. Soc. 2006;128:3130. doi: 10.1021/ja060291x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.