

Abstract
The identity of the cell type responsive to sclerostin, a negative regulator of bone mass, is unknown. Since sclerostin is expressed in vivo by mineral-embedded osteocytes, we tested the hypothesis that sclerostin would regulate the behavior of cells actively involved in mineralization in adult bone, the preosteocyte. Differentiating cultures of human primary osteoblasts exposed to recombinant human sclerostin (rhSCL) for 35 days displayed dose- and time-dependent inhibition of in vitro mineralization, with late cultures being most responsive in terms of mineralization and gene expression. Treatment of advanced (day 35) cultures with rhSCL markedly increased the expression of the preosteocyte marker E11 and decreased the expression of mature markers DMP1 and SOST. Concomitantly, matrix extracellular phosphoglycoprotein (MEPE) expression was increased by rhSCL at both the mRNA and protein levels, whereas PHEX was decreased, implying regulation through the MEPE-ASARM axis. We confirmed that mineralization by human osteoblasts is exquisitely sensitive to the triphosphorylated ASARM-PO4 peptide. Immunostaining revealed that rhSCL increased the endogenous levels of MEPE-ASARM. Importantly, antibody-mediated neutralization of endogenous MEPE-ASARM antagonized the effect of rhSCL on mineralization, as did the PHEX synthetic peptide SPR4. Finally, we found elevated Sost mRNA expression in the long bones of HYP mice, suggesting that sclerostin may drive the increased MEPE-ASARM levels and mineralization defect in this genotype. Our results suggest that sclerostin acts through regulation of the PHEX/MEPE axis at the preosteocyte stage and serves as a master regulator of physiologic bone mineralization, consistent with its localization in vivo and its established role in the inhibition of bone formation.
Keywords: OSTEOCYTE, SCLEROSTIN, SOST, MEPE, ASARM
Introduction
Sclerostin (SCL) is the product of the SOST gene, mutations in which cause the high-bone-mass disease sclerosteosis in humans.(1) Deletion of Sost in mice causes a similar high-bone-mass phenotype.(2) Partial deletion of a regulatory region approximately 35 kb distal to the SOST gene appears to be responsible for the high-bone-mass phenotype seen in Van Buchem disease.(3–5) Together these observations indicate that sclerostin has a key role in the regulation of bone mass. Neutralizing antibodies to SCL increase bone formation and strength dramatically in ovariectomized rats(6) and in intact aged male rats.(7) This anabolic effect of SCL was associated with a large increase in bone formation on quiescent bone surfaces (modeling).(6–8) Consistent with the effects of the SCL blocking antibodies, recent observations suggest that osteonal SCL is a strong determinant of whether osteoblasts actively produce bone(9) and underscore the need for a better understanding of its mode of action in human bone.
The cellular signaling of SCL is still to be fully elucidated.(10–14) Its inhibitory actions on bone morphogenetic protein (BMP) signaling have been attributed to a dominant effect on canonical Wnt signaling by virtue of binding to the Wnt coreceptor low-density lipoprotein receptors (LRP) 5 and 6.(15,16) More recently, LRP4 has been implicated as a major receptor for SCL.(17,18) The canonical Wnt signaling pathway fundamentally regulates osteoblast differentiation and bone formation.(1,19) Wnt ligands bind to frizzled (Fzd) and LRP5/6 coreceptors on target cells, preventing the proteosomal degradation of β-catenin and promoting the formation of transcription complexes with TCF/LEF transcription factors, resulting in the downstream transcription of osteogenesis-related genes. Several inhibitors of the Wnt pathway have been identified, including SCL, Dikkopf 1 (DKK-1), and secreted Fzd-related proteins (sFRPs).(1,19) Genetic models of under- and overexpression of factors that regulate the Wnt pathway demonstrate the central importance of this pathway in bone biology. Additionally, recent studies show that the anabolic action of parathyroid hormone (PTH) is due in part to the downregulation of SCL expression.(20–22)
Very little is known concerning the target cell type(s) for SCL and its effect on human osteoblast function. Sutherland and colleagues reported that SCL expression was increased in mineralized cultures of human mesenchymal stem cells (MSCs) and was increased further with stimulation of differentiation of both MSCs and primary human osteoblasts by BMP-4.(12) We have recently reported that SOST mRNA expression increased in cultures of human primary osteoblasts differentiated in the presence of strontium ranelate, a condition that increased the level of in vitro mineralization as well as expression of the osteocyte marker dentin matrix protein 1 (DMP1).(23) The increase in expression of SCL under conditions that increase osteogenesis is seemingly paradoxical, but this is consistent with the expression pattern of SCL in mineralized tissues during development(24) and in adult bone.(24–26) Irie and colleagues reported that SCL was expressed only by osteocytes if mineralization occurred and was coincident with the expression of the key osteoblast transcription factor osterix.(26) It is reasonable, therefore, to interpret the appearance of SCL expression by cells under conditions of deposited mineral as being indicative of an osteocyte-like phenotype, with the assumption that SCL expression is in response to the mineralized microenvironment. While it is evident that PTH treatment(20,22) and mechanical loading(27) likely exert an anabolic effect, at least in part, by suppressing SCL expression, other (catabolic) stimuli may increase SCL expression. We have reported recently that proinflammatory cytokines TWEAK and tumor necrosis factor α (TNF-α) induce SCL expression in human primary osteoblasts and in human bone, suggesting that this may be a mechanism by which bone formation is impaired in conditions of inflammatory bone loss, as in rheumatoid arthritis (RA).(28)
The mineralization of bone is a dynamic and actively regulated process. In lamellar bone, late osteoblasts/preosteocytes mineralize their organic matrix in a process concomitant with cell maturation.(29) Mineralization appears to be regulated by inhibitory peptides deriving from a group of extracellular matrix proteins, small integrin-binding ligand N-linked glycoproteins (SIBLINGs), that includes matrix extracellular phosphoglycoprotein (MEPE), DMP1, osteopontin (OPN), bone sialoprotein (BSP), enamelin, dentin sialo phosphoprotein (DSPP), and statherin, all of which are involved in the mineralization of bone and teeth.(30) A key feature of MEPE and several SIBLINGs, including DMP1 and OPN,(31) is that they contain an acidic serine aspartate-rich MEPE-associated (ASARM) motif.(32) This motif, when released as a protease-resistant phosphorylated peptide (ASARM-PO4 peptide), negatively affects mineralization and phosphate uptake.(31–33) It also has been shown that ASARM peptides are chiefly responsible for the in vitro mineralization defect in HYP mice and likely regulate mineralization in this mutant strain.(30,34) The protein phosphate-regulating gene with homologies to endopeptidases on the X-chromosome (PHEX) binds to full-length MEPE, inhibiting cleavage of MEPE by cathepsin B,(35) and the generation of MEPE-ASARM peptides and, importantly, is also able to cleave the otherwise protease-resistant MEPE-ASARM peptides, releasing the inhibitory effect of these on mineralization.(30,35–37)
The focus of this study was to investigate in more detail the cellular targets and downstream effects of SCL action. We examined the effects of SCL on human primary osteoblast mineralization and maturation. Our results suggest that SCL inhibits late osteoblast/preosteocyte progression to a mature osteocyte stage and inhibits matrix mineralization by regulating the PHEX/MEPE axis. Thus SCL appears to act as a potent local inhibitor in bone that regulates matrix mineralization by maturing osteocytes.
Materials and Methods
Recombinant cytokines, peptides, and antibodies
Full-length recombinant human sclerostin (rhSCL) was obtained from R&D Systems (Minneapolis, MN, USA). Rabbit antibodies to human PHEX, MEPE, and MEPE-ASARM were generated as described previously.(30) Lyophilized triphosphorylated peptides representing the human MEPE-ASARM peptide sequence [NH2-RDDSSESSDSGS(PO3H2)SS(PO3H2)ES(PO3H2)DGD-OH (ASARM-PO4)] and SPR4 peptide representing the human MEPE-ASARM-binding region of human PHEX (NH2-TVNAFYSAST-NYPRSLSYGAIGVIVGHEFTHGFDNNGRGENIADNG-OH; NeoMPS, Inc., San Diego, CA, USA), validated previously for their respective activities,(30) were reconstituted in PBS containing 0.1% bovine serum albumin (BSA).
Cells and culture medium
Adult human primary osteoblasts (normal human bone-derived cells, NHBCs) derived from cancellous bone explant cultures were used for this study because they have been characterized extensively to display the expected properties of osteoblasts, including the ability to differentiate into mature osteoblasts and form a mineralized matrix.(38–42) Furthermore, under certain culture conditions, NHBCs differentiate into osteocyte-like cells.(23,43,44) NHBCs were isolated from femoral neck trabecular bone, as described previously.(45) Cells were grown to confluence from bone chips and removed by digestion of the cell matrix with collagenase and dispase for either cryopreservation or immediate use in experiments.(45) In some cases, cells were recovered from cryopreservation and grown again to confluence prior to seeding into assay plates. In this way, we used primary cells that had not been passaged more than twice.
Immunofluorescence staining and flow cytometry
For in situ immunofluorescence, NHBCs were seeded into chamber slides (Lab-Tek, Nunc, Naperville, IL, USA) and cultured as indicated. Prior to immunostaining, cells were rinsed with PBS, fixed with 4% paraformaldehyde in PBS for 10 minutes on ice, rinsed with PBS, and permeabilized with 0.1% Triton X-100 for 5 minutes. Nonspecific binding sites were blocked with PBS containing 10% goat serum for 30 minutes at room temperature. The cells were incubated for 30 minutes with rabbit anti-human PHEX or rabbit anti-human MEPE-ASARM primary antibodies. Following three washes in PBS, cells were incubated with anti-rabbit IgG-FITC for 45 minutes in a dark humidified container. After washing again in PBS, cells were incubated for 1 minute with 4′,6-diamidino-2-phenyindole (DAPI; Sigma, St Louis, MO, USA). Cells then were washed in PBS and mounted (P olong Gold with DAPI antifade mounting medium; Invitrogen, Carlsbad, CA, USA). Samples were examined by confocal microscopy on a Radiance 2100 dual-laser confocal microscope (Bio-Rad Microscience, Ltd., Hemel Hemstead, UK).
Preparation of total RNA and RT-PCR from human cells
Total RNA and complementary DNA (cDNA) were prepared as described previously.(43) Relative expression between samples was calculated using the comparative cycle threshold (Ct) method (ΔCt) using GAPDH as the reference gene, as we have published previously.(43) Oligonucleotide primers were designed in-house to flank intron-exon boundaries and were purchased from Geneworks (Thebarton, South Australia, Australia). Real-time oligonucleotide primers for the amplification of human osteocalcin (OCN), type 1 collagen α1 (COL1α1), DMP1, E11, and SOST were described previously.(43) Sequences for the amplification of mouse E11 and Gapdh also were published previously.(44) Other primers for real-time polymerase chain reaction (PCR) amplification were human LRP4 mRNA (183-bp product): GGG CCA TTG CTG TTT TCC CCA G (sense) and GCG CAT CCA CCC AGT AGA TCC T (antisense); human PHEX mRNA (114-bp product): CAT CCA ATG AAC ATA TCT TGA AGC (sense) and ACT TGT AAA GGG CAT CCC GAT AA (antisense); human tissue-nonspecific alkaline phosphatase (TNAP) mRNA (215-bp product): TGC TCC CAC GCG CTT GTG CCT GGA (sense) and CTG GCA CTA AGG AGT TAG TAA G (antisense); human OPN mRNA (65-bp product): CTG GAA GTT CTG AGG AAA AGC AGC (sense) and GTT TAG CCA TGT GGC CAC AGC ATC (antisense); human ANK mRNA (172-bp product): TGC ATG GCT CTG TCA CTC ACG C (sense) and TGA GAT GCG CCC TCA CTG TGA C (antisense); and human ENPP1 mRNA (127-bp product): CTT CTT CCT GTT ATT AGC AAA CTA AA (sense) and CAT GAG ATT CTG GAT ACA ATC CG (antisense).
Sost expression in HYP mice
Total RNA was prepared from the femurs of 35-day-old male HYP mice and their wild-type littermates (Jackson Laboratories, Bar Harbor, ME, USA), and real-time reverse-transcriptase (RT) PCR was performed as described previously.(30) Sost expression was normalized to that of transferrin(30) because Gapdh expression is dysregulated in HYP mice (P Rowe, unpublished observations). The following primer sequences were used for amplification of mouse Sost: PR#63: sense primer 5′-CCA GGG CTT GGA GAG TAC C-3′ and antisense primer 5′-GCA GCT GTA CTC GGA CAC ATC-3′ (123-bp product).
In vitro mineralization
To determine the ability of NHBCs to form a mineralized matrix, a modification of a method reported previously(40) was used. NHBCs were plated in replicate cultures at 2.1 ± 104 cells/cm2 in mineralization medium consisting of α modified essential medium 10 (α-MEM-10) medium, dexamethasone (10−8 M), and KH2PO4 (1.8 mM) in the presence or absence of rhSCL or ASARM-PO4 with full medium changes twice weekly for the duration of the experiment. In some experiments, NHBCs were cultured for 35 days in mineralization medium, which we have shown previously promotes an osteocyte-like phenotype,(23,44) and then treated with combinations of rhSCL, α-ASARM Ab, SPR4, or ASARM-PO4 for 3 or 7 days, and then RNA was prepared as described earlier or measurement of cell layer–associated Ca2+ levels was performed as described previously.(40)
Western blot analysis
MEPE protein expression was analyzed in cultures maintained under mineralizing conditions for 35 days, as described earlier, and treated for 4 days with rhSCL. Cell lysates and Western blot analysis were performed as described previously(28) with some modifications. Since long-term cultures had developed an extensive type 1 collagenous matrix, 70 μg of total protein per track was electrophoresed on 4% to 12% SDS-PAGE gels (NuPage; Invitrogen, Mount Waverley, Victoria, Australia). Proteins then were transferred to polyvinyl-difluoroacetate filters (PVDF; Geneworks). Subsequent steps were performed using a Snap ID Western blotting system (Millipore) as per the manufacturer’s instructions with blocking for 10 minutes and primary antibodies for the detection of MEPE. Following washing, filters were incubated with either mouse or rabbit IgG-specific antibodies conjugated to AP (Amersham Biosciences, Poole, UK), and binding was detected with Attophos substrate (Promega Corp., Madison, WI, USA) on a FluorImager (Molecular Dynamics, Sunnyvale, CA, USA). Following this, filters were stripped using a Western Blot Recycling Kit (Alpha Diagnostic International, San Antonio, TX, USA) and reprobed for β-actin expression, as described previously.(28)
Statistical analysis
Unless stated otherwise, for experiments where two parameters were used, Student’s t test was employed to analyze statistical differences. For experiments with more than two parameters, one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc analysis was used (GraphPad Instat software, La Jolla, CA, USA). A value for p <.05 was considered to be significant.
Results
Effect of rhSCL on osteoblast in vitro mineralization
Under differentiating conditions, NHBCs begin to mineralize the cell layer from approximately days 14 to 21 of culture. Mineralization was highly sensitive to treatment with rhSCL, which inhibited calcium incorporation into the matrix of NHBC cultures at concentrations of rhSCL as low as 1 ng/mL (Fig. 1A). Commencement of treatment of mineralizing cultures at 21 and 28 days, after the onset of mineralization, resulted in increased efficacy of rhSCL relative to continuously treated cultures (Fig. 1B).
Fig. 1.

Effects of exogenously added SCL on NHBC in vitro mineralization. NHBCs were cultured under mineralizing conditions for up to 35 days in the absence or presence of rhSCL at 1, 10, or 50 ng/mL. The medium was replenished every 3 to 4 days. (A) Continuously treated cultures were assayed on days 21, 28, and 35 for cell layer–associated calcium levels, determined as described in “Materials and Methods.” (B) Comparison of in vitro mineralization when cells were treated with rhSCL for the entire 35-day period, from days 21 to 35, or from days 28 to 35. Note that NHBCs from different donors were used for the two experiments depicted. Data represent mean values from quadruplicate wells ± SD. *Difference Difference from control (no added rhSCL), p <.05.
Effect of exogenous SCL on gene expression during differentiation
To determine the effect of rhSCL on osteoblast gene expression, we performed real-time RT-PCR analysis on primary osteoblasts treated either continuously throughout differentiation for a period of up to 35 days or only after maturation. For most genes examined, the effect of continuous treatment was evident only at a relatively late stage of differentiation. As shown in Fig. 2A, OCN expression increased with time in all cultures. At the 35-day time point, its expression was reduced by SCL dose-dependently. E11, a preosteocyte marker, was increased in response to SCL at the 35-day time point (Fig. 2B). DMP1 and SOST, both markers of mature osteocytes, increased throughout the culture period in control cultures, but both were reduced relative to controls over the last 7 days of culture (Fig. 2C, D). The effects of rhSCL on osteocyte marker genes suggested that sclerostin was targeting late osteoblasts/preosteocytes in the cultures and arresting them at the preosteocyte stage, blocking or retarding their differentiation into mature osteocyte-like cells.
Fig. 2.

Effects on gene expression of continuous exposure of mineralizing NHBC cultures to exogenously added SCL. NHBCs were cultured under mineralizing conditions for up to 35 days in the absence or presence of rhSCL at 1, 10, or 50 ng/mL. The medium was replenished every 3 to 4 days. At the time points indicated, total RNA was prepared and real-time RT-PCR was performed to determine mRNA expression of (A) OCN, (B) E11, (C) DMP1, and (D) SOST. Data shown are means of triplicate reactions ± SD normalized to expression of GAPDH mRNA. a, b, and c indicate significant differences (p <.05) from untreated controls for rhSCL at 1, 10, and 50 ng/mL, respectively. Similar results were obtained from three independent experiments using NHBCs from different donors.
Effect of SCL on late-stage osteoblast/preosteocyte-like cells
To address the possibility that SCL targeted mature osteoblasts/preosteocytes, further experiments were performed, in which SCL treatment was initiated in NHBCs that had been cultured for a period of 35 days under mineralizing conditions. The pretreatment conditions resulted in an osteocyte-like phenotype, evident from the gene expression pattern shown in Fig. 2, and as we have reported previously.(23,43,44) At day 35, the cultures were treated with rhSCL, and gene expression was assessed after a further 3 and 7 days. Gene expression in the untreated control cultures over the 7-day period displayed increases in all of OCN, DMP1, SOST, PHEX, and MEPE, consistent with further differentiation toward a mature osteocyte phenotype (Fig. 3). These mature cultures were highly responsive to SCL, with concentrations as low as 1 ng/mL effective (Fig. 3A). OCN mRNA levels were strongly and dose-dependently inhibited by SCL, consistent with our previous report.(28) The expression of mRNA encoding COL1α1 also was suppressed by rhSCL (Fig. 3B). E11 mRNA expression was significantly upregulated following SCL exposure for 3 days but then returned to control levels (Fig. 3C). Similarly, the expression of TNAP mRNA was transiently upregulated by rhSCL (Fig. 3D). DMP1 mRNA expression increased in control cultures over the 7-day culture period but was strongly and dose-dependently suppressed by rhSCL (Fig. 3E). Additionally, SOST mRNA expression was suppressed dose-dependently by SCL (Fig. 3F), indicating a ligand-dependent negative regulation of the SOST gene. Results in these experiments were highly reproducible between the different donor cells tested. Expression of mRNA encoding the recently described receptor for SCL, LRP4,(18) was downregulated in response to rhSCL at all doses tested after 3 days and remained so after 7 days in the presence of 10 to 50 ng/mL of rhSCL (Fig. 3G). Expression of both LRP5 and LRP6, also proposed receptors for SCL, also tended to be downregulated by rhSCL treatment, but the effects of rhSCL on these genes was not as dose dependent (data not shown). These data together indicated that SCL was capable of acting on preosteocytes and inhibiting their transition into phenotypically mature osteocytes.
Fig. 3.

Effects on gene expression of acute exposure of mature NHBC cultures to exogenously added SCL. NHBCs were cultured under mineralizing conditions for 35 days. Cells then were cultured for a further 3 or 7 days in the absence or presence of rhSCL at 1, 10, or 50 ng/mL. The medium and supplements were replenished on day 3. Total RNA was prepared and real-time RT-PCR was performed to determine mRNA expression of (A) OCN, (B) COL1α1, (C) E11, (D) TNAP, (E) DMP1, (F) SOST, (G) LRP4, (H) PHEX, and (I) MEPE. Data shown are means of triplicate reactions ± SD normalized to expression of GAPDH mRNA. a, b, and c indicate significant differences (p <.05) from untreated controls for rhSCL at 1, 10, and 50 ng/mL, respectively. Near-identical results were obtained from three independent experiments using NHBCs from different donors.
Effect of SCL on mineralization-regulating genes
SCL appeared to potently regulate genes associated with matrix mineralization. At 10 or 50 ng/mL, SCL strongly inhibited PHEX mRNA expression after 3 days of exposure and continued to suppress PHEX at all doses tested after 7 days of exposure (Fig. 3F). In five independent experiments, the fold change in relative PHEX mRNA expression after 3 days in response to rhSCL (50 ng/mL) was 0.55 ± 0.12 (mean ± SEM; p =.03, one-sample t test). In contrast, mRNA levels of the SIBLING family member MEPE were correspondingly increased by rhSCL (Fig. 3G). In five independent experiments, the mean fold change in relative MEPE mRNA expression after 3 days in response to rhSCL (50 ng/mL) was 2.9 ± 0.50 (mean ± SEM; p =.017, one-sample t test). rhSCL had no reproducible effect on the expression of another SIBLING family member, OPN (data not shown). We also tested the effects of rhSCL on the genes ENPP1(46) and ANK,(47) associated with pyrophosphate production and secretion, respectively, but their expression was not regulated by rhSCL (data not shown).
Effect of SCL on MEPE-PHEX-regulated mineralization
We hypothesized that these effects of SCL would have functional consequences because PHEX has a demonstrated role in cleaving MEPE-derived ASARM peptides in the matrix surrounding embedded late osteoblasts/osteoid osteocytes, releasing their inhibitory effect on mineralization.(37) First, we tested whether human primary osteoblast mineralization was sensitive to inhibition by ASARM-PO4 peptides. For this, differentiating cultures were set up in the presence of increasing concentrations of ASARM-PO4. The level of matrix mineralization by NHBCs, as measured by either Von Kossa (VK) stain for phosphate incorporation or alizarin red (AR) stain for calcium, was exquisitely sensitive to ASARM-PO4, with complete inhibition achieved at a concentration of 10 μM (Fig. 4A). Analysis of the remaining calcium concentration in the culture supernatant confirmed that these cultures of mineralizing cells potently removed calcium, with just 60% remaining after 3 days, compared with the cell-free (CF) control. The addition of ASARM-PO4 at 10 to 20 μM completely blocked calcium apposition and removal from the medium by the cells (Fig. 4B).
Fig. 4.

Effect on in vitro mineralization of NHBCs of ASARM-PO4. NHBCs were cultured under mineralizing conditions for 28 days in the absence or presence of ASARM-PO4 peptide at 1, 10, or 20 μM final concentration. (A) Cell layers were assayed for phosphate incorporation by Von Kossa (VK) staining or calcium incorporation using alizarin red (AR) staining. VK stains were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Data are means of quadruplicate cultures ± SD. ***Difference from untreated control (p <.001). (B) Supernatant or cell-free (CF) medium taken from the day 28 time point (representing an incubation period of 3 days) was assayed for remaining calcium concentration, as described in “Materials and Methods,” and expressed as a ratio to the level present in the CF control. Data are means of quadruplicate cultures ± SD. ***Difference from CF control (p <.001).
Immunostaining of mineralizing NHBCs revealed that MEPE-ASARM levels increased with time (data not shown). rhSCL treatment for 3 days of NHBC cultures grown under the differentiating conditions described earlier to a stage resembling preosteocytes (day 35) showed increased immunoreactivity for MEPE-ASARM (Fig. 5A). Conversely, treatment of such cultures with rhSCL resulted in decreased immunoreactivity for PHEX (Fig. 5B). Western blot analysis of similar SCL-treated cultures confirmed that full-length MEPE protein levels increased dose-dependently in response to rhSCL (Fig. 5D).
Fig. 5.

The effect of rhSCL on endogenous levels of MEPE-ASARM. NHBCs were cultured under mineralizing conditions for 35 days in chamber slides and then treated for a further 3 days in the absence or presence of rhSCL (50 ng/mL). Cells were stained by immunofluorescence antibodies for (A) MEPE-ASARM and (B) PHEX and analyzed by confocal microscopy. Representative images of triplicate wells are shown for each treatment. The relative mean fluorescence intensity ± SEM (arbitrary units) of the cell layer for each treatment was quantified using ImageJ analysis of images (n =6) taken of the unmerged antibody-specific (FITC) signal. (C) The level of background staining using a normal rabbit IgG as the primary antibody. Nuclei were stained using DAPI, as described in “Materials and Methods.” (D) The effect of a 3-day treatment of day 35 differentiated NHBC cultures with rhSCL (0 to 50 ng/mL) on full-length MEPE expression assessed by Western blot. Relative MEPE levels were compared with those of β-actin, which served as a loading control, and represent four independent blots. In all cases, an asterisk indicates difference from untreated (p <.05).
To test the functional significance of SCL induction of MEPE-ASARM, 35-day cultures were treated with rhSCL in the presence or absence of neutralizing antibodies to MEPE-ASARM and allowed to differentiate for a further 7 days. The addition of neutralizing antibody to MEPE-ASARM did not affect the response to rhSCL at the level of gene expression (Fig. 6A). As shown in Fig. 6B, rhSCL at 50 ng/mL inhibited the further deposition of calcium over this 7-day time frame. The addition of anti-ASARM antibody reversed the inhibitory effect of rhSCL, an effect seen in 4 of 4 independent experiments and an example of which is shown in Fig. 6B, consistent with this being a major pathway by which sclerostin effects inhibition of mineralization. We also incubated cells with the PHEX peptide SPR4, which corresponds to the region of PHEX known to bind to MEPE and inhibit MEPE cleavage into MEPE-ASARM peptides.(30) SPR4 also reversed rhSCL inhibition of mineralization (Fig. 6B), consistent with cleavage of full-length MEPE and generation of MEPE-ASARM peptides contributing to the inhibitory action of SCL.
Fig. 6.

The effect of neutralizing antibody to ASARM-PO4 on the response to SCL. (A) NHBCs cultured under mineralizing conditions for 35 days then were cultured either untreated (control) or with rhSCL at 50 ng/mL, neutralizing antibody to ASARM-PO4 (10 μg/mL), or a combination for 3 days, and the effect on gene expression was measured for OCN, E11, SOST, and MEPE. Data are means of triplicate reactions ± SD and are representative of two independent experiments. (B) The inhibitory effect of SCL on mineralization is reversed by ASARM-PO4 and SPR4 peptides. NHBCs were cultured under mineralizing conditions for 35 days as earlier and then were cultured for a further 7 days either untreated (control) or with combinations of rhSCL at 10 and 50 ng/mL, neutralizing antibody to ASARM-PO4 (10 μg/mL), or SPR4 peptide (10 μM). The medium and supplements were replenished on day 3. Cell layer–associated calcium was determined after 7 days, as described in “Materials and Methods.” Data are expressed as means ± SD of quadruplicate observations. Significance, as indicated, was determined by one-way ANOVA. Similar results were obtained in four independent experiments.
Expression of Sost in HYP bone
Since our in vitro experiments indicated that an important activity of SCL in vivo may be to regulate MEPE-ASARM levels in bone, we examined Sost expression in the long bones of HYP mutant mice. HYP mice have been shown previously to exhibit increased MEPE-ASARM levels, which contribute to the bone mineralization defect in these animals.(30,37,48) Real-time RT-PCR analysis of 35-day-old male HYP mice compared with age- and sex-matched wild-type littermates revealed that Sost mRNA was elevated 3.2 ± 1.3-fold (mean ± SEM) in HYP femoral bone (p =.0078, Wilcoxon signed-rank test, two-tailed, n =5), consistent with the hypothesis that SCL contributes to the defective bone mineralization in these animals.
Discussion
Accumulating evidence suggests that SCL is a key determinant of bone mass in humans, and blockage of SCL activity is currently being trialed as a novel anabolic treatment option for osteoporosis.(49,50) While there is no doubt as to the effect of SCL, and the loss of SCL, on bone mass in both humans(1) and mice,(2,49) little is known regarding the responding or target cell type or the effect of SCL on the function of these cells. Instead, the vast majority of research to date has focused on proximal events of SCL action with respect to the signaling pathways affected. However, Winkler and colleagues found that SCL at relatively high concentrations (>1 μg/mL) inhibited alkaline phosphatase activity in the immature mouse cell line C3H10T1/2(11,14) and inhibited the proliferation and differentiation of immature human MSCs at similar concentrations.(11) We reported recently that human primary osteoblasts cultured in vitro respond to rhSCL by downregulating the expression of RUNX2 and OCN mRNAs and by activating ERK1/2 signaling.(28) In this study we investigated in more detail the cellular targets and downstream effects of SCL action. We found that SCL has marked effects on mineralization by human primary osteoblasts at concentrations as low as 1 ng/mL. Strong effects of rhSCL also were observed on gene expression in differentiated human osteocyte-like cultures, again at concentrations as low as 1 ng/mL, but with dose-dependent effects up to 50 ng/mL, with the higher concentrations having major effects on a number of genes associated with phenotypically mature cells. These concentrations of SCL are higher than but nevertheless near to the levels recently reported to be found in the circulation of adult humans—between 0.3 and 0.6 ng/mL.(51–53) The concentration of SCL in bone is unknown. These data suggest that cells at the late osteoblast/preosteocyte stage are likely to represent an important physiologic target for SCL. This is consistent with the restricted expression of SCL under steady-state conditions by mature, mineral-embedded osteocytes.(11,25,54,55)
We identified at least three effects of SCL treatment that are consistent with inhibition of bone formation and mass. First, SCL inhibited in vitro mineralization by human primary osteoblasts. It is known that mineralization in lamellar bone occurs during the osteoblast-to-osteocyte transition, a process reviewed and described in a comprehensive study by Barragan-Adjemian and colleagues,(29) and the pattern of mineralization and the corresponding gene expression profile we observed with our human primary osteoblasts were consistent with this. Second, SCL appeared to block the maturation of osteocytes, promoting instead a preosteocyte phenotype. This effect was suggested by the upregulation of E11, a hallmark of the preosteocyte,(56,57) by sclerostin, an effect we also observed in MLO-Y4 cells (not shown). Also consistent with maintenance by SCL of the preosteocyte phenotype, expression of the mature osteocyte markers DMP1 and SOST were decreased by SCL treatment. Third, SCL strikingly regulated the expression of key genes involved in bone matrix mineralization. The endopeptidase PHEX has been demonstrated to degrade SIBLING protein–derived ASARM peptides,(31,37) in particular, those derived from MEPE and OPN, which bind to nascent bone mineral and inhibit mineral deposition.(30,31,37) SCL treatment of human preosteocyte cultures resulted in the striking downregulation of PHEX mRNA, and a corresponding decrease in PHEX protein levels was evident by immunofluorescence. Conversely, the expression of MEPE, a product of both osteoblasts and osteocytes,(58,59) was increased at both the mRNA and protein levels in response to SCL, which corresponded to the observed increased staining for MEPE-ASARM in rhSCL-treated cultures. The question posed by these findings was whether the regulation of MEPE and PHEX was a mechanism by which SCL exerted an effect on mineralization. To address this question, we first confirmed that human osteoblast/preosteocyte mineralization was strongly inhibited by the addition of exogenous ASARM-PO4 peptides, similar to the effects observed previously in mineralizing cultures of mouse bone marrow stromal cells(30) and MC3T3-E1 cells.(37) We also confirmed by immunostaining that endogenous MEPE-ASARM peptides were deposited into human cell monolayers and found that these increased during mineral acquisition, perhaps as a control mechanism. Third, the coaddition of neutralizing antibodies to ASARM-PO4 reversed the inhibitory effect of rhSCL on mineral apposition without having effects on gene expression, consistent with an effect in the extracellular matrix. The addition of the MEPE-binding PHEX peptide SPR4, shown previously to inhibit ASARM-PO4 activity,(30,37) also reversed the effect of rhSCL on mineralization. Together these findings strongly suggest that SCL, by targeting late osteoblasts/preosteocytes, controls matrix mineralization at least in part in a MEPE- and PHEX-dependent manner, as depicted in Fig. 7. A recent study by Addison and colleagues(31) reported that OPN-ASARM is also a negative regulator of bone mineralization, implying that SIBLING family members in addition to MEPE may be important in this context. OPN-derived ASARM peptides strongly inhibited mineralization by MC3T3-E1 cells and were shown to bind hydroxyapatite.(31) However, while OPN mRNA was expressed by NHBCs at appreciable levels, SCL had no reproducible effect on the expression of this gene in the current study (data not shown). It should be noted that the expression of another SIBLING family member, DMP1, was decreased in response to SCL. While DMP1-derived ASARM peptides are also thought to inhibit mineralization,(30) this complex protein also has a described role as a (positive) nucleator of mineralization,(60) and gene deletion of Dmp1 results in disrupted mineralization patterns in vivo,(61) implying that the type of posttranslational processing of DMP1 may be critical in determining its role as either a positive or negative regulator. Taken together, at this stage it can be concluded that SIBLING family members are not redundant in their function owing simply to the presence of an ASARM motif and indeed seem to be differentially regulated by SCL.
Fig. 7.
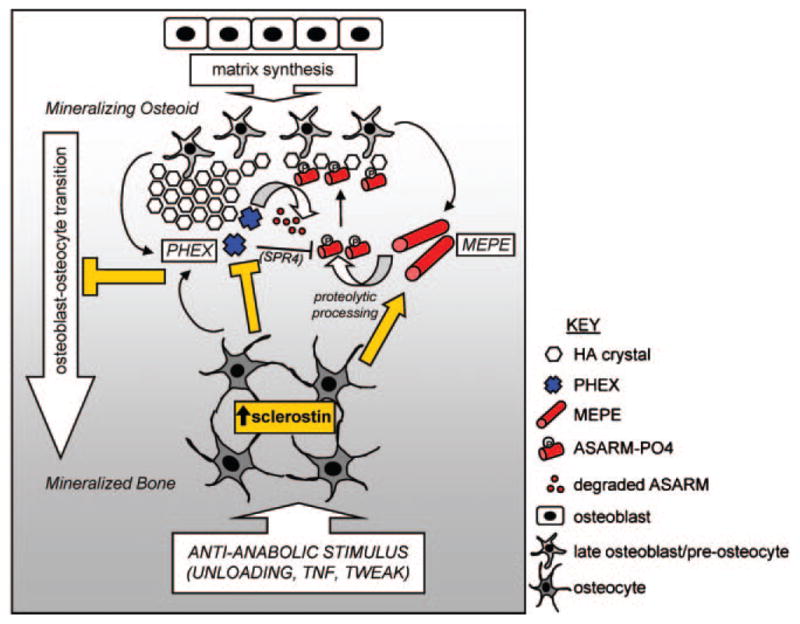
Cartoon representing the proposed effects of SCL as a master regulator of bone mineralization. Anti-anabolic stimuli, such as mechanical unloading or proinflammatory cytokines TNF-α and TWEAK, induce the expression of SCL by mineral-embedded osteocytes. As indicated (yellow arrow), SCL then acts on late osteoblasts/preosteocytes in local osteoid by stimulating the expression of phosphorylated MEPE, which is cleaved (by cathepsin B) into phosphorylated ASARM peptides (ASARM-PO4). ASARM-PO4 peptides attach to nascent hydroxyapatite-like (HA) crystals and prevent further mineralization. PHEX promotes mineralization by inhibiting MEPE cleavage into ASARM peptides and by degrading mineralization-inhibiting ASARM-PO4 peptides, reversing the inhibitory effect of MEPE on mineralization. SCL decreases PHEX expression (yellow bar), potentially by either late osteoblasts through to mature osteocytes, thereby decreasing this positive effect. The net inhibition of mineralization by SCL is accompanied by inhibition of late osteoblast/preosteocyte transition into mature osteocytes (yellow bar), as described in the text.
We infer from our study of HYP mice that a SCL-MEPE-PHEX axis exists in vivo. These animals carry a mutation in the Pex/Phex locus and have severe hypophosphatemia associated with elevated serum levels of the phosphaturic hormone fibroblast growth factor 23 (FGF-23) and renal wasting of phosphate. HYP mice are considered an animal model of the human condition X-linked hypophosphatemic rickets/osteomalacia (XLH).(62,63) Osteoblasts derived from HYP mice have an intrinsic mineralization defect,(48,64) demonstrating that the bone defects are not due entirely to either the elevated FGF-23, hypophosphatemia, hyperparathyroidism, or low levels of circulating 1,25-dihydroxyvitamin D in these animals.(65) Important to this study, HYP mice also have defective bone mineralization, which appears to be due to elevated levels of Mepe-ASARM peptides.(30,37) Treatment of HYP mice with the cathepsin inhibitor CA074, reduced serum levels of Mepe-ASARM peptide, consistent with cathepsin B being responsible for cleaving Mepe,(35) and corrected the bone mineralization defect.(48) This study shows that Sost mRNA levels are upregulated threefold in the femurs of young-adult HYP mice, which may precede the bone matrix accumulation of Mepe-ASARM peptide. This will need to be confirmed in future studies where SCL is neutralized in these animals. In addition, it will be of interest in future studies to examine in detail the nature of the osteocytes, and potentially other cells, contributing to the elevated expression of Sost in HYP bone. It remains to be seen if SOST is also upregulated and therefore a potential therapeutic target in human XLH. Consistent with our findings is a recent report in abstract(66) of increased Sost mRNA expression in the bones of HYP mice or in mice with osteocalcin promoter–driven osteoblast/osteocyte deletion of Phex. Of potential importance, Sost expression was not normalized with correction of the hypophosphatemia in these animals.(66) Interestingly, a second report found a reduction in Sost mRNA expression in the long bones of neonatal (12-day-old) HYP mice,(67) implying that SCL may have different roles in this condition at different stages of development in this genotype. As reviewed,(30) the Mepe null mouse displays an increase in bone mass with age. From the findings of our study, it is tempting to speculate that this may be due to loss of SCL efficacy in these animals.
In summary, we have identified for the first time that SCL targets cells at the late osteoblast/preosteocyte stage. In doing so, SCL inhibits their ability to mineralize their matrix and does so, at least in part, by increasing the level of MEPE-ASARM deposited into the matrix, both by increasing the production of full-length MEPE by a transcriptional mechanism and by downregulating expression of the MEPE inhibitor PHEX (Fig. 7). Our findings begin to explain the extraordinary anabolic effects observed of neutralizing SCL either by gene mutation or administration of neutralizing antibodies.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (NHMRC). GJA was supported by an NHMRC R Douglas Wright Fellowship. The authors gratefully acknowledge the technical help of C Vincent and S Hay and are grateful to the surgeons and nursing staff of the Department of Orthopaedics and Trauma, Royal Adelaide Hospital, for the provision of bone samples.
Footnotes
Essential findings of this study were presented at the 32nd Annual Meeting of the ASBMR and published in abstract: Atkins GJ et al. J Bone Miner Res. 2010; 25(Suppl 1):S186.
Disclosures
All authors state that they have no conflicts of interest.
References
- 1.Baron R, Rawadi G, Roman-Roman S. Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol. 2006;76:103–127. doi: 10.1016/S0070-2153(06)76004-5. [DOI] [PubMed] [Google Scholar]
- 2.Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 3.ten Dijke P, Krause C, de Gorter DJ, Lowik CW, van Bezooijen RL. Osteocyte-derived sclerostin inhibits bone formation: its role in bone morphogenetic protein and Wnt signaling. J Bone Joint Surg Am. 2008;90:S31–35. doi: 10.2106/JBJS.G.01183. [DOI] [PubMed] [Google Scholar]
- 4.Balemans W, Patel N, Ebeling M, et al. Identification of a 52-kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staehling-Hampton K, Proll S, Paeper BW, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–152. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Ominsky MS, Warmington KS, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24:578–588. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Warmington KS, Niu QT, et al. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res. 2010;25:2647–2656. doi: 10.1002/jbmr.182. [DOI] [PubMed] [Google Scholar]
- 8.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 9.Power J, Poole KE, van Bezooijen R, et al. Sclerostin and the regulation of bone formation: Effects in hip osteoarthritis and femoral neck fracture. J Bone Miner Res. 2010;25:1867–1876. doi: 10.1002/jbmr.70. [DOI] [PubMed] [Google Scholar]
- 10.Kusu N, Laurikkala J, Imanishi M, et al. Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem. 2003;278:24113–24117. doi: 10.1074/jbc.M301716200. [DOI] [PubMed] [Google Scholar]
- 11.Winkler DG, Sutherland MK, Geoghegan JC, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. Embo J. 2003;22:6267–6276. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutherland MK, Geoghegan JC, Yu C, Winkler DG, Latham JA. Unique regulation of SOST, the sclerosteosis gene, by BMPs and steroid hormones in human osteoblasts. Bone. 2004;35:448–454. doi: 10.1016/j.bone.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 13.van Bezooijen RL, Roelen BA, Visser A, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winkler DG, Sutherland MS, Ojala E, et al. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J Biol Chem. 2005;280:2498–2502. doi: 10.1074/jbc.M400524200. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 16.van Bezooijen RL, Svensson JP, Eefting D, et al. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res. 2007;22:19–28. doi: 10.1359/jbmr.061002. [DOI] [PubMed] [Google Scholar]
- 17.Oliver LLHC, Morvan F, Hu S, Lu C, Bauer M, Kneissel M. LRP4 is a novel osteoblast and osteocyte expressed specific facilitator of SOST-mediated inhibition of in vitro bone formation. J Bone Miner Res. 2009;24(Suppl 1) Available at http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=c8cba0d3-a82c-4036-b4034d4038-29979e29961e29977a29972. [Google Scholar]
- 18.Choi HY, Dieckmann M, Herz J, Niemeier A. Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS One. 2009;4:e7930. doi: 10.1371/journal.pone.0007930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–1209. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silvestrini G, Ballanti P, Leopizzi M, et al. Effects of intermittent parathyroid hormone (PTH) administration on SOST mRNA and protein in rat bone. J Mol Histol. 2007;38:261–269. doi: 10.1007/s10735-007-9096-3. [DOI] [PubMed] [Google Scholar]
- 21.Shoback D. Update in osteoporosis and metabolic bone disorders. J Clin Endocrinol Metab. 2007;92:747–753. doi: 10.1210/jc.2007-0042. [DOI] [PubMed] [Google Scholar]
- 22.Bellido T, Ali AA, Gubrij I, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–4583. doi: 10.1210/en.2005-0239. [DOI] [PubMed] [Google Scholar]
- 23.Atkins GJ, Welldon KJ, Halbout P, Findlay DM. Strontium ranelate treatment of human primary osteoblasts promotes an osteocyte-like phenotype while eliciting an osteoprotegerin response. Osteoporos Int. 2009;20:653–664. doi: 10.1007/s00198-008-0728-6. [DOI] [PubMed] [Google Scholar]
- 24.Ohyama Y, Nifuji A, Maeda Y, Amagasa T, Noda M. Spaciotemporal association and bone morphogenetic protein regulation of sclerostin and osterix expression during embryonic osteogenesis. Endocrinology. 2004;145:4685–4692. doi: 10.1210/en.2003-1492. [DOI] [PubMed] [Google Scholar]
- 25.Poole KE, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. Faseb J. 2005;19:1842–1844. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 26.Irie K, Ejiri S, Sakakura Y, Shibui T, Yajima T. Matrix mineralization as a trigger for osteocyte maturation. J Histochem Cytochem. 2008;56:561–567. doi: 10.1369/jhc.2008.950527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of sost/sclerostin. J Biol Chem. 2008;283:5866–5875. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 28.Vincent C, Findlay DM, Welldon KJ, et al. Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res. 2009;24:1434–1449. doi: 10.1359/jbmr.090305. [DOI] [PubMed] [Google Scholar]
- 29.Barragan-Adjemian C, Nicolella D, Dusevich V, Dallas MR, Eick JD, Bonewald LF. Mechanism by which MLO-A5 late osteoblasts/early osteocytes mineralize in culture: similarities with mineralization of lamellar bone. Calcif Tissue Int. 2006;79:340–353. doi: 10.1007/s00223-006-0107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin A, David V, Laurence JS, et al. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology. 2008;149:1757–1772. doi: 10.1210/en.2007-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Addison WN, Masica DL, Gray JJ, McKee MD. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J Bone Miner Res. 2010;25:695–705. doi: 10.1359/jbmr.090832. [DOI] [PubMed] [Google Scholar]
- 32.Rowe PS. The wrickkened pathways of FGF23, MEPE and PHEX. Crit Rev Oral Biol Med. 2004;15:264–281. doi: 10.1177/154411130401500503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowe PS, Kumagai Y, Gutierrez G, et al. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone. 2004;34:303–319. doi: 10.1016/j.bone.2003.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan B, Takaiwa M, Clemens TL, et al. Aberrant Phex function in osteoblasts and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest. 2008;118:722–734. doi: 10.1172/JCI32702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo R, Rowe PS, Liu S, Simpson LG, Xiao ZS, Quarles LD. Inhibition of MEPE cleavage by Phex. Biochem Biophys Res Commun. 2002;297:38–45. doi: 10.1016/s0006-291x(02)02125-3. [DOI] [PubMed] [Google Scholar]
- 36.Rowe PS, Garrett IR, Schwarz PM, et al. Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: a model for impaired mineralization in X-linked rickets (HYP) Bone. 2005;36:33–46. doi: 10.1016/j.bone.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Addison WN, Nakano Y, Loisel T, Crine P, McKee MD. MEPE-ASARM peptides control extracellular matrix mineralization by binding to hydroxyapatite: an inhibition regulated by PHEX cleavage of ASARM. J Bone Miner Res. 2008;23:1638–1649. doi: 10.1359/jbmr.080601. [DOI] [PubMed] [Google Scholar]
- 38.Robey PG, Termine JD. Human bone cells in vitro. Calcif Tissue Int. 1985;37:453–460. [PubMed] [Google Scholar]
- 39.Fedarko NS, Termine JD, Young MF, Robey PG. Temporal regulation of hyaluronan and proteoglycan metabolism by human bone cells in vitro. J Biol Chem. 1990;265:12200–12209. [PubMed] [Google Scholar]
- 40.Findlay DM, Welldon K, Atkins GJ, Howie DW, Zannettino AC, Bobyn D. The proliferation and phenotypic expression of human osteoblasts on tantalum metal. Biomaterials. 2004;25:2215–2227. doi: 10.1016/j.biomaterials.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Gronthos S, Zannettino AC, Graves SE, Ohta S, Hay SJ, Simmons PJ. Differential cell surface expression of the STRO-1 and alkaline phosphatase antigens on discrete developmental stages in primary cultures of human bone cells. J Bone Miner Res. 1999;14:47–56. doi: 10.1359/jbmr.1999.14.1.47. [DOI] [PubMed] [Google Scholar]
- 42.Stewart K, Walsh S, Screen J, et al. Further characterization of cells expressing STRO-1 in cultures of adult human bone marrow stromal cells. J Bone Miner Res. 1999;14:1345–1356. doi: 10.1359/jbmr.1999.14.8.1345. [DOI] [PubMed] [Google Scholar]
- 43.Atkins GJ, Welldon KJ, Holding CA, Haynes DR, Howie DW, Findlay DM. The induction of a catabolic phenotype in human primary osteoblasts and osteocytes by polyethylene particles. Biomaterials. 2009;30:3672–3681. doi: 10.1016/j.biomaterials.2009.03.035. [DOI] [PubMed] [Google Scholar]
- 44.Atkins GJ, Welldon KJ, Wijenayaka AR, Bonewald LF, Findlay DM. Vitamin K promotes mineralization, osteoblast-to-osteocyte transition, and an anticatabolic phenotype by {gamma}-carboxylation-dependent and -independent mechanisms. Am J Physiol Cell Physiol. 2009;297:C1358–1367. doi: 10.1152/ajpcell.00216.2009. [DOI] [PubMed] [Google Scholar]
- 45.Atkins GJ, Kostakis P, Pan B, et al. RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res. 2003;18:1088–1098. doi: 10.1359/jbmr.2003.18.6.1088. [DOI] [PubMed] [Google Scholar]
- 46.Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta. 2003;1638:1–19. doi: 10.1016/s0925-4439(03)00058-9. [DOI] [PubMed] [Google Scholar]
- 47.Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–270. doi: 10.1126/science.289.5477.265. [DOI] [PubMed] [Google Scholar]
- 48.Rowe PS, Matsumoto N, Jo OD, et al. Correction of the mineralization defect in hyp mice treated with protease inhibitors CA074 and pepstatin. Bone. 2006;39:773–786. doi: 10.1016/j.bone.2006.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan A, van Bezooijen RL, Lowik CW. A new paradigm in the treatment of osteoporosis: Wnt pathway proteins and their antagonists. Curr Opin Investig Drugs. 2007;8:293–298. [PubMed] [Google Scholar]
- 50.Rey JP, Ellies DL. Wnt modulators in the biotech pipeline. Dev Dyn. 239:102–114. doi: 10.1002/dvdy.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drake MT, Srinivasan B, Modder UI, et al. Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J Clin Endocrinol Metab. 2010;95:5056–5062. doi: 10.1210/jc.2010-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaudio A, Pennisi P, Bratengeier C, et al. Increased sclerostin serum levels associated with bone formation and resorption markers in patients with immobilization-induced bone loss. J Clin Endocrinol Metab. 2010;95:2248–2253. doi: 10.1210/jc.2010-0067. [DOI] [PubMed] [Google Scholar]
- 53.Modder UI, Clowes JA, Hoey K, et al. Regulation of circulating sclerostin levels by sex steroids in women and in men. J Bone Miner Res. 2011;26:27–34. doi: 10.1002/jbmr.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jager A, Gotz W, Lossdorfer S, Rath-Deschner B. Localization of SOST/sclerostin in cementocytes in vivo and in mineralizing periodontal ligament cells in vitro. J Periodontal Res. 2009;45:246–254. doi: 10.1111/j.1600-0765.2009.01227.x. [DOI] [PubMed] [Google Scholar]
- 55.van Bezooijen RL, Bronckers AL, Gortzak RA, et al. Sclerostin in mineralized matrices and van Buchem disease. J Dent Res. 2009;88:569–574. doi: 10.1177/0022034509338340. [DOI] [PubMed] [Google Scholar]
- 56.Wetterwald A, Hoffstetter W, Cecchini MG, et al. Characterization and cloning of the E11 antigen, a marker expressed by rat osteoblasts and osteocytes. Bone. 1996;18:125–132. doi: 10.1016/8756-3282(95)00457-2. [DOI] [PubMed] [Google Scholar]
- 57.Zhang K, Barragan-Adjemian C, Ye L, et al. E11/gp38 selective expression in osteocytes: regulation by mechanical strain and role in dendrite elongation. Mol Cell Biol. 2006;26:4539–4552. doi: 10.1128/MCB.02120-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Argiro L, Desbarats M, Glorieux FH, Ecarot B. Mepe, the gene encoding a tumor-secreted protein in oncogenic hypophosphatemic osteomalacia, is expressed in bone. Genomics. 2001;74:342–351. doi: 10.1006/geno.2001.6553. [DOI] [PubMed] [Google Scholar]
- 59.Gowen LC, Petersen DN, Mansolf AL, et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003;278:1998–2007. doi: 10.1074/jbc.M203250200. [DOI] [PubMed] [Google Scholar]
- 60.Qin C, D’Souza R, Feng JQ. Dentin matrix protein 1 (DMP1): new and important roles for biomineralization and phosphate homeostasis. J Dent Res. 2007;86:1134–1141. doi: 10.1177/154405910708601202. [DOI] [PubMed] [Google Scholar]
- 61.Feng JQ, Ward LM, Liu S, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Francis F, Hennig S, Korn B, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11:130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 63.Yamazaki Y, Okazaki R, Shibata M, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87:4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- 64.Xiao ZS, Crenshaw M, Guo R, Nesbitt T, Drezner MK, Quarles LD. Intrinsic mineralization defect in Hyp mouse osteoblasts. Am J Physiol. 1998;275:E700–708. doi: 10.1152/ajpendo.1998.275.4.E700. [DOI] [PubMed] [Google Scholar]
- 65.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291:E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 66.Yuan B, Meudt J, Zhang R, Xing Y, Feng J, Drezner MK. Increased SOST production: A factor underlying abnormal bone mineralization in X-linked hypophosphatemia. J Bone Miner Res. 2009;24(Suppl 1) Available at http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=78aee231-b395-234b267-284c239-00267e00287c00528. [Google Scholar]
- 67.Liu S, Tang W, Fang J, et al. Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol. 2009;23:1505–1518. doi: 10.1210/me.2009-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]