

Abstract
The C-type lectin dendritic cell-specific intercellular adhesion molecule 3–grabbing nonintegrin (DC-SIGN) can serve as a docking site for pathogens on the surface of dendritic cells. Pathogen binding to DC-SIGN can have diverse consequences for the host. DC-SIGN can facilitate HIV-1 dissemination, but the interaction of Mycobacterium tuberculosis with DC-SIGN is important for host immunity. The ability of pathogens to target DC-SIGN provides impetus to identify ligands that can perturb these interactions. Here, we describe the first stable small molecule inhibitors of DC-SIGN. These inhibitors were derived from a collection of quinoxalinones, which were assembled using a tandem cross metathesis-hydrogenation sequence. To assess the ability of these small molecules to block DC-SIGN-mediated glycan adhesion and internalization, we developed a sensitive flow cytometry assay. Our results reveal that the quinoxalinones are effective inhibitors of DC-SIGN–glycan interactions. These compounds block both glycan binding to cells and glycan internalization. We anticipate that these non-carbohydrate inhibitors can be used to elucidate the role of DC-SIGN in pathogenesis and immune function.
Introduction
Carbohydrate-binding proteins have diverse roles in physiological and pathophysiological processes. One critical function for lectins is to direct immune responses, and its importance is underscored by the prevalence of different C-type lectin family members on the surface of dendritic cells.1–4 As professional antigen presenting cells, dendritic cells function as critical monitors of immune responses.5, 6 They detect foreign entities using the lectins on their surface as detectors. Lectins engage glycosylated antigen, mediate internalization, and influence cell signaling.7, 8 One lectin that has been implicated in all of these roles is DC-SIGN.1 Upon binding glycans, DC-SIGN can mediate uptake as well as influence signaling pathways that lead to immunity or tolerance.3, 9, 10 As a result, the ability of pathogen glycans to interact with DC-SIGN has consequences for the host. For example, DC-SIGN interactions with HIV can facilitate its dissemination, while DC-SIGN binding to Mycobacterium tuberculosis mitigates host immune responses to this organism.11 Understanding the molecular mechanisms by which pathogens exploit DC-SIGN for binding and internalization could yield major benefits for human health. Consequently, we sought to develop molecular probes of DC-SIGN.
DC-SIGN preferentially binds to high mannose oligosaccharides found on the surface of pathogens including HIV-11, 12 and M. tuberculosis.13, 14 The lectin also interacts with fucose-containing structures that include the Lewis-type epitopes found on parasites such as Schistosoma mansoni cercariae15–18 and Helicobacter pylori (Fig. 1).11, 19 The structural requirements for carbohydrate binding to DC-SIGN have been elucidated using various approaches including NMR,20 X-ray crystallography,21–23 and carbohydrate arrays.24 Although these studies have contributed insight into the specificity of DC-SIGN, effective functional probes have been elusive. Indeed, the multitude of carbohydrates that bind to DC-SIGN do so with low affinity (Kd ~ 0.1–10 mM). Most efforts to find higher affinity ligands have focused on generating carbohydrate derivatives, but these compounds generally exhibit only modest increases in affinity.25 Alternatively, multivalent glycoconjugates have been designed that bind DC-SIGN with enhanced avidity, but the production of these can require considerable investment in multistep syntheses.21, 26–31 We therefore sought an alternative strategy. The utility of non-carbohydrate inhibitors of lectins provided impetus to search for effective small molecule probes.26, 32–34 Herein, we describe the generation of stable compounds that bind potently to DC-SIGN and are capable of inhibiting cell-surface DC-SIGN-glycan interactions.
Fig 1.

Representative carbohydrates that bind DC-SIGN including (A) mannose (B) Lewis-type epitopes and (C) high mannose oligosaccharides. Values represent binding constants adapted from aref (35), bref (23) and cref (19).
Results and Discussion
Inhibitor design and synthesis of quinoxalinones
We previously used a high-throughput assay to screen libraries of small molecules to identify inhibitors of DC-SIGN.35 This approach yielded two major classes of non–carbohydrate ligands: those containing a quinoxalinone core (1) and those possessing a pyrazolone scaffold (2) (Fig. 2). Although compounds of each type were effective at blocking DC-SIGN—mediated carbohydrate interactions, all had liabilities. Pyrazolones such as 2 are electrophiles, which complicate their use in cell-based assays.36 Though quinoxalinone 1 is not an electrophile, it undergoes degradation and inactivation.
Fig 2.

Compounds identified in a high-throughput screen as DC-SIGN inhibitors include quinoxalinone (1) and pyrazolone (2) scaffolds. Values represent binding constants adapted from ref (35).
We postulated that the oxidizable thioether functional group in heterocycle 1 was the source of its instability. This instability was especially problematic, as it precluded using compounds like 1 as DC-SIGN probes not only in vivo but also in cell-based assays. To design effective probes, we needed to ascertain what functionality contributes to DC-SIGN binding and whether the thioether could be replaced. We reasoned chemical synthesis could be used to address these issues.
The quinoxalinones represent an important class of compounds and members of this class have activity as anticancer, antibacterial, and antiviral agents.37 Despite the utility of quinoxalinones, relatively few synthetic routes have been described.38–40 Many of these rely on solid phase synthesis to generate the quinoxalinone core.38, 41–47 Alternatively, quinoxalinones have been prepared using transition-metal catalysts, but these innovative approaches impose limitations on substrate scope.48 Our objective was to implement an efficient synthesis of quinoxalinones that would allow for late-stage diversification. We reasoned that a divergent route could be used to elucidate the compound features that result in DC-SIGN binding. This information would allow us to convert the compounds identified in our initial screen into effective cellular probes. Installing the elements of diversity at a late stage in the synthesis would enhance efficiency and utility. Finally, we wanted to replace the sulfur with a methylene to test our hypothesis that this oxidizable functionality was the source of the instability of compound 1.
We envisioned generating allyl quinoxalinone 6 and then using tandem olefin cross metathesis-hydrogenation in the penultimate step of the synthesis (Scheme 1). There are several advantages of this approach. First, it provides a means to rapidly modify the common intermediate 6. Second, the remarkable functional group compatibility of the ruthenium carbene catalysts should allow for the installation of a wide range of functionality.49 Finally, the ruthenium carbene catalyst can be transformed from a species that promotes metathesis to one that is capable of transfer hydrogenation.50–53 This dual reactivity can be used in tandem to streamline the production of potential inhibitors. Moreover, the reaction sequence serves as a convenient method for incorporating the methylene-containing substituent.
Scheme 1.

Synthetic route to quinoxalinone inhibitors
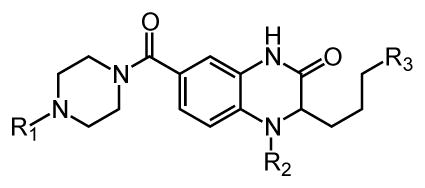
The utility of a tandem transformation in divergent synthesis depends on its compatibility with diverse functionality and on an effective route to the substrate. We chose allyl bearing quinoxalinone 6 as the key substrate for the tandem metathesis reaction because we envisioned accessing it via an efficient process. To this end, the assembly of this building block began with preparation of the heterocycle 5, which was generated from the commercially available benzoic acid derivative 3. Either enantiomer of the final product 7 can be synthesized from aminopentenoate 4; the latter can be generated as either the R or S isomer using the Schollkopf auxiliary.54 Amino acid 4 was used in an SNAr reaction, and subsequent zinc–mediated nitro group reduction was followed by cyclization to afford the allyl quinoxalinone 5 in 93% yield. The carboxylate was elaborated via amide coupling with piperazine building blocks. An R2 substituent could be incorporated through alkylation of the secondary amine to efficiently generate intermediate 6.
The key transformation in our strategy is appending the R3 group using a tandem metathesis–hydrogenation sequence. Cross metathesis reactions of quinoxalinones were unknown. We investigated, therefore, a variety of ruthenium catalysts and solvent conditions. To promote solubility of the allyl quinoxalinone 6, we conducted the reaction in a mixture of dichloromethane and methanol. We anticipated that this mixed solvent would also facilitate the subsequent hydrogenation reaction. Initiation of metathesis with the Grubbs first-generation catalyst55 failed, possibly because of catalyst deactivation. In contrast, the Grubbs second-generation catalyst56 and Hoveyda-Grubbs catalysts57, 58 both could initiate metathesis. Of these two, the Grubbs second-generation catalyst was the more efficient promoter of the tandem sequence. The effective ruthenium catalyst was compatible with a variety of functional groups, and the desired reduction process occurred in the presence of ester and ketone59 groups. In this way, the versatile yet chemoselective ruthenium catalyst could be exploited to achieve rapid diversification. Using this process, we synthesized over 20 compounds60 for evaluation as DC-SIGN inhibitors.
Quinoxalinones generated by metathesis are stable and bind potently to DC-SIGN
The synthetic route provided the means to elucidate what features of quinoxalinone 1 influence its affinity for DC-SIGN and what sites could be modified to generate tailored probes. To test the synthetic compounds for DC-SIGN binding, we employed a fluorescence assay. Specifically, we assessed the ability of each synthetic compound to compete with a fluorescein glycoconjugate for binding to the extracellular domain of DC-SIGN.35 Several compounds that block DC-SIGN–carbohydrate binding were identified (Table 1).
Table 1.
Synthetic small molecules that function as potent inhibitors of the lectin DC-SIGN. Values are reported as the concentration required to inhibit 50% (IC50) of the binding of a fluorescein mannosylated glycoconjugate to immobilized tetrameric DC-SIGN.62
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | IC50 (μM) |
| 8 | Pr | Et | Et | 370 ± 70.0 |
| 9 | Et | Me | Et | 360 ± 69.2 |
| 10 | Me | Me | Me | 329 ± 65.8 |
| 11 | Me | H | Me | 313 ± 47.7 |
| 12 | Boc | Et | Me | 270 ± 43.8 |
| 13 | 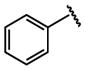 |
Bn | 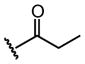 |
170 ± 28.5 |
| 14 | 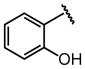 |
H | 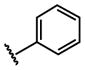 |
113 ± 20.4 |
| 15 | 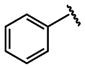 |
H | 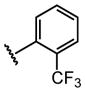 |
71 ± 11 |
| 16 | 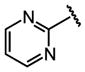 |
Et | 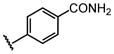 |
68 ± 11 |
| 17 | 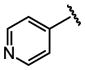 |
Bn | 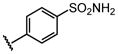 |
39 ± 6.2 |
| 18 | 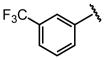 |
H | 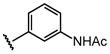 |
23 ± 3.4 |
| 19 | 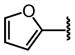 |
H | 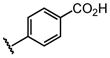 |
10 ± 1.3 |
| 20 | 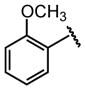 |
H | 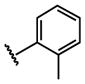 |
4.4 ± 0.64 |
| 21 | 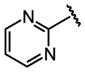 |
H | 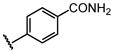 |
2.6 ± 0.28 |
| 22 | 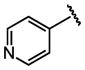 |
H | 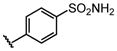 |
0.31 ± 0.13 |
From the set of compounds tested, approximately 25% were potent lectin inhibitors with IC50 values that ranged from 0.31–10 μM (ESI† S3). These results validate the choice of the quinoxalinone core as a scaffold for generating DC-SIGN ligands. Indeed, compared to the monosaccharides that bind to DC-SIGN, the non–carbohydrate ligands are approximately 1000-fold more active. The potency of these small molecules compares favorably even to that of higher molecular weight, multivalent DC-SIGN ligands.26–30
As we postulated, the methylene-containing compounds were stable over the course of several months. In contrast, compound 1 undergoes oxidation within several days at ambient temperature. Thus, the replacement of the sulfur atom by a methylene enhances compound stability. Indeed, the minor enhancement in binding observed for compound 20 over thioether 1 may be a reflection of the latter’s instability. These data suggest that other spacer units can be used to append the R3 substituent to the quinoxalinone core. Consistent with this observation, the immediate alkene precursors of 9, 12, 19 and 22 were also DC-SIGN ligands, with inhibitory activities that are similar to the products (i.e., IC50 values decreased by only 3- to 10-fold (data not shown)). These results highlight that compounds described herein address key liabilities of the initial lead compounds.
We also examined the influence of substituents at other positions on binding. The identity of the R1 moiety was critical. Piperazine derivatives functionalized with aliphatic substituents were less active, and those containing aromatic groups, especially nitrogen heterocycles, afforded the highest affinity. These data implicate the R1 group in DC-SIGN binding. The importance of the quinoxalinone ring system itself was emphasized by our observation that derivatization at R2 lead to a decrease in affinity (i.e., compounds 10, 16, and 17). The R3 substituent also impacts binding, and aromatic groups bearing polar functionality were favored. That the most active DC-SIGN ligands possess aromatic groups at both the R1 and R3 positions provides additional support that appending aromatic groups to a ligand can enhance its affinity for lectins. Indeed, DC-SIGN is one of many C-type lectins in which aromatic amino acids line the carbohydrate binding site, and it has been shown that sugars with pendant aromatic substituents can exhibit enhanced binding to their target lectins.61
Quinoxalinones inhibit glycoconjugate binding and internalization mediated by DC-SIGN
The activities of the compounds in the DC-SIGN binding assay prompted us to investigate their abilities to inhibit internalization. Upon engaging a ligand, DC-SIGN facilitates its uptake into the cell. Most strategies for assessing ligand binding to DC-SIGN rely on immobilization techniques such as glycan arrays24, 63 or conjugation of the lectin to surfaces for analysis by surface plasmon resonance (SPR).27, 64, 65 Alternatively, cell adhesion assays have been developed in which the binding of fluorescent DC-SIGN positive cells to a surface is evaluated in the presence of inhibitors.28, 35 Although useful, these approaches do not account for the ability of DC-SIGN to mediate ligand internalization. Therefore, we devised a competitive assay that assesses both cell surface binding and DC-SIGN-mediated internalization.
We used flow cytometry to evaluate the ability of the active small molecules to block the binding and uptake of a fluorescent mannosylated glycoconjugate (Man-Fl-BA) to DC-SIGN-displaying cells. Our assay was implemented using a Raji (derived from Burkitt’s lymphoma) cell line stably transfected with a vector encoding DC-SIGN.66 The presence of cell surface DC-SIGN was confirmed by exposing the transfected cells to the fluorescent anti-DC-SIGN antibody AZND1-phycoerythrin (PE) and the level of cell surface DC-SIGN was visualized by microscopy (Fig. 4A) and quantified by flow cytometry (Fig. 4B). Relative to untransfected Raji cells, the stable transfectants exhibit increased levels of fluorescence, which is indicative of the presence of cell-surface DC-SIGN. We next exposed a Man-Fl-BSA to cells and assessed the probe’s fate. When DC-SIGN-producing cells were treated, increased probe binding and internalization were observed. To confirm that probe binding was mediated by DC-SIGN–carbohydrate interactions, we added N-acetylmannosamine (ManNAc) as a competitor. ManNAc (100 mM) treatment led to a significant decrease in probe uptake by DC-SIGN-positive cells (ESI† S6). These studies demonstrate the utility of Man-Fl-BSA for monitoring ligand interactions with cell surface DC-SIGN. Moreover, because we could use the same DC-SIGN glycan probe in the protein binding assay and the flow cytometry assay, the relative activities of the compounds in each assay could be compared and contrasted.
Fig 4.

Measurement of cell surface expression of DC-SIGN transfected Raji cells. (A) Visualization of cell surface expression using the DC-SIGN–specific antibody AZND1-PE (Ab AZND1-PE) and (B) Assessment of the level of cell surface expression of DC-SIGN by flow cytometry using the DC-SIGN-specific antibody DCN46-FITC. Bars represent 10 μm.
Small molecules 21 and 22 were tested for their ability to inhibit DC-SIGN mediated cellular uptake of the mannosylated probe. These compounds were chosen for testing in the cell based assay because they both are water–soluble and excellent inhibitors in the binding assay. In addition, the differences in the structures of 21 and 22 provide the means to ascertain how subtle perturbations in the quinoxalinone scaffold influence DC-SIGN mediated glycan uptake. Specifically, the binding assay data indicate that compound 22 is approximately 10-fold more potent than is compound 21. We sought to investigate whether this difference in binding activity was preserved when internalization was monitored DC-SIGN can internalize glycans, and the receptor is readily recycled to the plasma membrane.67 Thus, the ability of compounds to block uptake of the glycan probe into DC-SIGN-displaying cells is a rigorous test of their ability to block a key DC-SIGN function. Compounds were assessed at doses devised to block probe binding without inducing cytotoxicity (i.e. <10 mM) (ESI† S4). The compounds had no effect on the fluorescent probe binding to Raji cells that do not produce DC-SIGN. Dose dependent inhibition of fluorescence, however, was observed with DC-SIGN-displaying cells in the presence of each heterocycle (Fig. 5). Specifically, the percentage inhibition for compound 21 was 53% and 77% at 0.10 and 1 mM. These values compare to the percentage inhibition of 79% and 92% for the more potent compound 22 at the same concentrations. Thus, the trends in the inhibition data from the DC-SIGN binding assay and the cell-based glycan uptake assay are similar.
Fig 5.

Extent of binding and internalization of a fluorescent mannosylated probe (Man-Fl-BSA) to DC-SIGN producing Raji cells in the presence of (A) compound 21 and (B) compound 22 as measured by flow cytometry. The percent inhibition was determined from the geometric mean normalized to the results obtained from the cells with no inhibitor present.
The concentrations required for blocking glycoconjugate binding to cells displaying DC-SIGN are higher than those needed to block probe binding to immobilized DC-SIGN. There are several reasons why our cell-based assay is a highly stringent test for DC-SIGN inhibition. Specifically, the concentration of DC-SIGN on Raji cells can vary widely and is heterogeneous within the cell population. Cells with high levels of DC-SIGN are likely to be more adept at internalizing the probe and uptake into these cells will be difficult to inhibit. Moreover, once the glycan probe has been internalized, it will remain within the cell. Thus, the reversibility of glycan-binding to immobilized DC-SIGN assay in the plate assay is not preserved in the internalization assay. A higher concentration of compound is needed in the cell-based assay to reach saturation of the DC-SIGN binding sites and thereby prevent internalization. Indeed, the ability of the small molecules to block DC-SIGN-mediated glycan uptake is notable. Taken together, these data highlight the utility of the synthetic compounds at blocking cell surface DC-SIGN function.
Conclusions
Our results demonstrate that highly potent and stable small molecule inhibitors of DC-SIGN can be used as probes of cell-surface DC-SIGN binding and internalization. A divergent and modular synthetic strategy for the synthesis of quinoxalinones using a tandem cross metathesis–hydrogenation sequence allowed for the incorporation of diversity elements to explore the effect of ligand structure on binding to DC-SIGN. The development of a sensitive flow cytometry inhibition assay revealed that the compounds were active at preventing mannosylated glycans from interacting with cells displaying DC-SIGN. To the best of our knowledge, this is the first description of a small molecule capable of blocking not only DC-SIGN-glycan binding but also internalization. Moreover, small molecule probes of lectins have properties (i.e, ease of synthesis and enhanced affinity) that render them complementary to carbohydrate based probes. We anticipate that the non-carbohydrate lectin inhibitors described herein will continue to provide insight into the role of DC-SIGN in physiologically and medically relevant cell–surface binding events.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (GM049975). We thank Prof. K. Drickamer for providing the DC-SIGN expression vector. Stably transfected DC-SIGN Raji cells were kindly provided by the NIH AIDS Research Program, Division of AIDS, NIAID, NIH courtesy of Drs. Li Wu and Vineet N. Kewal-Ramani. Confocal microscopy was performed at the W.M. Keck Laboratory for Biological Imaging at the University of Wisconsin-Madison. Flow cytometry was performed at the Carbone Cancer Center at the University of Wisconsin-Madison. The UW-Madison Chemistry NMR facility is supported by the NSF (CHE-0342998 and CHE-9629688) and the NIH (1-S10-RR13866). L.R.P. is an NIH postdoctoral fellow (GM089084).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and characterization data for compounds. See DOI: 10.1039/b000000x/
Notes and references
- 1.Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, Cornelissen IL, Nottet HS, KewalRamani VN, Littman DR, Figdor CG, van Kooyk Y. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 2.Kerrigan AM, Brown GD. Immunobiology. 2009;214:562–575. doi: 10.1016/j.imbio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osorio F, Sousa GRE. Immunity. 2011;34:651–664. doi: 10.1016/j.immuni.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 4.van Kooyk Y, Rabinovich GA. Nat Immunol. 2008;9:593–601. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- 5.Banchereau J, Steinman RM. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 6.Steinman RM, Cohn ZA. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lis H, Sharon N. Chem Rev. 1998;98:637–674. doi: 10.1021/cr940413g. [DOI] [PubMed] [Google Scholar]
- 8.van Kooyk Y, Geijtenbeek TB. Nat Rev Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Vallejo JJ, van Kooyk Y. Immunol Rev. 2009;230:22–37. doi: 10.1111/j.1600-065X.2009.00786.x. [DOI] [PubMed] [Google Scholar]
- 10.Svajger U, Anderluh M, Jeras M, Obermajer N. Cell Signal. 2010;22:1397–1405. doi: 10.1016/j.cellsig.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Geijtenbeek TBH. Nat Immunol. 2009;10:1081–1088. doi: 10.1038/ni.1778. [DOI] [PubMed] [Google Scholar]
- 12.Wu L, KewalRamani VN. Nat Rev Immunol. 2006;6:859–868. doi: 10.1038/nri1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda N, Nigou J, Herrmann JL, Jackson M, Amara A, Lagrange PH, Puzo G, Gicquel B, Neyrolles O. J Biol Chem. 2003;278:5513–5516. doi: 10.1074/jbc.C200586200. [DOI] [PubMed] [Google Scholar]
- 14.Tailleux L, Schwartz O, Herrmann JL, Pivert E, Jackson M, Amara A, Legres L, Dreher D, Nicod LP, Gluckman JC, Lagrange PH, Gicquel B, Neyrolles O. J Exp Med. 2003;197:121–127. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Appelmelk BJ, van Die I, van Vliet SJ, Vandenbroucke-Grauls CM, Geijtenbeek TB, van Kooyk Y. J Immunol. 2003;170:1635–1639. doi: 10.4049/jimmunol.170.4.1635. [DOI] [PubMed] [Google Scholar]
- 16.Van Liempt E, Bank CM, Mehta P, Garcia-Vallejo JJ, Kawar ZS, Geyer R, Alvarez RA, Cummings RD, Kooyk Y, Van Die I. FEBS Lett. 2006;580:6123–6131. doi: 10.1016/j.febslet.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 17.Van Liempt E, Imberty A, Bank CM, Van Vliet SJ, Van Kooyk Y, Geijtenbeek TB, Van Die I. J Biol Chem. 2004;279:33161–33167. doi: 10.1074/jbc.M404988200. [DOI] [PubMed] [Google Scholar]
- 18.Van Die I, van Vliet SJ, Nyame AK, Cummings RD, Bank CM, Appelmelk B, Geijtenbeek TB, van Kooyk Y. Glycobiology. 2003;13:471–478. doi: 10.1093/glycob/cwg052. [DOI] [PubMed] [Google Scholar]
- 19.Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth HP, Kapsenberg ML, Vandenbroucke-Grauls CM, van Kooyk Y, Appelmelk BJ. J Exp Med. 2004;200:979–990. doi: 10.1084/jem.20041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reina JJ, Diaz I, Nieto PM, Campillo NE, Paez JA, Tabarani G, Fieschi F, Rojo J. Org Biomol Chem. 2008;6:2743–2754. doi: 10.1039/b802144a. [DOI] [PubMed] [Google Scholar]
- 21.Feinberg H, Castelli R, Drickamer K, Seeberger PH, Weis WI. J Biol Chem. 2007;282:4202–4209. doi: 10.1074/jbc.M609689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feinberg H, Mitchell DA, Drickamer K, Weis WI. Science. 2001;294:2163–2166. doi: 10.1126/science.1066371. [DOI] [PubMed] [Google Scholar]
- 23.Guo Y, Feinberg H, Conroy E, Mitchell DA, Alvarez R, Blixt O, Taylor ME, Weis WI, Drickamer K. Nat Struct Mol Biol. 2004;11:591–598. doi: 10.1038/nsmb784. [DOI] [PubMed] [Google Scholar]
- 24.Taylor ME, Drickamer K. Glycobiology. 2009;19:1155–1162. doi: 10.1093/glycob/cwp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Timpano G, Tabarani G, Anderluh M, Invernizzi D, Vasile F, Potenza D, Nieto PM, Rojo J, Fieschi F, Bernardi A. ChemBioChem. 2008;9:1921–1930. doi: 10.1002/cbic.200800139. [DOI] [PubMed] [Google Scholar]
- 26.Garber KC, Wangkanont K, Carlson EE, Kiessling LL. Chem Commun. 2010;46:6747–6749. doi: 10.1039/c0cc00830c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Avila O, Bedoya LM, Marradi M, Clavel C, Alcami J, Penades S. Chembiochem. 2009;10:1806–1809. doi: 10.1002/cbic.200900294. [DOI] [PubMed] [Google Scholar]
- 28.Obermajer N, Svajger U, Jeras M, Sattin S, Bernardi A, Anderluh M. Anal Biochem. 2010;406:222–229. doi: 10.1016/j.ab.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 29.Reina JJ, Bernardi A, Clerici M, Rojo J. Future Med Chem. 2010;2:1141–1159. doi: 10.4155/fmc.10.203. [DOI] [PubMed] [Google Scholar]
- 30.Rojo J, Delgado R. J Antimicrob Chemother. 2004;54:579–581. doi: 10.1093/jac/dkh399. [DOI] [PubMed] [Google Scholar]
- 31.Wang SK, Liang PH, Astronomo RD, Hsu TL, Hsieh SL, Burton DR, Wong CH. Proc Natl Acad Sci U S A. 2008;105:3690–3695. doi: 10.1073/pnas.0712326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ernst B, Magnani JL. Nat Rev Drug Discov. 2009;8:661–677. doi: 10.1038/nrd2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grim JC, Garber KC, Kiessling LL. Org Lett. 2011;13:3790–3793. doi: 10.1021/ol201252x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiessling LL, Splain RA. Annu Rev Biochem. 2010;79:619–653. doi: 10.1146/annurev.biochem.77.070606.100917. [DOI] [PubMed] [Google Scholar]
- 35.Borrok MJ, Kiessling LL. J Am Chem Soc. 2007;129:12780–12785. doi: 10.1021/ja072944v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlson EE, May JF, Kiessling LL. Chem Biol. 2006;13:825–837. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Carta A, Piras S, Loriga G, Paglietti G. Mini-Rev Med Chem. 2006;6:1179–1200. doi: 10.2174/138955706778742713. [DOI] [PubMed] [Google Scholar]
- 38.Abou-Gharbia M, Freed ME, McCaully RJ, Silver PJ, Wendt RL. J Med Chem. 1984;27:1743–1746. doi: 10.1021/jm00378a039. [DOI] [PubMed] [Google Scholar]
- 39.Rueping M, Tato F, Schoepke FR. Chem Eur J. 2010;16:2688–2691. doi: 10.1002/chem.200902907. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Tempest P. Tetrahedron Lett. 2004;45:6757–6760. [Google Scholar]
- 41.Chanda K, Kuo J, Chen CH, Sun CM. J Comb Chem. 2009;11:252–260. doi: 10.1021/cc800137p. [DOI] [PubMed] [Google Scholar]
- 42.Krchnak V, Szabo L, Vagner J. Tetrahedron Lett. 2000;41:2835–2838. [Google Scholar]
- 43.Laborde E, Peterson BT, Robinson L. J Comb Chem. 2001;3:572–577. doi: 10.1021/cc010025+. [DOI] [PubMed] [Google Scholar]
- 44.Lee J, Murray WV, Rivero RA. J Org Chem. 1997;62:3874–3879. [Google Scholar]
- 45.Mahaney PE, Webb MB, Ye F, Sabatucci JP, Steffan RJ, Chadwick CC, Harnish DC, Trybulski EJ. Bioorg Med Chem. 2006;14:3455–3466. doi: 10.1016/j.bmc.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 46.Tung CL, Sun CM. Tetrahedron Lett. 2004;45:1159–1162. [Google Scholar]
- 47.Zaragoza F, Stephensen H. J Org Chem. 1999;64:2555–2557. doi: 10.1021/jo001735p. [DOI] [PubMed] [Google Scholar]
- 48.Luo X, Chenard E, Martens P, Cheng YX, Tomaszewski MJ. Org Lett. 2010;12:3574–3577. doi: 10.1021/ol101454x. [DOI] [PubMed] [Google Scholar]
- 49.Vougioukalakis GC, Grubbs RH. Chem Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]
- 50.Bielawski CW, Louie J, Grubbs RH. J Am Chem Soc. 2000;122:12872. doi: 10.1021/ja016431e. [DOI] [PubMed] [Google Scholar]
- 51.Camm KD, Castro NM, Liu Y, Czechura P, Snelgrove JL, Fogg DE. J Am Chem Soc. 2007;129:4168–4169. doi: 10.1021/ja071047o. [DOI] [PubMed] [Google Scholar]
- 52.Cossy J, Bargiggia F, BouzBouz S. Org Lett. 2003;5:459–462. doi: 10.1021/ol027347m. [DOI] [PubMed] [Google Scholar]
- 53.Schmidt B, Pohler M. Org Biomol Chem. 2003;1:2512–2517. doi: 10.1039/b303441k. [DOI] [PubMed] [Google Scholar]
- 54.Schollkopf U, Groth U, Deng C. Angew Chem Int Ed. 1981:20. [Google Scholar]
- 55.Schwab P, Grubbs RH, Ziller JW. J Am Chem Soc. 1996;118:100–110. [Google Scholar]
- 56.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 57.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
- 58.Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. J Am Chem Soc. 1999;121:791–799. [Google Scholar]
- 59.We observed ~10% reduction of ketone groups under the reported conditions
- 60.See Supporting Information for a list of the compounds
- 61.Jimenez-Barbero J, Asensio JL, Canada FJ, Poveda A. Curr Opin Struct Biol. 1999;9:549–555. doi: 10.1016/s0959-440x(99)00004-4. [DOI] [PubMed] [Google Scholar]
- 62.The measured Kd of the Man-Fl-BSA probe to immobilized tetrameric DC-SIGN was 2 mM.
- 63.Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH, Paulson JC. Proc Natl Acad Sci U S A. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Becer CR, Gibson MI, Geng J, Ilyas R, Wallis R, Mitchell DA, Haddleton DM. J Am Chem Soc. 2010;132:15130–15132. doi: 10.1021/ja1056714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reina JJ, Maldonado OS, Tabarani G, Fieschi F, Rojo J. Bioconjug Chem. 2007;18:963–969. doi: 10.1021/bc060369z. [DOI] [PubMed] [Google Scholar]
- 66.Wu L, Martin TD, Carrington M, KewalRamani VN. Virology. 2004;318:17–23. doi: 10.1016/j.virol.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 67.Engering A, Geijtenbeek TBH, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, Lanzavecchia A, Fransen J, Figdor CG, Piguet V, van Kooyk Y. J Immunol. 2002;168:2118–2126. doi: 10.4049/jimmunol.168.5.2118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.