

Abstract
We report the synthesis and controlled radical homo- and block copolymerization of 3-guanidinopropyl methacrylamide (GPMA) utilizing aqueous reversible addition-fragmentation chain transfer (aRAFT) polymerization. The resulting homopolymer and block copolymer with N-(2-hydroxypropyl) methacrylamide (HPMA) were prepared to mimic the behavior of cell penetrating peptides (CPPs) and poly(arginine) (> 6 units) which have been shown to cross cell membranes. The homopolymerization mediated by 4-cyano-4-(ethylsulfanylthiocarbonylsulfanyl)pentanoic acid (CEP) in aqueous buffer exhibited pseudo-first-order kinetics and linear growth of molecular weight with conversion. Retention of the “living” thiocarbonylthio ω-end-group was demonstrated through successful chain extension of the GPMA macroCTA yielding GPMA37-b-GPMA61 (Mw/Mn =1.05). Block copolymers of GPMA with the non-immunogenic, biocompatible HPMA were synthesized yielding HPMA271-b-GPMA13 (Mw/Mn = 1.15). Notably, intracellular uptake was confirmed by fluorescence microscopy, confocal laser scanning microscopy, and flow cytometry experiments after 2.5 h incubation with KB cells at 4 °C and at 37 °C utilizing FITC-labeled, GPMA-containing copolymers. The observed facility of cellular uptake and the structural control afforded by aRAFT polymerization suggest significant potential for these synthetic (co)polymers as drug delivery vehicles in targeted therapies.
Keywords: aqueous reversible addition-fragmentation chain transfer polymerization, aRAFT, cell-penetrating peptides, CPPs, guanidine-containing methacrylamido monomers and copolymers, water-soluble copolymers
In recent years, there has been extensive research regarding the unique cellular uptake properties of cell penetrating peptides (CPPs).1–4 Common CPPs, such as Tat and poly(arginine), are small (< 20 nm), cationic, and can cross the plasma membrane of most mammalian cells.5 Significantly, cell entry of both Tat and poly(arginine) oligopeptides can occur via an endocytotic-independent pathway, although the precise mechanism is still debated.5–12 The enhanced cellular uptake of these peptides is reported to depend on the presence of basic amino acid sequences rich in arginine residues and not on peptide secondary structure.5, 12, 13 Wender and coworkers have further demonstrated the cell penetrating properties of arginine by synthesizing a D-arginine oligomer (9 units) that exhibited >100 fold increase in cellular uptake over Tat49–57.12
One of the most attractive features of CPPs is their ability to transport macromolecules easily across cellular membranes. Additionally, analysis of in vivo tissue samples reveals uptake into most tissues including the brain.2, 14 Based on observed trans-membrane transport alone, modification of synthetic drug delivery vehicles with CPPs appears to hold great promise in targeted therapies. Cellular uptake of synthetic drug delivery vehicles may occur through one of several endocytotic pathways. In order for efficacious delivery, the internalized vehicle and/or the delivered cargo must escape the endosome prior to lysosomal degradation or exocytosis. Some suggested mechanisms for assisting escape involve membrane disruption (i.e. proton sponge effect, fusogenic peptides). Another potential strategy for efficacious delivery involves bypassing endocytosis altogether, a process that conceptually would avoid lysosomal degradation and/or exocytosis of the packaged therapeutic. For example, Kopecek and coworkers conjugated a Tat peptide to a biocompatible copolymer that was subsequently internalized into ovarian mammalian cancer cells through both endocytotic and non-endocytotic pathways. 15, 16 However, despite the success in cell uptake, the difficulty in synthesis and low conjugation efficiency demonstrated the need for a more direct route. Funhoff et al. polymerized a guanidine-containing methacrylate by classical free radical polymerization and condensed plasmid DNA into small polyplexes that successfully transfected COS-7 cells in the absence of serum. In the latter case, however, cellular uptake of the free (uncontrolled) polymer and its complexes was mainly endocytotic in nature rather than via direct cell penetration. 17 Inspired by the above work and drawing from our previous success at controlled aqueous polymerization, we targeted (co)polymer architecture that might mimic the cell-uptake behavior of Tat, poly(arginine), or other guanidine-pendent polyplexes.
Controlled polymerization techniques now allow tailored block lengths and advanced architectures while maintaining narrow molecular weight distributions. Here we report the aqueous reversible addition-fragmentation chain transfer (aRAFT) polymerization of guanidine-containing monomers directly in water without protecting groups, thus adding an additional synthetic pathway for highly functional systems.18–22 Previously, RAFT polymerizations have been reported with a variety of functional monomers including anionic,23–25 zwitterionic, 26,27 and neutral28–40 in both organic and aqueous media. 41–48 For example, our group conducted the initial controlled polymerization of the cationic methacrylamide monomer, N-[3-(dimethylamino)propyl]methacrylamide (DMAPMA), using aqueous media and 4-cyanopentanoic acid dithiobenzoate (CTP) as the chain transfer agent.18 An acidic environment was necessary in order to obtain controlled molecular weight (Mn) and low Mw/Mn values.
In order to provide a controlled synthetic mimic for cell penetration, we first prepared 3-guanidinopropyl methacrylamide (GPMA) and subsequently conducted its polymerization via aRAFT in an acetate buffer solution and mediated by cyano-4-(ethylsulfanylthiocarbonylsulfanyl)pentanoic acid (CEP) as the chain transfer agent. The polymerization showed linear molecular weight dependence with conversion, yielding control over both Mn and Mw/Mn. Chain extension of the polyGPMA macroCTA was successfully accomplished by adding GPMA, as was block copolymerization by adding GPMA to the poly HPMA macroCTA. HPMA was chosen as a comonomer since poly(HPMA) has the reported attributes of being biocompatible, non-immunogenic, and sufficiently hydrophilic to promote the enhanced permeability and retention (EPR) effect that allows accumulation within tumoral tissue. 49,50
In our work we adapted a one-step approach to the synthesis of GPMA first reported by Shea et al.51 We utilized amino propyl methacrylamide (APMA) and 2-ethyl-2-thiopseudourea hydrobromide (Scheme 1) to prepare the methacrylamide monomer in 72% yield (full experimental description and characterization (Schemes S1–S3), Figures S1–S4 can be found in the electronic supporting information). The guanidinium group, while not a strong nucleophile, is very basic with the guanidinium cation having a pka of around 13. The polymerization kinetics, shown in Figure 1, were determined using CEP as the chain transfer agent and V-501 as the initiator at two initial monomer concentrations ([M]0: 0.5 and 1.0 M). The linearity of the kinetic plots (Figure 1, top) up to 97% conversion demonstrates pseudo-first order behavior for the polymerization. The refractive index traces were symmetrical and shifted to lower elution volumes as the reaction proceeded (Figure 1, bottom). As often observed for CEP-mediated RAFT polymerization of acrylamido monomers, after an early initialization period, experimental molecular weights (close to those theoretically predicted) and narrow Mw/Mn values were observed with conversion (Figure 1, middle graph and Table 1).
Scheme 1.

Synthesis of 3-guanidinopropyl methacrylamide (GPMA) and subsequent aRAFT polymerization of the monomer to form a GPMA homopolymer and HPMA block copolymer.
Figure 1.

Plot of ln([M]0/[M]t) vs. conversion of the growing GPMA homopolymer fitted to a linear prediction (top). Plot of Mn vs. conversion of the growing GPMA homopolymer with Mn theoretical and Mw/Mn(middle) and Refractive Index traces (bottom).
Table 1.
GPMA homopolymers synthesized at both 0.5 and 1.0 M initial monomer concentration ([M]0) in acetate buffer at 70 °C with V-501 as free radical initiator and CEP as chain transfer agent.
| Time (h) | [M]0 | Conversion (%) | Mn (expt) | Mn (theory) | Mw/Mn |
|---|---|---|---|---|---|
| 1 | 0.5 | 0 | 300 | 300 | -- |
| 2 | 0.5 | 12 | 2800 | 1500 | 1.61 |
| 3 | 0.5 | 20 | 3200 | 2300 | 1.32 |
| 4 | 0.5 | 29 | 3700 | 3200 | 1.30 |
| 5 | 0.5 | 36 | 4500 | 3900 | 1.35 |
| 7 | 0.5 | 49 | 4600 | 5200 | 1.15 |
| 9 | 0.5 | 55 | 5000 | 5800 | 1.11 |
| 1 | 1.0 | 20 | 9800 | 5900 | 1.39 |
| 2 | 1.0 | 41 | 13800 | 11600 | 1.06 |
| 3 | 1.0 | 59 | 18200 | 16600 | 1.12 |
| 4 | 1.0 | 72 | 20000 | 20300 | 1.16 |
| 5 | 1.0 | 80 | 21800 | 22500 | 1.17 |
| 5.8 | 1.0 | 87 | 25700 | 24300 | 1.15 |
| 9.4 | 1.0 | 94 | 26900 | 26400 | 1.19 |
| 11.4 | 1.0 | 97 | 28300 | 27100 | 1.18 |
To further verify the livingness of the aRAFT polymerization of the GPMA monomer, we conducted blocking experiments with a PGPMA macro-CTA (Figure 2, top) and a PHPMA macro-CTA (Figure 2, bottom). PGPMA macro-CTA was synthesized in acetate buffer at pH 5.2 with CEP as the chain transfer agent and V-501 as the initiator. The polymerization was quenched after 6 h (17% conversion) and an aliquot was taken for GPC analysis (Figure 2, top). Additional V-501 was added and the reaction was allowed to proceed overnight (98% conversion). The shift in elution volume (Figure 2, top) of the GPMA37-b-GPMA61 homopolymer shows that the chain ends remained active with no detectable hydrolysis or aminolysis.
Figure 2.

Aqueous size exclusion chromatography trace of GPMA macroCTA chain extension with GPMA to form GPMA-b-GPMA copolymers (top). Aqueous size exclusion chromatography trace of HPMA macroCTA chain extension with GPMA to form HPMA-b-GPMA copolymer (bottom).
An HPMA-b-GPMA copolymer was also synthesized in order to prepare copolymers that structurally mimic CPPs or poly(arginine). The HPMA macroCTA was synthesized as previously reported.21 Following purification via dialysis and lyophilization, the macro-CTA was chain extended with GPMA using V-501 as the initiator. Since poly(arginine) exhibits optimum cellular uptake at lower (>6 repeat units)8, 9 segmental lengths, block copolymers consisting of a long HPMA block (Xn = 271) and a short GPMA block length (Xn = 13) were targeted. Chain extension was successful as demonstrated by the shift in elution volume (Figure 2, bottom) of the block copolymer.
The ability of GPMA98 and HPMA271-b-GPMA13 to enter cells via both endocytotic and non-endocytotic pathways was probed by incubating the polymers with KB cells at both 37 °C and 4 °C. At 37 °C, all energy dependent endocytotic pathways (clathrin-mediated endocytosis, caveolae, macropinocytosis, and phagocytosis) are operational.52 However, at 4 °C, ATP production is slowed considerably, and these pathways are thus inhibited.53 Therefore, uptake of any macromolecular structure should occur via an alternative uptake mechanism. Polymers were labeled with an amine-containing FITC dye via the carboxylic acid end group of the polymer using EDC coupling with sulfo-NHS in acetate buffer at pH 6. The polymer samples (HPMA458, GPMA98, and HPMA271-b-GPMA13) were incubated with KB cells for 2.5 h at both 37 °C and 4 °C. Each polymer sample was examined using fluorescence microscopy (Figure 3), confocal scanning laser microscopy (Figure S6) and flow cytometry (Figure S7). As expected, the HPMA homopolymer did not enter cells at 37 °C or 4 °C for the short incubation time period (Panels A & B in Figure 3) since there is no moiety for direct uptake.15, 16 Both the GPMA homopolymer and the cell penetrating peptide mimic (HPMA271-b-GPMA13) showed significant uptake into KB cells after incubating 2.5 h at 37 °C and 4 °C. In addition, flow cytometry results indicated that the HPMA271-b-GPMA13 copolymer had increased uptake compared to the GPMA homopolymer at 37 °C (Figure S7B). Fluorescence microscopy and flow cytometry of KB cells incubated with polymer at 4 °C showed both GPMA98 and HPMA271-b-GPMA13 entering cells (Panels D & F, Figure 3, and Figure S7B). The cell count and mean fluorescence for these cells were significantly lower than those observed for tests conducted at 37 °C (Table S1). To confirm polymer entry into the cells, cellular cross sections from top to bottom were examined by Z-stack confocal laser scanning microscopy (Figure S6). Together the above results suggest that GPMA98 and HPMA271-b-GPMA13 may enter the cell through both endocytotic and energy-independent pathways. Although not the primary focus of this communication, further studies will be necessary to fully understand the cellular uptake behavior and capabilities of these synthetic copolymers.
Figure 3.
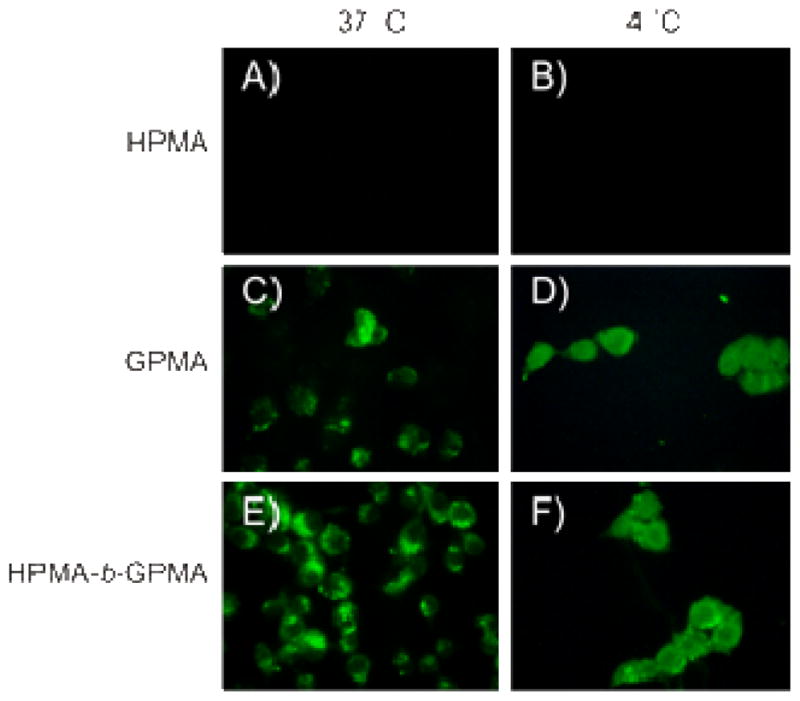
Fluorescent microscopy images of FITC-labeled polymers at 37 °C (A, C, E) and 4 °C (B, D, F) in KB cells. FITC-labeled HPMA (Mn = 60,000 g/mol) is shown in A, B with no fluorescence. GPMA (Mn = 18,100 g/mol) is shown in C and D, transfecting cells at 37 °C and at 4 °C. HPMA-b-GPMA (Mn = 39,810 g/mol) is shown in E and F to transfect cells at both 37°C and 4 °C.
In conclusion, we successfully synthesized homo- and block copolymers of a guanidinium-containing methacrylamide monomer using aRAFT polymerization. The block copolymer HPMA271-b-GPMA13 and homopolymer GPMA98 were incubated with KB cells at both 37 °C and 4 °C to investigate the mechanism of uptake. Fluorescence microscopy and flow cytometry results indicate intracellular uptake via both endocytotic and energy-independent pathways. The ability to tailor precise architectures, the molecular weight, and molecular weight distribution directly in water opens the door for guanidinium-functional polymers to be used as pro-drugs, in gene delivery or for other applications as advanced bio-materials.22
Supplementary Material
Acknowledgments
The authors would like to acknowledge financial support from the National Science Foundation through the MRSEC (DR-0213883) and NSF EPSCoR (EPS-0903787) programs and from NIH (R15CA152822). We thank Mississippi Functional Genomics Network for the use of Confocal Laser Scanning Microscope. Finally, we would like to thank Baobin Kang for technical assistance with the confocal microscope.
Footnotes
Paper #153 in a series entitled “Water-Soluble Polymers”
Supporting Information. Experimental details and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Derossi D, Joliot AH, Chassaing G, Prochiantz A. J Biol Chem. 1994;269:10444–50. [PubMed] [Google Scholar]
- 2.Snyder EL, Dowdy SF. Pharm Res. 2004;21:389–393. doi: 10.1023/B:PHAM.0000019289.61978.f5. [DOI] [PubMed] [Google Scholar]
- 3.Vives E. J Controlled Release. 2005;109:77–85. doi: 10.1016/j.jconrel.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 4.Vives E, Brodin P, Lebleu B. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 5.Silhol M, Tyagi M, Giacca M, Lebleu B, Vives E. Eur J Biochem. 2002;269:494–501. doi: 10.1046/j.0014-2956.2001.02671.x. [DOI] [PubMed] [Google Scholar]
- 6.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Proc Natl Acad Sci U S A. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Futaki S. Int J Pharm. 2002;245:1–7. doi: 10.1016/s0378-5173(02)00337-x. [DOI] [PubMed] [Google Scholar]
- 8.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. J Biol Chem. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 11.Vives E, Granier C, Prevot P, Lebleu B. Lett Pept Sci. 1997;4:429–436. [Google Scholar]
- 12.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pouton CW, Lucas P, Thomas BJ, Uduehi AN, Milroy DA, Moss SH. J Controlled Release. 1998;53:289–299. doi: 10.1016/s0168-3659(98)00015-7. [DOI] [PubMed] [Google Scholar]
- 14.Rousselle C, Clair P, Lefauconnier JM, Kaczorek M, Scherrmann JM, JT Mol Pharmacol. 2000;57:679–686. doi: 10.1124/mol.57.4.679. [DOI] [PubMed] [Google Scholar]
- 15.Nori A, Jensen KD, Tijerina M, Kopeckova P, Kopecek J. J Controlled Release. 2003;91:53–59. doi: 10.1016/s0168-3659(03)00213-x. [DOI] [PubMed] [Google Scholar]
- 16.Nori A, Jensen KD, Tijerina M, Kopeckova P, Kopecek J. Bioconjugate Chem. 2003;14:44–50. doi: 10.1021/bc0255900. [DOI] [PubMed] [Google Scholar]
- 17.Funhoff AM, Van Nostrum CF, Lok MC, Fretz MM, Crommelin DJA, Hennink WE. Bioconjugate Chem. 2004;15:1212–1220. doi: 10.1021/bc049864q. [DOI] [PubMed] [Google Scholar]
- 18.Scales CW, Huang F, Li N, Vasilieva YA, Ray J, Convertine AJ, McCormick CL. Macromolecules. 2006;39:6871–6881. [Google Scholar]
- 19.York AW, Huang F, McCormick CL. Biomacromolecules. 2009;11:505–514. doi: 10.1021/bm901249n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.York AW, Kirkland SE, McCormick CL. Adv Drug Delivery Rev. 2008;60:1018–1036. doi: 10.1016/j.addr.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 21.York AW, Zhang Y, Holley AC, Guo Y, Huang F, McCormick CL. Biomacromolecules. 2009;10:936–943. doi: 10.1021/bm8014768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunt JN, Feldman KE, Lynd NA, Deek J, Campos LM, Spruell JM, Hernandez BM, Kramer EJ, Hawker CJ. Advanced Materials. 2011;23:2327–2331. doi: 10.1002/adma.201004230. [DOI] [PubMed] [Google Scholar]
- 23.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS. J Controlled Release. 2009;133:221–229. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumerlin BS, Donovan MS, Mitsukami Y, Lowe AB, McCormick CL. Macromolecules. 2001;34:6561–6564. [Google Scholar]
- 25.Sumerlin BS, Lowe AB, Thomas DB, McCormick CL. Macromolecules. 2003;36:5982–5987. [Google Scholar]
- 26.Donovan MS, Lowe AB, Sanford TA, McCormick CL. J Polym Sci, Part A: Polym Chem. 2003;41:1262–1281. [Google Scholar]
- 27.Yusa S, Shimada Y, Mitsukami Y, Yamamoto T, Morishima Y. Macromolecules. 2003;36:4208–4215. [Google Scholar]
- 28.An Z, Shi Q, Tang W, Tsung CK, Hawker CJ, Stucky GD. J Am Chem Soc. 2007;129:14493–14499. doi: 10.1021/ja0756974. [DOI] [PubMed] [Google Scholar]
- 29.Bernard J, Hao X, Davis TP, Barner-Kowollik C, Stenzel MH. Biomacromolecules. 2006;7:232–238. doi: 10.1021/bm0506086. [DOI] [PubMed] [Google Scholar]
- 30.Boyer C, Liu J, Wong L, Tippett M, Bulmus V, Davis TP. J Polym Sci, Part A: Polym Chem. 2008;46:7207–7224. [Google Scholar]
- 31.Convertine AJ, Lokitz BS, Lowe AB, Scales CW, Myrick LJ, McCormick CL. Macromol Rapid Commun. 2005;26:791–795. [Google Scholar]
- 32.Donovan MS, Sanford TA, Lowe AB, Sumerlin BS, Mitsukami Y, McCormick CL. Macromolecules. 2002;35:4570–4572. [Google Scholar]
- 33.Favier A, Charreyre MT, Pichot C. Polymer. 2004;45:8661–8674. [Google Scholar]
- 34.Joso R, Stenzel MH, Davis TP, Barner-Kowollik C, Barner L. Aust J Chem. 2005;58:468–471. [Google Scholar]
- 35.Kakwere H, Chun CKY, Jolliffe KA, Payne RJ, Perrier S. Chem Commun. 2010;46:2188–2190. doi: 10.1039/b924112d. [DOI] [PubMed] [Google Scholar]
- 36.Millard PE, Barner L, Reinhardt J, Buchmeiser MR, Barner-Kowollik C, Mueller AHE. Polymer. 2010;51:4319–4328. [Google Scholar]
- 37.Mori H, Kato I, Matsuyama M, Endo T. Macromolecules. 2008;41:5604–5615. [Google Scholar]
- 38.Ouyang L, Wang L, Schork FJ. Polymer. 2011;52:63–67. [Google Scholar]
- 39.Ouyang L, Wang L, Schork FJ. Macromol Chem Phys. 2010;211:1977–1983. [Google Scholar]
- 40.Thomas DB, Convertine AJ, Myrick LJ, Scales CW, Smith AE, Lowe AB, Vasilieva YA, Ayres N, McCormick CL. Macromolecules. 2004;37:8941–8950. [Google Scholar]
- 41.Favier A, Charreyere MT, Chaumont P, Pichot C. Macromolecules. 2002;35:8271–8280. [Google Scholar]
- 42.Lowe AB, McCormick CL. Prog Polym Sci. 2007;32:283–351. [Google Scholar]
- 43.McCormick CL, Lowe AB. Acc Chem Res. 2004;37:312–325. doi: 10.1021/ar0302484. [DOI] [PubMed] [Google Scholar]
- 44.Moad G, Rizzardo E, Thang SH. Aust J Chem. 2006;59:669–692. [Google Scholar]
- 45.Moad G, Rizzardo E, Thang SH. Polymer. 2008;49:1079–1131. [Google Scholar]
- 46.Boyer C, Granville A, Davis TP, Bulmus V. J Polym Sci, Part A: Polym Chem. 2009;47:3773–3794. [Google Scholar]
- 47.Min EH, Ting SRS, Billon L, Stenzel MH. J Polym Sci, Part A: Polym Chem. 2010;48:3440–3455. [Google Scholar]
- 48.Liu Z, Hu J, Sun J, He G, Li Y, Zhang G. J Polym Sci, Part A: Polym Chem. 2010;48:3573–3586. [Google Scholar]
- 49.Kopecek J, Kopeckova P. Adv Drug Delivery Rev. 2010;62:122–149. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsumura Y, Maeda H. Cancer Res. 1986;46:6387–92. [PubMed] [Google Scholar]
- 51.Spivak D, Shea KJ. J Org Chem. 1999;64:4627–4634. doi: 10.1021/jo982118s. [DOI] [PubMed] [Google Scholar]
- 52.Jones AT. J Cell Mol Med. 2007;11:670–684. doi: 10.1111/j.1582-4934.2007.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duncan R, Lloyd JB. Biochim Biophys Acta, Gen Subj. 1978;544:647–655. doi: 10.1016/0304-4165(78)90339-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.