

Abstract
Especially mild, organic solvent-free conditions have been found that allow for allylic ethers to undergo Pd-catalyzed aminations.
Allylic ethers are not commonly regarded as reactive electrophiles in allylic substitution reactions. Indeed, unlike activated systems such as sulfonate, phosphate, halide, acetate, and carbonate groups, ethers (other than allylic epoxides) tend to be viewed more as hydroxyl protecting groups than as cross-coupling partners.1 Characteristics of allylic ethers, such as limited susceptibility to direct (usually high temperature) displacement by nucleophiles, reduced tendencies to undergo transition metal-catalyzed insertion into an electron-rich C–O σ-bond, and no option for hydrolysis, offer significant synthetic advantages. Examples of allylic ethers as electrophiles matched with highly reactive nucleophiles (e.g., Grignard reagents)2 certainly exist; however, there appears to be no general methodology for allylic amination by this functional group under mild conditions. Literature examples of net substitution by nitrogen involve relatively special circumstances (e.g., diphosphinidenecyclo-butene complexes, and use of phenolic resins).3 We have recently described an approach to allylic aminations4 of free allylic alcohols, which could be effected in water as the only medium in the presence of the non-ionic amphiphile PTS.5 To avoid, in general, the hydroxyl protecting group issue, we now present our studies on allylic amination reactions that rely on allylic alcohol-derived phenyl ether derivatives as leaving group (Fig. 1).6
Fig. 1.
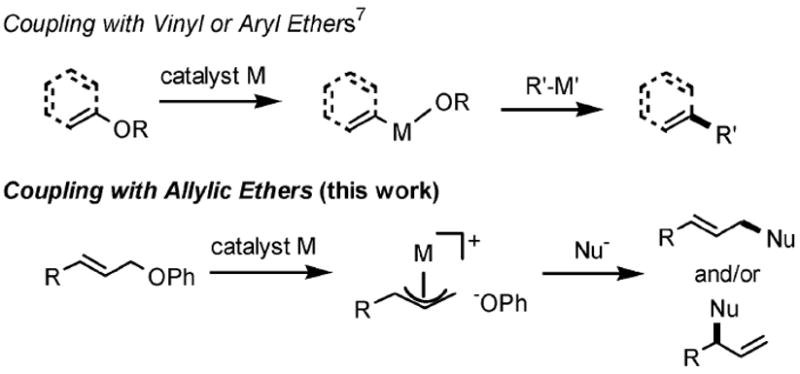
Use of phenyl ethers of allylic alcohols in coupling reactions.
Palladium-catalyzed allylic aminations occurred readily in water as the only medium containing only 2 wt% of nanomicelle-forming PTS (global concentration = 0.5 M).8 Optimization using cinnamyl phenyl ether (1a, 0.5 mmol) as a model educt, along with the sterically demanding nucleophile dibenzylamine (2a, 0.75 mmol), led to a very efficient coupling at room temperature (Table 1). Catalytic [Pd(allyl)Cl]2 (0.0025 mmol, 0.5 mol%) exposed to DPEphos as ligand (0.005 mmol, 1 mol%) appears to be the most effective combination (Run 11). Addition of methyl formate was also crucial, as its presence had been found previously as well to increase reaction rates of allylic alcohols toward allylic aminations.5 In the absence of this ester the amination was sluggish (Run 7) affording only 3% product after 30 minutes, and 24% after six hours (Run 11). Although dppf was quite effective as ligand among the mono- and bidentate ligands screened (Runs 1–9), rigid P–P ligand DPEphos gave the highest (quantitative) yield within the allotted 30 minute time frame (Run 11). By contrast, a biaryl bidentate P–N ligand was totally ineffective (Run 10).
Table 1.
Ligand effects
 | |||||
|---|---|---|---|---|---|
| Run | Ligand | Yield (%) | Run | Ligand | Yield (%) |
| 1 | PPh3a | 10 | 10 | 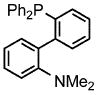 |
trace |
| 2 | P(o-tol)3a | trace | |||
| 3 | dppm | 7 | |||
| 4 | dppe | 8 | 11 | 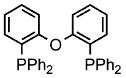 |
99 (3,b 24,b,c 53d) |
| 5 | dppp | 8 | |||
| 6 | dppb | 76 | |||
| 7 | dppf | 90 (5)b | 12 | 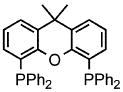 |
trace |
| 8 | biphep | 57 | |||
| 9 | rac-binap | 14 | |||
2 mol% of ligand was used.
In the absence of HCO2Me.
After 6 h.
Reaction was carried out “on water”.
Increasing rigidity in going from DPEphos to Xantphos resulted in only trace amounts of allylic amination (Run 12). The catalytic nanomicelle reactors formed spontaneously by PTS in water8,9 appear to play an important role, as a corresponding reaction run in its absence, or essentially “on water”, dropped the yield significantly (to 53%; Run 11). Surprisingly, there was no difference between reactions run under an inert argon atmosphere and those conducted fully exposed to air, notwithstanding the known instability of [Pd(allyl)Cl]2 under such conditions.
Under optimized conditions, the antifungal drug naftifine10 could be successfully synthesized from simple precursors in 98% yield in minutes at room temperature in water (Table 2, Run 1). Several analogs of naftifine derived from allylic phenyl ether derivatives were also prepared, as illustrated in Table 2. Methallyl phenyl ether (1b) reacted with N-methyl-N-naphthylmethylamine (2b) to afford 3c in 81% yield in one hour (Run 2). Unhindered allyl phenyl ether was by far the most reactive of the allylic ethers tested, leading to allyl amine 3d (91% yield) in five minutes (Run 3). Allylic phenyl ether 1d possessing a highly lipophilic alkyl chain gave derivative 3e in 90% yield with excellent linear- and E-selectivity (Run 4). Secondary phenyl ether 1e also reacted with amine 2b to produce 3f in 80% yield after five hours at room temperature (Run 5).
Table 2.
Reactions of allylic ethers (1a–e) with amine 2ba
| Run | Ether | Time (h) | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 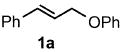 |
[20 min] | 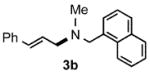 |
98 |
| 2 | 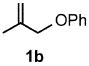 |
1.0 | 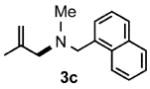 |
81 |
| 3 | 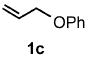 |
[5 min] | 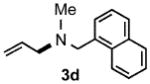 |
91 |
| 4 | 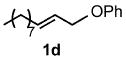 |
1.5 | 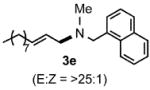 |
90 |
| 5 | 5.0 | 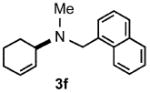 |
80 |
Reactions were carried out under air at rt in 2 wt% PTS–water in the presence of [Pd(allyl)Cl]2 (0.5 mol%), DPEphos (1 mol%), ether 1 (1 equiv.), amine 2b (1.5 equiv.), K2CO3 (1.5 equiv.) and HCO2Me (4 equiv.). Isolated yields.
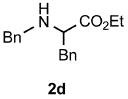
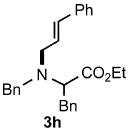
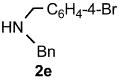
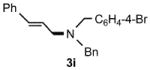
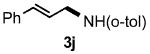
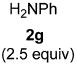
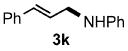
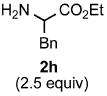
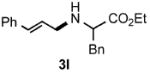
Functional group tolerance with respect to the amine component was also examined using cinnamyl phenyl ether 1a as the allylating agent (Table 3). Cross-coupling with the bulky secondary amine 2c derived from phenylalanine gave the desired trans-cinnamylated product 3g in 83% isolated yield. It is noteworthy that the aryl bromide present, normally a good coupling partner in Pd-catalyzed reactions, remained fully intact (Run A). Brominated product 3g could then be used in a subsequent cross-coupling also in PTS–water at room temperature with 4-anisylboronic acid to produce the biphenyl-containing amino acid ester 4 in 80% yield (Scheme 1).11 In the presence of both [Pd(allyl)Cl]2 and amines, an aromatic bromide is known to be reactive in water.12 Nonetheless, under our conditions allylation of the amine with an allylic phenyl ether occurred chemoselectively. The non-halogenated analog, phenylalanine derivative 2d, reacted much faster than did 2c with 1a (Run B). Reaction of 1a with N-4-bromobenzyl substituted benzylamine unexpectedly took longer, but allylic amine 3i was obtained again chemoselectively in over 80% yield (Run C). Mono-substituted amines were too reactive to selectively give mono-substituted allylated products. However, increasing the steric demand of the aryl ring, as in the case of o-toluidine (2f), led to only traces of the doubly allylated by-product (Run D). To suppress double substitution of the allylic fragment, 2.5 equivalents of amines were used thereby affording an 80% yield of mono-cinnamylated amine 3k (Run E). Ethyl phenyalanate (2h) gave mainly mono-allylated amine 3l (Run F).
Table 3.
Reaction of trans-cinnamyl phenyl ether (1a) with aminesa
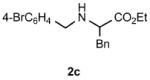
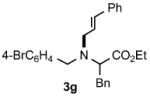
Reactions were carried out in air at rt in 2 wt% PTS–water in the presence of [Pd(allyl)Cl]2 (0.5 mol%), DPEphos (1 mol%), ether 1a (1 equiv.), amine 2 (1.5 equiv. or 2.5 equiv.), K2CO3 (1.5 equiv.) and HCO2Me (4 equiv.).
Isolated yields.
Doubly allylated product.
Scheme 1.
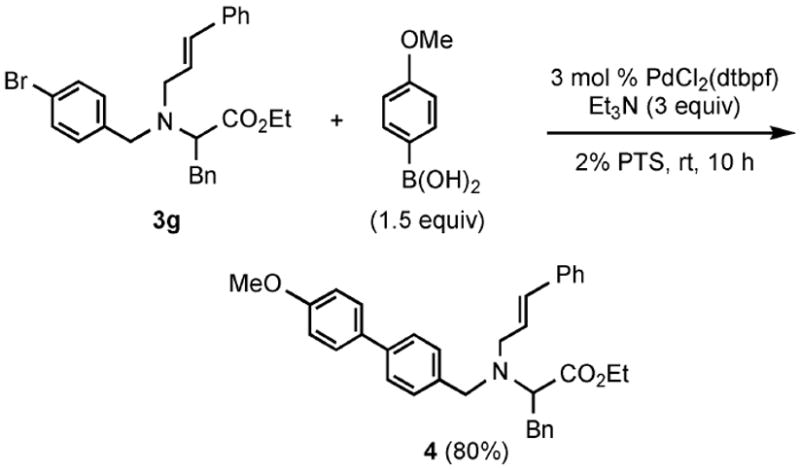
Suzuki–Miyaura coupling of allylated product 3g to 4.
While functionalized amino acid 2i was unreactive toward 1a, allyl phenyl ether itself (1c) reacted smoothly to produce 3l in 85% yield (Scheme 2). In the case of the allylic substrate 1f having both acetate and ether moieties (Scheme 3), coupling first occurred chemoselectively (in the absence of methyl formate) at the acetate in the presence of Xantphos to give 3n. That is, no product (3p) resulting from phenyl ether displacement was detected, nor was potential product 3o observed from double substitution. While the resulting amine 3n formed could be isolated (83%), its in situ generation allows for a second, single-flask coupling at the allylic phenyl ether site, once methyl formate and DPEphos has been added to the mixture. Thus, an unsymmetrical diamine such as 3q could be realized in excellent overall yield (Scheme 4).
Scheme 2.
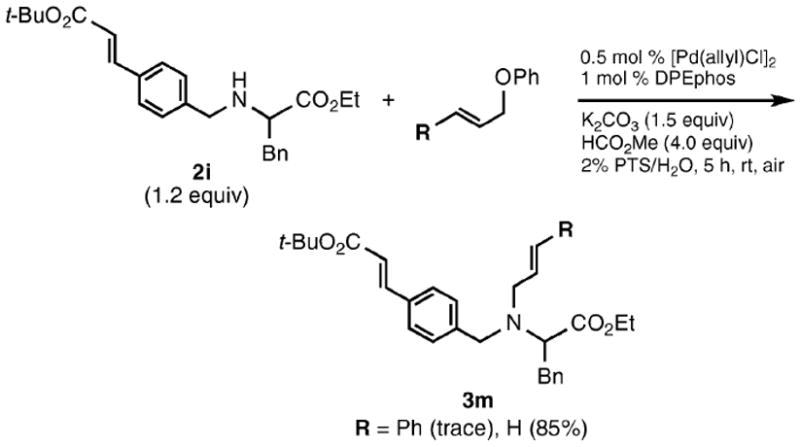
Limits of allylation.
Scheme 3.
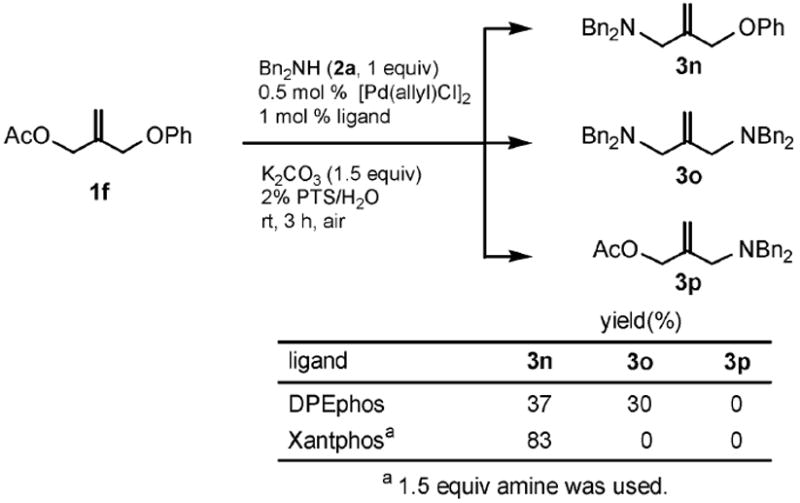
Chemoselective reaction of 1f.
Scheme 4.
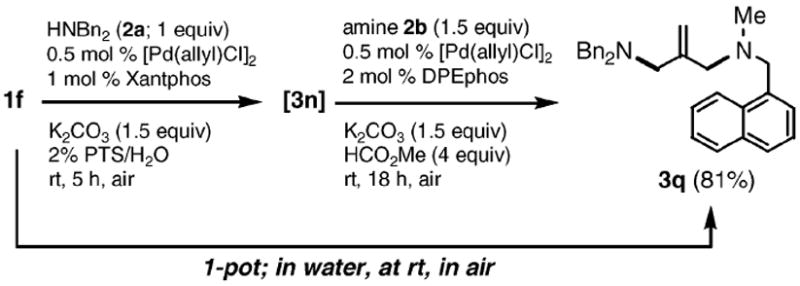
One-pot synthesis of diamine 3q from 1f.
In summary, the first general method for Pd-catalyzed aminations of allylic ethers has been developed. These reactions occur in excellent yields, without inert atmosphere conditions, in water as the only medium,13 and at room temperature. Further applications from our laboratory will be described in due course.
Supplementary Material
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds.
Notes and references
- 1.Lu Z, Ma S. Angew Chem Int Ed. 2008;47:258. doi: 10.1002/anie.200605113., and references cited therein; Weissman SA, Zewge D. Tetrahedron. 2005;61:7833.; Greene TW, Wuts PGM, editors. Protective Groups in Organic Synthesis. 3. John Wiley and Sons; New York: 1999. p. 17.; Takahashi K, Miyake A, Hata G. Bull Chem Soc Jpn. 1972;45:230.
- 2.Terao J, Watabe H, Watanabe H, Kambe N. Adv Synth Catal. 2004;346:1674. [Google Scholar]
- 3.(a) Mora G, Piechaczyk O, Le Goff XF, Le Floch P. Organometallics. 2008;27:2565. [Google Scholar]; (b) Murakami H, Minami T, Ozawa F. J Org Chem. 2004;69:4482. doi: 10.1021/jo049732q. [DOI] [PubMed] [Google Scholar]; (c) Fisher M, Brown RCD. Tetrahedron Lett. 2001;42:8227. [Google Scholar]; (d) Bergbreiter DE, Chen B, Lynch TJ. J Org Chem. 1983;48:4179. [Google Scholar]; (e) Inoue Y, Taguchi M, Toyofuki M, Hashimoto H. Bull Chem Soc Jpn. 1984;57:3021. [Google Scholar]
- 4.Muzart J. Eur J Org Chem. 2007:3077. [Google Scholar]
-
5.Nishikata T, Lipshutz BH. Org Lett. 2009;11:2377. doi: 10.1021/ol900235s.
- 6.Other leaving groups were not examined, although from related studies, it is expected to react far more slowly under similar conditions; see Nishikata T, Lipshutz BH. J Am Chem Soc. 2009;131:12103. doi: 10.1021/ja905082c.
- 7.Arylboron reagents: Tobisu M, Shimasaki T, Chatani N. Angew Chem Int Ed. 2008;47:4866. doi: 10.1002/anie.200801447.; Grignard reagents with aryl ether: Dankwardt JW. Angew Chem Int Ed. 2004;43:2428. doi: 10.1002/anie.200453765.; Grignard reagents with vinyl ether: Wenkert E, Michelotti EL, Swindell CS. J Am Chem Soc. 1979;101:2246.
- 8.Lipshutz BH, Ghorai S. Aldrichimica Acta. 2008;41:58. [PMC free article] [PubMed] [Google Scholar]
- 9.Dwars T, Paetzold E, Oehme G. Angew Chem Int Ed. 2005;44:7174. doi: 10.1002/anie.200501365. [DOI] [PubMed] [Google Scholar]
- 10.(a) Feuerstein M, Laurenti D, Doucet H, Santelli M. J Mol Catal A Chem. 2002:182. [Google Scholar]; (b) Feuerstein M, Laurenti D, Doucet H, Santelli M. Tetrahedron Lett. 2001;42:2313. [Google Scholar]
- 11.Lipshutz BH, Abela AR. Org Lett. 2008;10:5329. doi: 10.1021/ol801712e. [DOI] [PubMed] [Google Scholar]
- 12.Lipshutz BH, Chung DW, Rich B. Adv Synth Catal. 2009;351:1717. doi: 10.1002/adsc.200900323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Herrerías CI, Yao X, Li Z, Li C-J. Chem Rev. 2007;107:2546. doi: 10.1021/cr050980b. [DOI] [PubMed] [Google Scholar]; (b) Shaughnessy KH, DeVasher RB. Curr Org Chem. 2005;9:585. [Google Scholar]; (c) Li C-J. Chem Rev. 2005;105:3095. doi: 10.1021/cr030009u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.