

Abstract
Macrocycles are key structural elements in numerous bioactive small molecules and are attractive targets in the diversity-oriented synthesis of natural product-based libraries. However, efficient and systematic access to diverse collections of macrocycles has proven difficult using classical macrocyclization reactions. To address this problem, we have developed a concise, modular approach to the diversity-oriented synthesis of macrolactones and macrolactams involving oxidative cleavage of a bridging double bond in polycyclic enol ethers and enamines. These substrates are assembled in only 4–5 synthetic steps and undergo ring expansion to afford highly functionalized macrocycles bearing handles for further diversification. In contrast to macrocyclization reactions of corresponding seco-acids, the ring expansion reactions are efficient and insensitive to ring size and stereochemistry, overcoming key limitations of conventional approaches to systematic macrocycle synthesis. Cheminformatic analysis indicates that these macrocycles access regions of chemical space that overlap with natural products, distinct from currently-targeted synthetic drugs.
Macrocycles are large ring structures found in myriad natural products, and over 100 of these molecules have been developed into approved drugs1,2. Macrolactone and macrolactam structures are particularly prevalent (Fig. 1a), often derived from polyketide or peptide biosynthetic pathways. The macrocyclic constraint is a key structural element that organizes the overall molecular scaffold to present functional groups to biological targets in appropriate pharmacophoric conformations3–5. This conformational restriction can also provide increased binding affinity6 (although not necessarily entropic in origin7,8) and bioavailability9. Notably, a variety of macrocyclic natural products modulate challenging targets that are difficult to address with conventional drug-like molecules1, which are often comprised of small heteroaromatic structures.
Figure 1. Macrocyclic natural products and overall strategy for the diversity-oriented synthesis of macrolactones and macrolactams.

(a) Examples of macrocyclic natural products with diverse chemical structures and biological activities. (b) Overall approach to the diversity-oriented synthesis of functionalized macrocycles using oxidative ring expansion reactions. Polycyclic substrates are prepared using concise, modular reactions that allow for the introduction of chemical diversity both prior to and after ring expansion.
Accordingly, macrocycles are compelling targets in the diversity-oriented synthesis of natural product-based libraries for probe and drug discovery screening10,11. However, macrocycles are severely underexploited in this regard due to challenges associated with their synthesis. Indeed, a recent substructure search of the >360,000-compound NIH Molecular Libraries Small Molecule Repository returned only 22 macrolactones and macrolactams with 10-, 11-, or 12-membered ring carbon scaffolds of the sort treated herein (Supplementary Fig. 1). Thus, while individual macrocycles can commonly be synthesized by macrocyclization of an appropriate linear precursor, such reactions are highly sensitive to substrate effects that impact precursor conformation, such as ring size, substituent pattern, and stereochemical configuration12–15. As a result, this class continues to pose major challenges in the context of diversity-oriented synthesis, in which efficient, flexible, and ideally systematic access to a range of macrocyclic scaffolds is required. Indeed, previous efforts to synthesize macrocycle libraries by macrocyclization of diverse linear precursors have been hampered by the highly variable and unpredictable efficiency of these reactions16–19. The high-dilution conditions typically required for macrocyclizations are also poorly suited for use in library synthesis. Thus, macrocycles remain underrepresented in current diversity libraries and new approaches to macrocycle synthesis must be developed to capitalize fully upon the biological potential of these molecules1.
To address this chemical challenge, we posited that ring expansion reactions that are insensitive to substrate conformational effects would provide an attractive alternative to conventional macrocyclization-based strategies for systematic macrocycle library synthesis20–22. Such approaches have received very limited attention in diversity-oriented synthesis, restricted to the synthesis of individual core scaffolds23–25. We envisioned the general synthetic strategy outlined in Fig. 1b, which features rapid, modular substrate synthesis, leading to an oxidative ring expansion approach to macrolactones pioneered in the 1960s26–28 that builds upon the classic oxidative cleavage of Δ9,10-octalin to 1,6-cyclodecanedione29. Cyclic 1,3-diketone starting materials 1, which are readily available with a variety of backbone functionalities and ring sizes, would first be converted to the corresponding 1,3-disiloxydienes 2. Diels–Alder cyclocondensation reactions with various dienophiles (e.g., aldehydes, imines) would then provide the bicyclic enones 3. Enantioselective versions of this transformation can be readily envisioned, and the resulting ketone functionality could undergo stereoselective transformations, leveraging cyclic stereocontrol, with introduction of additional diversity elements as desired. This would provide the key bicyclic precursors 4 having a bridging double bond for the pivotal oxidative ring expansion in only 4–5 synthetic steps. The ring expansion reaction would then ideally proceed to macrocycles 5 efficiently and independent of substrate conformational effects that would impact conventional macrocyclizations of the corresponding linear precursors (e.g., seco-acids). Subsequent transformations could be used for additional downstream functionalization of these macrocyclic scaffolds.
We report herein the development of a concise, modular approach to the systematic synthesis of functionalized macrolactone and macrolactam scaffolds related to those found in numerous natural products, using oxidative ring expansion of polycyclic substrates having a bridging double bond. We further demonstrate that this ring expansion approach provides superior reaction efficiency and independence from substrate conformational effects compared to conventional macrocyclizations of related seco-acid substrates. We have successfully applied this strategy to the synthesis of a first-generation library of macrocycles having diverse ring sizes and substituent patterns. Cheminformatic analyses of structural and physicochemical parameters using PCA and of three-dimensional molecular shapes using principal moment of inertia (PMI) analysis indicate that this library accesses regions of chemical space that overlap with natural products, including known macrolactones and macrolactams, and are complementary to those addressed by high-profile synthetic drugs currently targeted by the pharmaceutical industry and related drug-like libraries.
RESULTS
Modular synthesis of polycyclic ring expansion substrates
The requisite polycyclic enol ether and enamine substrates for ring expansion were synthesized as shown in Fig. 2. Cyclic ketones 6 were converted to β-diketones 7 by α-C-acetylation30,31 (7a,d are commercially available). Two sequential treatments with LDA and TMSCl in one pot provided the corresponding bis(trimethylsilyloxy)dienes 8 in ≥90% purity after filtration through basic alumina32,33. Hetero-Diels–Alder reactions with aldehydes and N-nosyl imines afforded dihydropyrones 9 and dihydropyridones 10, respectively34. The linear Mukaiyama aldol adduct was observed by TLC prior to TFA-induced cyclodehydration in all cases, and was isolated in the case of 8a → 9a. The intermediate Lewis acidity of Yb(OTf)335 (vs. ZnCl2 or BF3·OEt2) proved well-suited for the initial Mukaiyama aldol step and 4 Å MS were required for high efficiency36, presumably by preventing hydrolysis of the diene substrate. The ketone functionality then underwent diastereoselective reductions to 11 and 1237, followed by silylation to afford ring expansion substrates 13 and 14 in excellent overall yields (see Table 2 below). Notably, these reactions could be carried out on multigram scale to enable scale-up of individual scaffolds as necessary.
Figure 2. Modular approach to diverse polycyclic substrates for oxidative ring expansion.

Average yields en route to 13 and 14 are shown in brackets (cf. Table 2). a) LDA (inverse addition), THF, −78°C, 1 h; AcCl (inverse addition), −78°C, 1 h or Ac2O, BF3(AcOH)2, 4.5 h; NaOAc, MeOH, H2O, 85 °C, 2.5 h; [85%]. b) LDA, THF, −78 °C, 1 h; TMSCl, −78 °C → rt, 1.5 h; LDA, −78°C, 1 h; TMSCl, −78 °C → rt, 1.5 h; [96%]. c) R1CHX, Yb(OTf)3, 4 Å MS, THF, 12 h; TFA, CH2Cl2, rt, 20 min; [86%]. d) NaBH4, CeCl3, CH2Cl2, MeOH, −78°C, 2 h or LiEt3BH, toluene, −78°C, 40 min; [94%]. e) TBSCl, imidazole, DMF, 50°C; [87%]. LDA = lithium diisopropylamide; MS = molecular sieves; Ns = 2-nitrobenzenesulfonyl; TBS = tert-butyldimethylsilyl; TFA = trifluoroacetic acid; TMS = trimethylsilyl. (See Supplementary Methods for full details).
Table 2.
Synthesis of diverse macrolactones and macrolactams by oxidative ring expansion
| Entry | Macrocycle Product | Precursor Yield (4 steps) [%][a] | Product Yield [%] | |
|---|---|---|---|---|
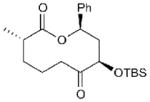
| 1 |
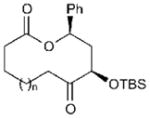
|
15a (n = 1) | 81 (13a) | 71[b] |
| 2 | 15b (n = 2) | 78 (13b) | 70[b] | |
| 3 | 15c (n = 3) | 73 (13c) | 71[b] | |
| 4 |
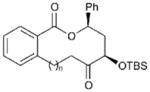
|
15d (n = 1) | 68 (13d) | 56[c] |
| 5 | 15e (n = 2) | 52 (13e) | 69[c] | |
| 6 | 15f (n = 3) | 67 (13f) | 59[c] | |
| 7 |
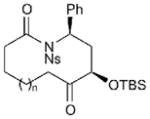
|
16a (n = 1) | 73 (14a) | 68[d] |
| 8 | 16b (n = 2) | 75 (14b) | 65[d] | |
| 9 | 16c (n = 3) | 66 (14c) | 66[d] | |
| 10 |
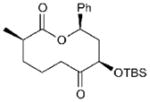
|
17 | 60 | 69[c] |
| 11 |
|
18 | 56 | 60[c] |
| 12 |
|
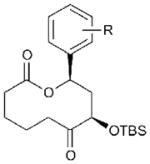
19 (R = m-I) | 71 | 70[c] |
| 13 | 20 (R = p-CH2-OMOM) | 45 | 71[c] | |
| 14 |
|
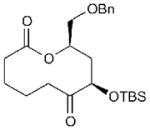
21 | 53 | 55[c] |
Overall yield of ring expansion substrates (e.g., 13,14) over four steps from diketones 7, which were commercially available (7a,d) or synthesized from 6 as shown in Fig. 2. (See Supplementary Methods for full details).
3.5 mol% RuCl3, NaIO4, 1:1 H2O/1,2-DCE, rt, 20–24 h.
3.5 mol% RuCl3, Oxone (DuPont), NaHCO3, 1:1.45 H2O/CH3CN, rt, 2–18 h
17 mol% RuO2, NaIO4, 1:2.3:1:1 H2O/CH2Cl2/acetone/CH3CN, rt, 1–2 h.
Ring expansions to form macrolactones and macrolactams
Initial studies on the pivotal ring expansion reactions were carried out with bicyclic dihydropyran derivative 13a (Table 1). Pioneering earlier work demonstrated the utility of m-CPBA and ozone for the oxidative cleavage of bridging olefins in ring expansion reactions of cyclic enol ethers26–28, building upon the classic oxidative cleavage of Δ9,10-octalin to 1,6-cyclodecanedione29. Unfortunately, while m-CPBA was effective for 13a (entry 1), it proved less efficient when applied to other substrates, such as enamine 14a, for which a complex mixture resulted. Initial attempts at ring expansion of 13a using ozonolysis were also disappointing, yielding only 11% of the desired macrolactone 15a (entry 2).
Table 1.
Oxidants for ring expansion of 13a to 15a.[a]
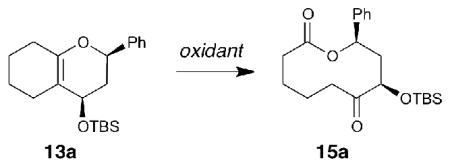
| |||
|---|---|---|---|
| Entry | Oxidant | Conditions | Yield [%] |
| 1 | m-CPBA | CH2Cl2, rt, 1h | 66 |
| 2 | O3 | CH2Cl2, −78 °C, 5 min; PPh3 | 11 |
| 3 | 0.5 mol% RuCl3, NaIO4 | H2O/1,2-DCE, rt, 24 hb | 62 |
| 4 | 3.5 mol% RuCl3, NaIO4 | H2O/1,2-DCE, rt, 20 h | 71 |
| 5 | 10 mol% RuCl3, NaIO4 | H2O/1,2-DCE, rt, 20 h | 37 |
| 6 | 3.5 mol% RuCl3, Pb(OAc)4 | H2O/1,2-DCE, rt,1h | 35 |
| 7 | 3.5 mol% RuCl3, Oxone | NaHCO3, H2O/CH3CN, rt, 2 h | 82 |
| 8 | 17 mol% RuO2, NaIO4 | H2O/CH2Cl2/acetone/CH3CN, rt, 1 h | 69 |
1,2-DCE = 1,2-dichloroethane; m-CPBA = 3-chloroperbenzoic acid; Oxone (DuPont) = potassium peroxomonosulfate;
conversion not complete after 1 day.
We were encouraged to find that a variety of RuO4-mediated reactions provided promising results38,39. Catalytic RuCl3 with NaIO4 as the stoichiometric oxidant was most effective at 3.5 mol% catalyst (entries 3–5)38–40. Although use of Pb(OAc)4 as the stoichiometric oxidant provided lower yields (entry 6), a buffered Oxone (DuPont)-based system was highly effective (entry 7)38,39. Furthermore, RuO2 could be substituted for RuCl3 as the precatalyst (entry 8)41,42.
Based on these results, we evaluated the most effective Ru-based oxidant systems across a wider range of substrates to produce both macrolactone and macrolactam scaffolds (Table 2). We were gratified to find that the ring expansion reactions proceeded with consistent yields across multiple ring sizes (entries 1–3, 4–6, 7–9), independent of ring size-based conformational effects that limit the utility of classical macrocyclization-based approaches in diversity-oriented synthesis. While slightly different oxidant systems proved optimal for the enol (13a–c), aryl enol (13d–f), and enamine substrate series (14a–c), this is not surprising given the electronically distinct nature of the reactive olefins in each series. More importantly, while substrate stereochemistry often has a large influence on the efficiency of classical macrocyclization reactions19, the ring expansion reaction was relatively insensitive to these effects, affording the diastereomeric methyl-substituted macrocycles 17 and 18 in similar efficiencies (entries 10,11; see Supplementary Methods for full details).
Related macrocycles 19–21 were synthesized by the analogous processes (entries 12–14; see Supplementary Methods for full details) and further highlight the tolerance of our ring expansion approach to latent functionalities that allow further downstream diversification. Macrocycle 19 includes an aryl iodide moiety suitable for subsequent palladium-catalyzed cross-coupling reactions. Macrocycles 20 and 21 contain protected alcohols and demonstrate the feasibility of including Ru-oxidation-sensitive benzyl ether moieties, albeit with somewhat reduced yield in the latter case. Macrocycle 21 also demonstrates the compatibility of the route with incorporation of aliphatic aldehydes at the Diels–Alder stage of the substrate synthesis.
Comparison to classical macrocyclization reactions
Next, to compare the efficiency of this ring expansion approach to conventional macrocyclizations, we investigated macrolactonization and macrolactamization reactions of the corresponding seco-acid substrates. We recognized that such substrates might be accessed systematically and conveniently for such studies by saponification of our macrocyclic ring expansion products. However, in initial experiments, we found that the seco-acid precursors that would lead directly to macrocycles 15a and 16a appear to be inherently unstable due to the reactive ketone functionality (see Supplementary Methods for full details). Accordingly, these macrocycles would be, at best, very difficult to access directly by classical macrocyclization.
To investigate seco-acid substrates that have closely related structures and might also reasonably be converted to our macrocyclic products after cyclization, the reactive ketone functionality in 15a–d was masked by methylenation (23a–d), after which saponification provided seco-acids 22a–d (See Supplementary Methods for full details). We then attempted macrocyclizations of these substrates with Shiina’s MNBA reagent, which is often effective for larger (≥13-membered) ring systems and is the preferred reagent in such macrolactonizations43,44. Strikingly, none of these substrates underwent efficient macrocyclization under the standard conditions (high-dilution, slow inverse addition over 22 h), with large amounts of starting material remaining (Table 3). Attempted macrolactamization of a related N-nosyl amino acid substrate derived from 16a under conditions that are effective for similar N-sulfonylamino acid substrates (DCC, 4-pyrrolidinopyridine45) led only to a linear byproduct (see Supplementary Methods for full details). Conditions that have been used in other macrolactamizations (HATU, DBU or Et3N46) led to complex mixtures. These results further highlight the unique effectiveness of our ring expansion approach in accessing these 10- to 12-membered macrolactones and macrolactams.
Table 3.
Macrocyclization of corresponding seco-acid substrates.[a]
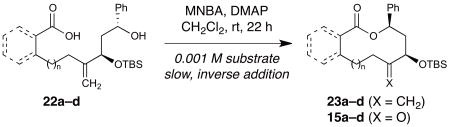
| ||||
|---|---|---|---|---|
| Entry | Substrate | Unreacted 22 [%][b] | Lactone 23 [%][b] | Corresponding Ring Expn Yield (15) [%][c] |
| 1 | 22a (n = 1) | 63 | 14 | 71 (15a) |
| 2 | 22b (n = 2) | 63 | ≤5 | 70 (15b) |
| 3 | 22c (n = 3) | 56 | 14 | 71 (15c) |
| 4 | 22d (n = 1, benzo) | 44[d] | 31 | 56 (15d) |
DMAP = 4-dimethylaminopyridine; MNBA = 2-methyl-6-nitrobenzoic anhydride.
Yields determined by 1H-NMR analysis of crude products after aqueous workup, based on an internal standard.
Isolated yields of 15a–d from Table 2.
Corresponding mixed anhydride observed intact after aqueous workup by 1H-NMR and MS.
Further diversification of macrocyclic scaffolds
With a variety of macrocyclic scaffolds in hand, we investigated several downstream reactions to provide additional structural diversity and functional group handles for coupling reactions (Fig. 3). As hoped, aryl iodide 19 proved an effective partner in several palladium-catalyzed cross-coupling reactions, including Sonogashira, Negishi, and Stille reactions (24–26). The nascent ketone functionalities in 15a–f can also serve as handles for additional diversification, as highlighted by diastereoselective reductions to provide the corresponding alcohols 27a–f. Alternatively, the ketone functionality of 15a–d could be modified by Petasis methylenation to afford olefins 23a–d. Subsequent hydroboration oxidation or epoxidation of 23a provided alcohol 28 or epoxide 29, respectively. In addition, the TBS-protected secondary alcohol 15a could be desilylated to yield α-hydroxy ketone 30 and acylated to give α-acetoxy ketone 31, illustrating the utility of this handle for further diversification.
Figure 3. Functionalization of macrocyclic scaffolds.

a) TBSOCH2CH2C≡CH, Pd(PPh3)4, CuI, Et3N, DMF, 60°C, 18 h, 81%. b) 4-MeOPhZnCl, Pd(PPh3)4, THF, rt, 16 h, 72%. c) Bu3SnCH=CH2, Pd2(dba)3, AsPh3, THF, 50°C, 18 h, 81%. d) NaBH4, CeCl3, MeOH, CH2Cl2, −78°C, 2 h; 27a: 86%, ≥98:2 dr, 27b: 93%, 1:1 dr; 27c: 99%, 90:10 dr; 27d: 55%, ≥98:2 dr; 27e: 73%, 96:4 dr; 27f: 67%, ≥98:2 dr. e) Cp2TiMe2, toluene, 65°C, 12 h; 23a: 55%; 23b: 55%; 23c: 50%; 23d: 71%. f) 9-BBN, THF, 0°C→ rt, 12 h, then NaOH, H2O2, 0°C→ rt, 1.5 h, 70%, ≥98:2 dr. g) DMDO, CH2Cl2, 0°C→ rt, 12 h, 88%, 63:37 dr. h) TBAF, AcOH, THF, rt, 3.5 h, 72%. i) AcCl, pyridine, CH2Cl2, 0°C, 10 min, 79%. 9-BBN = 9-bora-bicyclo[3.3.1]nonane; DMDO = dimethyldioxirane; DMF = N,N-dimethylformamide; TBAF = tetrabutylammonium fluoride; TBS = tert-butyldimethylsilyl; THF = tetrahydrofuran.
Cheminformatic analysis of macrocycle library
To assess the ability of our macrocycle library to access underrepresented regions of chemical space, we carried out principal component analysis (PCA) on a collection of 32 macrocyclic structures (see Supplementary Fig. 2) derived from the scaffolds shown in Table 2 and Fig. 3 by in silico desilylation as necessary. Using PCA, we compared the macrocycle library with our established reference sets of 40 top-selling brand name drugs, 60 diverse natural products, and 20 drug-like library members10. The drug reference set was selected to illustrate the structural bias in high-profile synthetic drugs currently targeted by the pharmaceutical industry. The broad natural product reference set was selected to represent a diverse range of biological activities and biosynthetic origins. This set includes all 24 compounds identified previously as having led to an approved drug in 1981–200647, as well as 13 other clinically used drugs. The drug-like library reference set is comprised of 10 molecules in one scaffold class each from ChemBridge and ChemDiv, two of the earliest and most widely used commercial library suppliers. These drug-like compounds are all present in the NIH Molecular Libraries Small Molecule Repository. An additional reference set of 24 macrocyclic natural products (see Supplementary Fig. 3) was also included to assess the ability of the macrocycle library to target a similar region of chemical space.
Each compound was analyzed for our established set of 20 calculated structural and physicochemical properties10 (see Supplementary Methods for full details on parameter selection), with PCA resulting in rotation of the complete 20-dimensional dataset onto three unitless, orthogonal axes that represent linear combinations of the original 20 parameters. Because several of the 20 properties are highly correlated with one another (see Supplementary Fig. 4 for loading plots and table), especially parameters that scale with molecular size, this three-dimensional plot is able to represent 75% of the information in the full 20-dimensional dataset (Fig. 4a–c and Supplementary Fig. 5). Similarly, >90% of the information in the full dataset is represented in the first six principal components.
Figure 4. Cheminformatic analyses of macrocycle library.

Principal component analysis (PCA) and principal moment of inertia (PMI) plots of 32 macrocycle library members, 24 macrocyclic natural products, and established reference sets of 40 top-selling brand name drugs, 60 diverse natural products, and 20 ChemBridge and ChemDiv drug-like library members. (a) PCA plot of PC1 vs. PC2 generated from 20 structural and physicochemical parameters. (b) PCA plot of PC3 vs. PC2. (c) PCA plot of PC1 vs. PC3. (d) PMI plot and representative macrocycle library members with lowest energy conformations having characteristic shapes. Structures 15f′, 16c′, 20′, and 26′ are derived from macrocycles 15f, 16c, 20, and 26, respectively, via in silico desilylation (see Supplementary Fig. 2). See Supplementary Figs. 5 and 6 for expanded PCA and PMI plots and Supplementary Datasets 1 and 2 for complete data. See Supplementary Fig. 7 for PMI plots with additional conformers within 3 kcal/mol of the minimum.
As hoped, the macrocycle library accesses a distinct region of chemical space compared to our reference sets containing current, high-profile drugs and related drug-like libraries, overlapping to a significant extent with the macrocyclic natural products. Examination of the component loadings in the PCA (see Supplementary Fig. 4) indicates that molecular weight and other parameters associated with molecular size shift molecules to the left along the x-axis (PC1). Hydrophobicity and aromatic ring count shift molecules upward along the y-axis (PC2), while aqueous solubility shifts molecules downward. Notably, larger ring systems shift molecules rearward (negative) along the z-axis (PC3), and stereochemical complexity shifts molecules left, downward, and rearward. Because the macrocyclic natural product reference set and our macrocycle library are comprised of relatively smaller molecules having similar molecular sizes to those of the drug and drug-like library reference sets, these last two properties appear to be important in differentiating the macrocycles from our reference sets of current, high-profile synthetic drugs and related drug-like libraries (Fig. 4b).
We next carried out a molecular shape analysis using normalized ratios of principal moments of inertia (PMI) to visualize the three-dimensional shape diversity of the various compound datasets in a chemically intuitive manner, as previously reported19,48 (Fig. 4d and Supplementary Fig. 6). In this analysis, a stochastic conformational search is carried out for each molecule (see Supplementary Methods for full details). Low-energy conformations are identified for each molecule in the data set and three PMI values (I1, I2, and I3; where I3 ≥ I2 ≥ I1) are calculated for each conformer. Normalized ratios of PMI (I1/I3 and I2/I3) are then calculated and plotted on a triangular graph, with the vertices (0,1), (0.5,0.5), and (1,1) representing a perfect rod, disc, and sphere, respectively48. To account for the fact that molecules may not always bind to proteins in energy-minimized conformations49, a collection of conformers within 3 kcal/mol of the minimum energy conformer was generated for each of the structures. For ease of visualization, the lowest energy conformers are plotted in Fig. 4d; additional PMI plots including all conformers up to various energy cutoffs above the minimum are shown in Supplementary Fig. 7.
While the distinctions between the different reference sets are less obvious in this PMI plot, the drug reference set tends to occupy rod-like space with some disc-like character (Fig. 4d). In contrast, our natural product reference set spans a much wider range of shapes, extending well into sphere-like space. Notably, both the macrocyclic natural products, including several approved drugs, and our macrocycle library occupy similar regions of the PMI plot, displaying decreased rod-like character in comparison to the drug reference set. The structures of four macrocycle library members are shown in Fig. 4d to illustrate the differences in molecular shape between the lowest energy conformers. Structures 15f′, 16c′, 20′, and 26′ are derived from macrocycles 15f, 16c, 20, and 26, respectively, via in silico desilylation (see Supplementary Fig. 2). Macrocycle 20′ falls closest to the rod vertex of the triangular plot (0,1) and its elongated, rod-like shape is evident when compared to the more flat, disc-like shape of macrocycle 15f′. In comparison, the substitution patterns of functional groups in the scaffolds of macrocycles 16c′ and 26′ impart more sphere-like character to these ring systems.
DISCUSSION
Despite their rich biological pedigree and distinctive molecular recognition capabilities, macrocycles remain severely underrepresented in current probe and drug discovery efforts1. The influence of conformational effects based on ring size, stereochemistry, and backbone substitution patterns on the efficiency of macrocyclization reactions, as well as the need for long reaction times under high-dilution conditions, generally limit the utility of macrocyclization-based approaches in the synthesis of macrocycle libraries. Indeed, previous efforts to synthesize natural product-inspired macrocycle libraries using macrocyclization reactions have displayed considerable dependence on the substitution patterns of the linear precursors16–19. For example, in a recent study, Marcaurelle and coworkers reported the synthesis of a collection of stereochemically diverse 8- to 14-membered rings using an aldol-based ‘build/couple/pair’ strategy19. Several different cyclization reactions were used (nucleophilic aromatic substitution, Huisgen [3+2] cycloaddition, ring-closing metathesis), and all were influenced considerably by the stereochemistry of the corresponding linear substrates. In some cases, changing a single stereochemical configuration in the linear substrate led to as much as 43% variation in yields of isolated cyclic products.
To overcome these limitations of classical macrocyclization strategies in the context of diversity-oriented synthesis, we have developed a ring expansion approach to systematic macrocycle synthesis that provides efficient access to a variety of macrocyclic scaffolds that comprise the core structures of numerous natural products. Our polycyclic ring expansion substrates are synthesized via a concise, 4–5 step route in high overall yields, requiring only two chromatographic steps (Figs. 1b and 2). This modular synthetic approach provides a range of ring sizes and substitution patterns and also leverages cyclic stereocontrol during substrate assembly. While the carbonyl functionalities of 4 and 5 were reduced herein, a variety of other diastereoselective transformations can be envisioned to provide additional diversity in the future (Fig. 1b). The oxidative ring expansion reactions proceed efficiently and predictably, independent of ring size and stereochemistry-based substrate conformational effects. In addition, our ring expansion approach uses mild conditions that allow for the inclusion of a wide array of latent functionalities in the macrocyclic products. Thus, these macrocycles are poised for a variety of downstream transformations that provide both additional structural diversity and functional group handles for coupling other building blocks in the future (Fig. 3).
Moreover, we have demonstrated explicitly that this ring expansion approach provides 10- to 12-membered lactones and lactams that are not readily accessible via classical macrocyclizations of seco-acid substrates (Table 3). Macrolactonization reactions of seco-acids 20a–d using Shiina’s MNBA reagent led to low conversion to the desired macrocycles after 22 h, with unreacted starting material and oligomeric byproducts observed. In contrast, the oxidative ring expansion reactions used to synthesize macrolactones 15a–d proceeded to complete conversion in 2–24 h, affording the target molecules in good isolated yields. Notably, the ring expansion reactions are carried out in aqueous solution and do not require inert atmosphere, slow addition or high substrate dilution, overcoming major technical limitations of classical macrocyclization reactions in the context of library synthesis. Taken together, these aspects of the ring expansion approach illustrate the utility of our method for the diversity-oriented synthesis of macrocycle libraries.
Cheminformatic analyses using PCA suggest that the macrocyclic scaffolds generated via our ring expansion method address regions of chemical space that are distinct from those addressed by our reference sets of current, high-profile synthetic drugs and related drug-like libraries (Fig. 4a–c). The component loadings (Supplementary Fig. 4) can be used to understand the relative influence of the original 20 physicochemical parameters upon the positioning of molecules in the PCA plot. These data indicate that hydrophobicity and aromatic character are among the most important parameters contributing to the positioning of the drug and drug-like library reference sets. Meanwhile, stereochemical complexity and aqueous solubility are two of the key parameters that differentiate the macrocycle library from these reference sets. Insights gleaned from such analyses can now be leveraged in the design and planning of future libraries that may probe more deeply into natural product-like space.
While PCA highlights differences in structural and physicochemical properties between compound classes, PMI analysis illustrates differences in the three-dimensional molecular shapes of macrocycles compared to our reference sets of current, high-profile synthetic drugs and related drug-like libraries (Fig. 4d). The drug reference set spans the region of rod- and disc-like shapes, with a bias toward the rod vertex of the triangle. This observation is consistent with previous PMI analyses of drugs and drug-like molecules48,50. Relative to the drug reference set, both the macrocyclic natural products and our macrocycle library members cluster farther from the rod vertex of the triangle and indicate a greater degree of sphere-like character. This difference in three-dimensional shapes may impact the abilities of these molecules to address challenging biological targets1.
In conclusion, we have developed an efficient, modular ring expansion approach to the systematic synthesis of macrocyclic scaffolds similar to those found in numerous natural products. This chemistry has been applied to the production of a first-generation macrocycle library that has been submitted to the NIH Molecular Libraries Program (PubChem Substance search term: DST_MC1_*). These macrocyclic scaffolds are now being screened against a wide range of challenging biological targets to evaluate further the functional capabilities of this important class of molecules. Biological screening results and further expansion of this synthetic approach will be reported in due course.
METHODS
General procedures for key steps in the synthesis of macrolactones 15a–f and macrolactams 16a–c are outlined below. Unless otherwise noted, reactions were performed in flame-dried glassware under an anhydrous Ar atmosphere. See Supplementary Methods for additional protocols, detailed experimental and cheminformatic procedures, and complete analytical data for all compounds.
General protocol for hetero-Diels–Alder reactions
Ground 4 Å molecular sieves (660 mg/mmol dienophile) were placed in a roundbottom flask and flame dried under high vacuum. After cooling to rt, anhydrous THF (3.0 mL/mmol dienophile) was added, followed by the bis(trimethylsiloxy)diene 8a–f (1.05–2.00 equiv) and benzaldehyde (1.00 equiv). Neat Yb(OTf)3 (2.0 mol%) was added, the flask was sealed with a glass stopper, and the mixture was stirred overnight at rt. TLC analysis of a reaction aliquot (diluted with EtOAc) indicated complete consumption of the dienophile and formation of the intermediate Mukaiyama aldol product. The reaction mixture was then filtered through a pad of celite and the pad was rinsed thoroughly with EtOAc and CH2Cl2. The solvents were then removed by rotary evaporation.
The resulting residue was dissolved in CH2Cl2 (9.5 mL/mmol dienophile) in an Ar-flushed roundbottom flask. TFA (0.5 mL/mmol dienophile) was added and the mixture was stirred at rt until the reaction was judged complete by TLC. The reaction was quenched with satd aq NaHCO3 and the mixture was stirred vigorously for 20 min. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3×). The combined organic extracts were dried (MgSO4), filtered, and concentrated by rotary evaporation. Purification by silica flash chromatography (hexanes/EtOAc) afforded the dihydropyrone 9a–f or dihydropyridone 10a–c.
General protocol for oxidative ring expansion using RuCl3 with NaIO4 stoichiometric oxidant
TBS-protected alcohol 13a–c was dissolved in 1,2-dichloroethane (5.50 mL/mmol substrate). Water (4.50 mL/mmol) was added, followed by a solution of RuCl3 (0.035 M in water, 3.5 mol%). Neat NaIO4 (2.00 equiv) was added in one portion, and the resulting biphasic mixture was stirred until the reaction was judged complete by TLC analysis of a reaction aliquot (quenched with satd aq Na2S2O3, extracted with EtOAc). Satd aq Na2S2O3 was added and the resulting mixture was stirred for 10 min, then extracted with CH2Cl2 (5×). The combined organic extracts were washed with water and brine, and dried (MgSO4). To the desiccant slurry, a spatula of celite was added and the mixture was agitated for 5 min. The suspension was then filtered through a plug of celite and the solvents were removed by rotary evaporation. Purification by silica flash chromatography (hexanes/EtOAc, 0.5 vol% Et3N) afforded macrolactone 15a–c.
General protocol for oxidative ring expansion using RuCl3 with Oxone (DuPont) stoichiometric oxidant
TBS-protected alcohol 13d–f was dissolved in acetonitrile (16.0 mL/mmol). Water (10.0 mL/mmol substrate) was added, which in most cases resulted in the precipitation of substrate from solution. RuCl3 (0.035 M in water, 3.5 mol%) was added and, after stirring for several minutes, the starting material partially redissolved. A solid mixture of Oxone (DuPont) (1.50 equiv) and NaHCO3 (4.60 equiv, 1.02 equiv relative to Oxone protons) was then added in portions. The mixture was stirred until the reaction was judged complete by TLC analysis of a reaction aliquot (quenched with satd aq Na2S2O3, extracted with EtOAc). Satd aq Na2S2O3 was added, and the resulting mixture was stirred for 10 min, then extracted with EtOAc (3×). The combined organic extracts were washed with water and brine, dried (MgSO4), filtered, and concentrated by rotary evaporation. Purification by silica flash chromatography (hexanes/EtOAc, 0.5 vol% Et3N) afforded macrolactone 15d–f.
General protocol for oxidative ring expansion using RuO2 with NaIO4 stoichiometric oxidant
TBS-protected alcohol 14a–c was dissolved in CH2Cl2 (14.0 mL/mmol). Acetonitrile (6.00 mL/mmol substrate) and acetone (6.00 mL/mmol substrate) were added. In a separate flask, NaIO4 (3.00 equiv) was dissolved in H2O (5.3 mL/mmol substrate) and RuO2 (17 mol%) was added. The activated catalyst was then added to the solution of TBS-protected alcohol in two portions over 5 min and stirred at until the reaction was complete as judged by TLC analysis (ca. 40 min). Satd aq Na2S2O3 was added and the resulting mixture was extracted with CH2Cl2 (3×). The combined organic extracts were washed with brine, dried (MgSO4), and filtered over a pad of celite covered by a thin layer of activated charcoal. The filtrate was concentrated by rotary evaporation. Purification by silica flash chromatography (hexanes/EtOAc) afforded the macrolactam 16a–c.
Cheminformatic analyses
The PCA and PMI analyses were carried out on a total of 176 compounds: 40 top-selling brand-name small molecule drugs by revenue in 2006, 10 drug-like pyrrazolecarboxamides in the MLSMR from ChemBridge, 10 drug-like dihydrotriazolopyrimidines in the MLSMR from Chem Div, 60 natural products with diverse structures, 24 macrocycle natural products, and 32 macrocycle library members.
In the PCA, each molecule was analyzed for a set of 20 physicochemical properties using free online cheminformatics tools, ChemDraw, or manual inspection. These data were assembled in a spreadsheet with the molecules in rows and physicochemical descriptors in columns. The mean value and standard deviation of each column was calculated and mean values were also calculated within each compound series. PCA was performed on mean-centered, normalized data using the R open source statistical computing package. The results from R were used to generate PCA plots (see Supplementary Methods for full details).
In the PMI analysis, minimum energy conformers were generated for each compound using the stochastic conformational search algorithm, as implemented in the MOE molecular modeling software package. The Merck Molecular Force Field (MMFF94) combined with the generalized Born (GB) solvation model of aqueous solvation were used. All conformers within 3 kcal/mol of the minimum energy conformation were recorded. Three principal moments of inertia (Ixx, Iyy, Izz) and normalized principal moments of inertia, npr1 (Ixx/Izz) and npr2 (Iyy/Izz) were calculated using MOE for each conformer. The npr1 and npr2 values were used to generate PMI plots (see Supplementary Methods for full details).
Supplementary Material
Acknowledgments
Dedicated to the memory of our colleague and mentor, Prof. David Y. Gin (1967–2011). We thank Prof. Isamu Shiina (Tokyo University of Science) for a generous gift of MNBA, Dr. George Sukenick, Dr. Hui Liu, Hui Fang, and Dr. Sylvi Rusli (MSKCC Analytical Core Facility) for expert mass spectral analyses, and Dr. Kirstin Kirschbaum (University of Toledo) for x-ray crystallographic analysis. D.S.T. is an Alfred P. Sloan Research Fellow and F.K. is a DAAD postdoctoral fellow. Financial support from the NIH (P41 GM076267) and The Starr Foundation is gratefully acknowledged.
Footnotes
AUTHOR CONTRIBUTIONS
F.K., C.F.S., and D.S.T. designed the experiments. F.K. and C.F.S. carried out the synthetic experiments. C.F.S carried out the PCA analysis. L.B.A. carried out the PMI analysis. F.K., C.F.S., and D.S.T. analyzed the data. F.K., C.F.S., and D.S.T. wrote the manuscript.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing financial interests.
Note: Supplementary Figures and Methods and chemical compound information is available on the Nature Chemical Biology website.
References
- 1.Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery - An underexploited structural class. Nat Rev Drug Discov. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 2.Wessjohann LA, Ruijter E, Garcia-Rivera D, Brandt W. What can a chemist learn from nature’s macrocycles?- A brief, conceptual view. Mol Div. 2005;9:171–186. doi: 10.1007/s11030-005-1314-x. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez-Pedregal VM, et al. The tubulin-bound conformation of discodermolide derived by NMR studies in solution supports a common pharmacophore model for epothilone and discodermolide. Angew Chem, Int Ed. 2006;45:7388–7394. doi: 10.1002/anie.200602793. [DOI] [PubMed] [Google Scholar]
- 4.Canales A, et al. The bound conformation of microtubule-stabilizing agents: NMR insights into the bioactive 3D structure of discodermolide and dictyostatin. Chem Eur J. 2008;14:7557–7569. doi: 10.1002/chem.200800039. [DOI] [PubMed] [Google Scholar]
- 5.Knust H, Hoffmann RW. Synthesis and conformational analysis of macrocyclic dilactones mimicking the pharmacophore of aplysiatoxin. Helv Chim Acta. 2003;86:1871–1893. [Google Scholar]
- 6.Khan AR, et al. Lowering the entropic barrier for binding conformationally flexible inhibitors to enzymes. Biochemistry. 1998;37:16839–16845. doi: 10.1021/bi9821364. [DOI] [PubMed] [Google Scholar]
- 7.Benfield AP, et al. Ligand preorganization may be accompanied by entropic penalties in protein–ligand interactions. Angew Chem, Int Ed. 2006;45:6830–6835. doi: 10.1002/anie.200600844. [DOI] [PubMed] [Google Scholar]
- 8.Udugamasooriya DG, Spaller MR. Conformational constraint in protein ligand design and the inconsistency of binding entropy. Biopolymers. 2008;89:653–667. doi: 10.1002/bip.20983. [DOI] [PubMed] [Google Scholar]
- 9.Veber DF, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 10.Bauer RA, Wurst JM, Tan DS. Expanding the range of ‘druggable’ targets with natural product-based libraries: An academic perspective. Curr Opin Chem Biol. 2010;14:308–314. doi: 10.1016/j.cbpa.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan DS. Diversity-oriented synthesis: Exploring the intersections between chemistry and biology. Nat Chem Biol. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 12.Illuminati G, Mandolini L. Ring closure reactions of bifunctional chain molecules. Acc Chem Res. 1981;14:95–102. [Google Scholar]
- 13.Woodward RB, et al. Asymmetric total synthesis of erythromycin. 2 Synthesis of an erythronolide A lactone system. J Am Chem Soc. 1981;103:3213–3215. [Google Scholar]
- 14.Blankenstein J, Zhu J. Conformation-directed macrocyclization reactions. Eur J Org Chem. 2005:1949–1964. [Google Scholar]
- 15.Gradillas A, Perez-Castells J. Macrocyclization by ring-closing metathesis in the total synthesis of natural products: Reaction conditions and limitations. Angew Chem, Int Ed. 2006;45:6086–6101. doi: 10.1002/anie.200600641. [DOI] [PubMed] [Google Scholar]
- 16.Su Q, Beeler AB, Lobkovsky E, Porco JA, Panek JS. Stereochemical diversity through cyclodimerization: Synthesis of polyketide-like macrodiolides. Org Lett. 2003;5:2149–2152. doi: 10.1021/ol034608z. [DOI] [PubMed] [Google Scholar]
- 17.Beeler AB, et al. Synthesis of a library of complex macrodiolides employing cyclodimerization of hydroxy esters. J Comb Chem. 2005;7:673–681. doi: 10.1021/cc050064b. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt DR, Kwon O, Schreiber SL. Macrolactones in diversity-oriented synthesis: Preparation of a pilot library and exploration of factors controlling macrocyclization. J Comb Chem. 2004;6:286–292. doi: 10.1021/cc020076m. [DOI] [PubMed] [Google Scholar]
- 19.Marcaurelle LA, et al. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: Discovery of macrocyclic histone deacetylase inhibitors. J Am Chem Soc. 2010;132:16962–16976. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stach H, Hesse M. Synthesis of macrocyclic compounds by ring enlargement. Tetrahedron. 1988;44:1573–1590. [Google Scholar]
- 21.Roxburgh CJ. The syntheses of large-ring compounds. Tetrahedron. 1995;51:9767–9822. [Google Scholar]
- 22.Roxburgh CJ. Syntheses of medium sized rings by ring expansion reactions. Tetrahedron. 1993;49:10749–10784. [Google Scholar]
- 23.Baeurle S, et al. From rigidity to conformational flexibility: Macrocyclic templates derived from ansa-steroids. Angew Chem, Int Ed. 2003;42:3961–3964. doi: 10.1002/anie.200351385. [DOI] [PubMed] [Google Scholar]
- 24.Baeurle S, et al. Novel macrocyclic templates by ring enlargement of ansa-steroids. Tetrahedron Lett. 2004;45:9569–9571. [Google Scholar]
- 25.Kumar N, Kiuchi M, Tallarico JA, Schreiber SL. Small-molecule diversity using a skeletal transformation strategy. Org Lett. 2005;7:2535–2538. doi: 10.1021/ol0504345. [DOI] [PubMed] [Google Scholar]
- 26.Borowitz IJ, Gonis G. The synthesis and oxidation of tetrahydrochroman. A new lactone synthesis. Tetrahedron Lett. 1964:1151–1155. [Google Scholar]
- 27.Borowitz IJ, Williams GJ. Medium ring compounds. II. Synthesis of 6-oxodecanolide. Tetrahedron Lett. 1965:3813–3817. [Google Scholar]
- 28.Borowitz IJ, Rapp RD. Ozonolysis of tetrahydrochromans. Formation of glycols and normal ozonolysis products. J Org Chem. 1969;34:1370–1373. [Google Scholar]
- 29.Hückel W, Danneel R, Schwartz A, Gercke A. Stereochemistry of bicyclic ring systems. V Δ9,10-Octalin. Justus Liebigs Ann Chem. 1929;474:121–144. [Google Scholar]
- 30.Seebach D, Weller T, Protschuk G, Beck AK, Hoekstra MS. Preparation of 1,3-diketones and of nitrodiketones by (1:1)-acylation of lithium enolates with acyl chlorides. Helv Chim Acta. 1981;64:716–735. [Google Scholar]
- 31.Patil ML, Borate HB, Ponde DE, Deshpande VH. Total synthesis of (±)-brasiliquinone B. Tetrahedron. 2002;58:6615–6620. [Google Scholar]
- 32.Sestelo JP, del Mar Real M, Sarandeses LA. Synthesis of polycyclic structures by the Diels-Alder reaction of inner-outer-ring 1,3-bis(trimethylsilyloxy)dienes. J Org Chem. 2001;66:1395–1402. doi: 10.1021/jo0015319. [DOI] [PubMed] [Google Scholar]
- 33.Sestelo JP, Del Mar Real M, Mourino A, Sarandeses LA. Synthesis of polycyclic structures by Diels–Alder reaction of inner-outer-ring 1,3-bis[(trimethylsilyl)oxy]dienes. Tetrahedron Lett. 1999;40:985–988. [Google Scholar]
- 34.Danishefsky S, Kerwin JF, Jr, Kobayashi S. Lewis acid catalyzed cyclocondensations of functionalized dienes with aldehydes. J Am Chem Soc. 1982;104:358–360. [Google Scholar]
- 35.Inokuchi T, Okano M, Miyamoto T. Catalyzed Diels-Alder reaction of alkylidene-or arylideneacetoacetates and Danishefsky’s dienes with lanthanide salts aimed at selective synthesis of cis-4,5-dimethyl-2-cyclohexenone derivatives. J Org Chem. 2001;66:8059–8063. doi: 10.1021/jo010575u. [DOI] [PubMed] [Google Scholar]
- 36.Dossetter AG, Jamison TF, Jacobsen EN. Highly enantio- and diastereoselective hetero-Diels–Alder reactions catalyzed by new chiral tridentate chromium (III) catalysts. Angew Chem, Int Ed. 1999;38:2398–2400. doi: 10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 37.Gemal AL, Luche JL. Lanthanoids in organic synthesis. 6 Reduction of α-enones by sodium borohydride in the presence of lanthanoid chlorides: Synthetic and mechanistic aspects. J Am Chem Soc. 1981;103:5454–5459. [Google Scholar]
- 38.Yang D, Zhang C. Ruthenium-catalyzed oxidative cleavage of olefins to aldehydes. J Org Chem. 2001;66:4814–4818. doi: 10.1021/jo010122p. [DOI] [PubMed] [Google Scholar]
- 39.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. A greatly improved procedure for ruthenium tetroxide catalyzed oxidations of organic compounds. J Org Chem. 1981;46:3936–3938. [Google Scholar]
- 40.Brown DS, Elliott Mark C, Moody CJ, Mowlem TJ. Preparation of oxonanes and azonanes by oxidative ring expansion: Synthesis of obtusan. J Chem Soc, Perkin Trans. 1995;1:1137–1144. [Google Scholar]
- 41.Torii S, Inokuchi T, Kondo K. A facile procedure for oxidative cleavage of enolic olefins to the carbonyl compounds with ruthenium tetroxide (RuO4) J Org Chem. 1985;50:4980–4982. [Google Scholar]
- 42.Rincon S, et al. A new route for the preparation of the 22,23-dioxocholestane side chain from diosgenin and its application to the stereocontrolled construction of the 22R,23S-diol function. Tetrahedron. 2006;62:2594–2602. [Google Scholar]
- 43.Shiina I, Kubota M, Ibuka R. A novel and efficient macrolactonization of ω-hydroxycarboxylic acids using 2-methyl-6-nitrobenzoic anhydride (MNBA) Tetrahedron Lett. 2002;43:7535–7539. [Google Scholar]
- 44.Shiina I, Kubota M, Oshiumi H, Hashizume M. An effective use of benzoic anhydride and its derivatives for the synthesis of carboxylic esters and lactones: A powerful and convenient mixed anhydride method promoted by basic catalysts. J Org Chem. 2004;69:1822–1830. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]
- 45.Tanner D, Somfai P. A mild and efficient method for the preparation of N-tosyl amides and lactams. Tetrahedron. 1988;44:613–618. [Google Scholar]
- 46.Dumez E, et al. Synthesis of macrocyclic, potential protease inhibitors using a generic scaffold. J Org Chem. 2002;67:4882–4892. doi: 10.1021/jo025615o. [DOI] [PubMed] [Google Scholar]
- 47.Ganesan The impact of natural products upon modern drug discovery. Curr Opin Chem Biol. 2008;12:306–317. doi: 10.1016/j.cbpa.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 48.Sauer WHB, Schwarz MK. Molecular shape diversity of combinatorial libraries: A prerequisite for broad bioactivity. J Chem Inf Comput Sci. 2003;43:987–1003. doi: 10.1021/ci025599w. [DOI] [PubMed] [Google Scholar]
- 49.Bostrom J, Norrby PO, Liljefors T. Conformational energy penalties of protein-bound ligands. J Comput Aided Mol Design. 1998;12:383–396. doi: 10.1023/a:1008007507641. [DOI] [PubMed] [Google Scholar]
- 50.Akritopoulou-Zanze I, Metz JT, Djuric SW. Topography-biased compound library design: The shape of things to come? Drug Discov Today. 2007;12:948–952. doi: 10.1016/j.drudis.2007.08.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.