

Abstract
The natural product Gambogic Acid (GA) has shown significant potential as an anti-cancer agent being able to induce apoptosis in multiple tumor cell lines, including multidrug resistant cell lines, as well as displaying antitumor activity in animal models. Despite the fact that GA has entered phase I clinical trials, the primary cellular target and mode of action of this compound remain unclear, although many proteins have been shown to be affected by it. Via a thorough analysis of several cellular organelles, both at a morphological and a functional level, we demonstrate that the primary effect of GA is directed at the mitochondria. We found that GA induces mitochondrial damage within minutes of incubation at low micromolar concentrations. Moreover, a fluorescent derivative of GA was able to specifically localize to the mitochondria and was displaced from these organelles after competition with unlabeled GA. These findings indicate that GA directly targets the mitochondria to induce the intrinsic pathway of apoptosis thus representing a new member of mitocans.
Keywords: mitochondria, mitocan, apoptosis, caged Garcinia xanthone, membrane depolarization
Introduction
The tropical trees of the genus Garcinia, found mainly in Southeast Asia, are widely known for their use in folk medicines.[1] Efforts to identify the bioactive ingredients from these plants have led to the identification of Gambogic Acid (GA),[2] the most studied member of an intriguing family of natural products that are structurally characterized by a xanthone backbone where the C ring has been converted into a caged tricyclic structure. [3] The ability of several caged Garcinia xanthones to inhibit tumor cell proliferation, combined with the capability to undergo structural modifications, renders these compounds attractive candidates for innovative anti-cancer drug design.[4] Particularly well documented is the antitumor activity of GA: in addition to inducing apoptosis in a variety of tumor cell lines including multidrug-resistant cell lines,[5,6] GA was shown to display antitumor activity in animal models.[7] Consequently, GA represents an appropriate candidate for clinical applications as an anticancer agent, and has recently entered phase I human tolerability trials in China.[8]
It is well established that nearly all traditional anticancer drugs exploit apoptosis pathways to exert their cytotoxic effects.[9] The process of apoptosis can be induced by two fundamentally different but intertwined ways, referred to as the extrinsic and the intrinsic pathways.[10] In the intrinsic pathway, mitochondrial outer membrane permeabilization leads to the release of pro-apoptotic proteins (such as cytochrome C) from the mitochondrial intermembrane space. This event results in the formation of a caspase activation platform termed the apoptosome.[11] The apoptosome activates an initiator caspase, caspase-9, which subsequently activates the executioner caspases, caspase -3 and caspase-7. In the extrinsic pathway, cell death receptors at the cell surface, upon receiving a death stimulus, initiate a signaling cascade that eventually recruits and activates caspase-8; active caspase-8 then directly cleaves and activates caspase-3 and caspase-7.[12] Caspase-8 is also able to cleave and induce mitochondrial translocation of Bid, a Bcl-2 family member. Truncated Bid then initiates the release of mitochondrial factors, resulting in apoptotic signaling via the intrinsic pathway.[13]
Despite its established role in inducing apoptosis in cancer cells, the primary direct molecular target of GA is still under investigation.[14] One of the first proposed targets was the Transferrin Receptor-1 (TfR-1).[15] Binding of GA to TfR was shown to activate the apoptosis cascade by activating both caspase-8 and the mitochondrial pathway. This hypothesis was, however, challenged by another study in which the activity of GA was not reduced in cells deficient in endogenous TfR-1.[16] Other studies have shown that GA inhibits the catalytic activity of human topoisomerase IIα,[17] modulates the nuclear factor-κB signaling pathway[18] and induces G2/M phase arrest of dividing cells, [19] suggesting that proteins involved in cell cycle may also be targeted by this natural product.[20] Additionally, GA was proposed to trigger apoptosis via the down-regulation of mdm2, which resulted in the up-regulation of p53 protein expression.[21] A strong link between GA and the mitochondrial-dependent induction of apoptosis has emerged from the finding that GA induced reactive oxygen species (ROS) accumulation and collapse of mitochondrial membrane potential, leading to release of cytochrome C from the mitochondria and ultimately resulting in cell death. [22] Moreover, GA was shown to be an antagonist of antiapoptotic Bcl-2 family proteins, which suppress mitochondria-initiated cell death by inhibiting proapoptotic Bcl-2 members Bax and Bak.[23] Importantly, it was also observed that Bax/Bak double knockout cells were still sensitive to GA, suggesting that this compound induces cytotoxicity via mechanisms that are only partly dependent on the Bcl-2 family of proteins.[23b]
Given the multitude of putative targets and mode of actions ascribed to GA, we decided to perform a thorough analysis of its localization and activity at a subcellular level. We analyzed the morphological as well as functional effects of GA towards the main intracellular compartments, such as the endoplasmic reticulum (ER), the endocytic compartment, the Golgi apparatus and the mitochondria. We were able to show that, at low micromolar concentrations and short incubation times, GA does not affect ER, Golgi nor endosomes, while inducing dramatic morphological changes in mitochondrial structure and function. Moreover, a fluorescent derivative of GA was shown to specifically localize in mitochondria and could be competed out by incubation with unlabeled GA. These studies unambiguously demonstrate that mitochondria are the main subcellular target of GA and that this compound affects cell viability by inducing the mitochondria-dependent pathway of apoptosis.
Results
Evaluation of the kinetics of GA-induced mitochondrial fragmentation
Since apoptosis is linked to mitochondrial damage, our first study aimed to evaluate the effect of GA on mitochondria as a function of time. To this end, we incubated HeLa cells with GA (2μM) and followed both the mitochondrial fragmentation and the production of caspase-3 (executioner of apoptosis) over a period of 3 hours. The effect of GA (2μM) on mitochondria was addressed by immunofluorescence (IF) analysis of the mitochondrial protein Tom20 (Figure 2). We observed partial loss of mitochondrial integrity and mitochondrial swelling after 30 min of treatment (Fig. 2b). After 1h of treatment (Fig. 2c), mitochondrial disruption was evident: the mitochondrial tubular network, that typically spreads across the cytoplasm, was lost while there was a predominant condensed perinuclear staining. Interestingly, at this point caspase-3 was not yet activated, which only appeared after 2–3h treatment (Figure 2f). Further exposure to GA induced continuous collapse of the mitochondria in the perinuclear area (Figure 2d–e). Apoptotic cells with pyknotic nuclei started to be visible after 3h treatment (Figure 2e). These studies show that GA has an effect on mitochondrial structure within the first 30 min of incubation and well before caspase-3 activation.
Figure 2. GA induces immediate mitochondrial fragmentation which results in cell apoptosis.

HeLa cells were treated with 2μM GA for 0 min (a), 30 min (b), 1h (c), 2h (d) and 3h (e). A first group of cells was processed for IF to visualize the status of the mitochondria (mitochondria are shown in red; nuclei are shown in blue). (f): A second group of HeLa cells was lysed and the level of apoptosis was measured by the presence of cleaved caspase-3 on WB. (g): Human normal skin fibroblast cells (BJ cells) were treated and processed as in (f) and the level of apoptosis was measured by the presence of cleaved caspase-3 on WB. An apoptotic cell, recognizable by the condensed nucleus, is also visible in (e).
We performed the same analysis in human skin fibroblasts (BJ cells), to assess whether any difference could be observed between fibroblasts of cancer origin (HeLa cells) and non-cancer primary fibroblasts. Both mitochondrial fragmentation (see Supporting Information), induction of caspase-3 and subsequent apoptosis was observed in BJ cells (Figure 2g). These results suggest that the GA-induced mitochondrial fragmentation is not cell-specific.
We also evaluated the concentration-dependent mitochondrial fragmentation in both HeLa and BJ cells. We found extensive loss in mitochondrial network of HeLa cells after 2 h of incubation with 0.5 μM GA, while in BJ cells 2 μM of GA are needed to induce comparable level of fragmentation after 2 h of incubation (see Supporting Information).
Effect of GA on the morphology of cellular organelles
To address whether the effect of GA on the mitochondrial network was selective, we assessed the structural organization of other intracellular organelles during the first hour of incubation. To this end, cells were treated with GA at 2μM concentration for 1h, well before caspase activation, to avoid any perturbation of organelles due to secondary effects of GA. Endoplasmic reticulum (Figure 3a,d), endosomes (Figure 3b,e) and Golgi apparatus (Figure 3c,f) were imaged using specific protein markers (calreticulin for ER, EEA1 for endosomes and mannosidase II for Golgi) in the presence of the mitochondrial marker Tom20. Figure 3 shows that, under conditions where mitochondria were clearly fragmented (Figure 3d,e,f), the structures of other organelles were not affected.
Figure 3. GA acts selectively on mitochondria and does not affect other intracellular organelles.

HeLa cells were treated with DMSO (top row) or 2μM GA (bottom row) for 1h and then processed for IF to reveal the status of Endoplasmic Reticulum (a, d), endosomes (b, e) and Golgi apparatus (c, f). Mitochondria are shown in red, while the others organelles are in green. Nuclei are in blue.
Effect of GA on the function of cellular organelles
The IF analysis of intracellular structures (Fig. 3) shows that no morphological changes other than to mitochondria could be visible after GA treatment. To verify the status of the intracellular organelles at a functional level, cells treated with GA for 1h were also analyzed to assess the status of protein trafficking and endosomal activity.
To functionally analyze the secretory pathway, cells were transfected with a temperature sensitive mutant of VSVG, a model secretory protein. At 40 °C, unfolded VSVG is trapped in the endoplasmic reticulum. Upon shifting the cells to the permissive temperature (32 °C), VSVG reaches the Golgi within 50 min, and then the plasma membrane in 180 min. [24] Cells transfected with VSVG were incubated overnight at 40 °C to accumulate VSVG in the ER. GA was added at 2μM for 30 min at 40 °C. The cells were then transferred to 32 °C (in the presence of GA) for 50 min (to allow VSVG to reach the Golgi apparatus) and 180 min (VSVG arrives at the plasma membrane). Cells were fixed after each time point and stained for VSVG as well as for mitochondria. As shown in Figure 4, VSVG was transported from the ER (Figure 4a,d) to the Golgi (Figure 4b,e) and to the plasma membrane (Figure 4c,f) in control cells (DMSO treated), as well as in GA treated cells. Mitochondrial staining shows that, as expected, GA induces fragmentation of mitochondria, while not affecting protein trafficking.
Figure 4. Protein trafficking is not affected by GA.

HeLa cells expressing VSVG-tsO45-GFP are incubated o/n at 40 °C to block VSVG in the Endoplasmic Reticulum. DMSO (a) or GA 2μM (d) is then added for 1h. The cells are switched to 32 °C to allow VSVG to be exported from the ER. VSVG reaches the Golgi apparatus in 50 min both in the absence (b) or presence (e) of GA. In 120 min VSVG arrives at the plasma membrane in control cells (c) as well as in GA treated cells (f). VSVG-GFP is in green; mitochondria are shown in red; nuclei are in blue.
To functionally test the endocytic compartment of cells treated with GA, we examined the binding of transferrin (Tf) to its receptor (TfR) and its subsequent internalization into endosomes. HeLa cells were incubated with DMSO (control) or GA (2 μM) for 1h at 37 °C. Tf was added at 4 °C for 1h to allow the binding to TfR at the plasma membrane (Figure 5a,d). The cells were then placed at 37 °C for 5 min (Figure 5b,e) or 10 min (Figure 5c,f) to allow Tf internalization in the endosomal compartment. Mitochondrial staining was performed as before to verify the activity of GA. As shown in Figure 5, Tf is able to bind its receptor and then to be transported to the endosomes in control and GA treated cells, in the presence of fragmented mitochondria. A previous study supporting our results has also shown that the binding of Tf to TfR and its internalization are not affected by GA.[16] Collectively, these results indicate that the primary organelle affected by GA is the mitochondria, since the structural integrity and function of other organelles are not altered by GA during the first hour of incubation when mitochondria are already broken down.
Figure 5. Transferrin uptake in GA treated cells.

HeLa cells were treated with DMSO (top row) or GA (bottom row) for 1h and then are incubated at 4 °C for 5 min. Transferrin (Tf-488) was added at 4 °C for 45 min to allow binding to the Transferrin receptor at the plasma membrane (a, d). To allow Tf-488 internalization within the endocytic compartment, the cells are transferred at 37 °C for 5 min or 10 min in the absence (b, c) or presence (e, f) of GA. Tf-488 is in green; mitochondria are in red; nuclei are in blue.
A previous study, reported that the primary target of GA was the TfR.[14] For this reason, we also examined the binding of radio-labeled GA (GA-ETA, for the structure see Figure 1) to purified TfR from two independent sources (commercially available and purified in house). At the onset of this study we determined that GA has comparable activity to GA-ETA (IC50 of GA-ETA is 0.86μM while that of GA is 0.4 μM). However, radio-ligand binding assays did not reveal any binding to TfR by the 14C-labeled GA-ETA (data not shown). Moreover, as shown in Figure 6, GA-ETA can bind to TRVb cells, that are known to lack endogenous TfR1,[16] similarly to CHO and PC3 cells that are known to express TfR. This finding indicates that TfR is not required for cells to take up GA, provides further evidence that the direct target of GA resides in mitochondria and suggests that TfR is not a direct target of GA.
Figure 1.
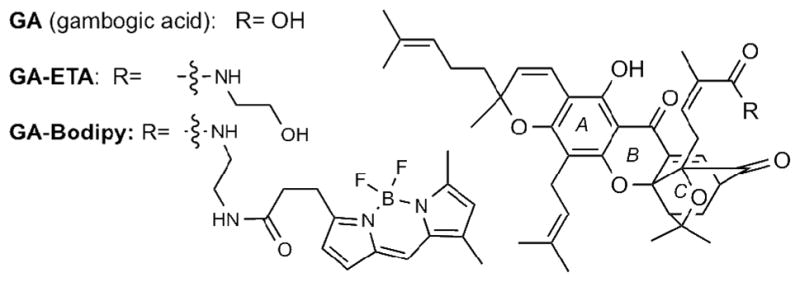
Chemical structures of Gambogic Acid (GA) and synthetic probes GA-ETA and GA-Bodipy.
Figure 6. Binding of 14C-GA-ETA to cells is independent of TfR expression.

Cells were incubated with increasing concentrations of 14C-GA-ETA for 2h at 4 °C and then washed. Membranes were solubilised and bound 14C-GA-ETA was determined by scintillation counting.
Effect of GA on mitochondrial depolarization
Agents that target mitochondria disrupt mitochondrial function resulting in the release of cytochrome c and activation of caspase-9, essential elements in the intrinsic pathway of apoptosis. To determine whether GA affected not only mitochondria structure but function as well, mitochondrial membrane potential was qualitatively measured using the mitochondrial membrane potential (MMP) sensitive dye JC-1 (Figure 7a,b). JC-1 exists as aggregates that emit at 590 nm (red fluorescence) when concentrated in mitochondria having intact membrane potential. In depolarized mitochondria JC-1 exists as a monomer and yields green fluorescence (530 nm). As shown in Figure 7a, in control cells treated with DMSO, mitochondria contained a red form of the dye; in GA treated cells, however, mitochondria appeared yellow, indicating the accumulation of monomeric (green) JC-1. The results of fluorescence microscopy experiments showed that cells treated with GA (Figure 7b) display a greater level of green fluorescence and less red fluorescence compared to control cells (Figure 7a) after 1 h treatment. These observations indicate that mitochondria in cells treated with GA have lost MMP and have become depolarized. Similar observations have been reported in SMMC-7721 cells.[21]
Figure 7. GA induces mitochondrial depolarization.

HeLa cells are treated with DMSO (a) or GA (b) for 1h and then are incubated with JC-1 5μM for 15 min. Cells treated as in (b) are analyzed by immunoblot for the presence of cleaved active caspase-9 (c).
The loss of MMP pointed towards a direct role of mitochondria-dependent apoptosis in GA-induced cell death. To confirm that indeed GA induced pro-apoptotic proteins release from the mitochondria to the cytosol, thereby triggering a downstream cascade reaction resulting in apoptosis, the presence of active caspase-9 was examined after GA treatment. As shown in Figure 7c, while control cells showed no cleaved caspase-9 (first lane), GA-treated cells had the distinctive band (Figure 7c, arrow) indicating the presence of active cleaved caspase-9. Together, our results show that GA is able to induce loss of MMP and activation of caspase-9, confirming activation of the mitochondrial pathway of apoptosis. Moreover, we demonstrate that GA damages mitochondria at both morphological and functional levels.
Subcellular localization of GA
The activity of GA on mitochondria could be direct or indirect. In other words, GA could induce mitochondrial fragmentation either by binding directly to a mitochondrial target or via an indirect pathway, for example by the translocation of Bid to mitochondria after cleavage by caspase-8.[13] In order to gain information on the mode of action and the possible intracellular targets of GA, we utilized of a fluorescent derivative of GA (GA-Bodipy, see Figure 1). We have previously shown, using a 3H-thymidine incorporation assay, that the bioactivity of GA-Bodipy is comparable to that of GA (IC50 of GA-Bodipy is 0.6 μM).[6b] To determine the intracellular localization of GA-Bodipy, the cells were processed for IF to visualize the mitochondrial network. GA-Bodipy was then added and the cells were imaged to reveal both mitochondria and the fluorescent compound. As shown in Figure 8, the green fluorescence due to GA-Bodipy clearly co-localized with the mitochondrial marker Tom20. The presence of an additional perinuclear bright staining is also indicative of mitochondrial disruption.
Figure 8. GA-Bodipy localizes in mitochondria.

HeLa cells labeled to visualize mitochondria (b) were then incubated with GA-Bodipy (a). Merged image is shown in (c). Nuclei are in blue. Respective insets are shown in (d), (e) and (f). Correlation plot is derived from a field of view shown in panel (g). Colocalzation efficiency was measured using ImageJ software and shown by Pearson’s coefficient (g,h) and Mander’s coefficient (i). The average and standard deviation were obtained by the analysis of at least 5 images.
We also quantified the percentage of localization of GA-Bodipy in the mitochondria using the colocalization finder and JaCoP plug-in of ImageJ software. Pearson’s coefficient (Figure 8g, h) is a measure of the degree of linear relationship between two variables. The correlation coefficient ranges from −1 to 1. A value of 1 implies that a linear equation describes the relationship between X and Y perfectly, with all data points lying on a line for which Y increases as X increases. A value of −1 implies that all data points lie on a line for which Y decreases as X increases. A value of 0 implies that there is no linear correlation between the variables. In our experiment the degree of colocalization between mitochondria and GA-Bodipy gave a value of 0.7, indicating a strong degree of correlation between the two variables. Manders’ M1 and M2 coefficients (Figure 8i) measure the portion of the pixels in each channel (here red and green) that coincides with a signal in the other channel. The M coefficient varies from 0 to 1; if all green pixels matched a red signal, the M value would be 1. In our study, we have obtained a M1 value of 0.94, indicating that virtually all mitochondria (red) are labelled by GA-bodipy (green). The M2 value of 0.7 indicates that the majority of GA-bodipy does colocalize with the mitochondria, while only a minor part localizes elsewhere in the cell. Comparable results were obtained when BJ cells were treated with GA-Bodipy, confirming that the ability of GA to bind mitochondria does not depend on the cell type (see Supporting Information).
The specificity of mitochondrial binding was further assessed by competition experiments. Cells stained with Tom20 and GA-Bodipy were incubated with either DMSO or GA, to show whether GA could displace its fluorescent analogue from the mitochondria. Indeed, as shown in Figure 9, in the presence of DMSO, GA-Bodipy maintained mitochondrial localization (Figure 9a–c) while, in the presence of GA, GA-Bodipy was significantly displaced (Figure 9d–f). The localization of GA in mitochondria, together with the effect of GA on mitochondrial morphology and function, strongly indicates that the main direct target of GA is a mitochondrial protein.
Figure 9. GA can displace GA-Bodipy from mitochondria.

HeLa cells labeled with Tom20 antibody to visualize mitochondria (a, d) are then treated with 1 μM GA-Bodipy for 30 min followed by washing. Cells were then further incubated with 0.1% DMSO (a, b, c) or 10 μM cluvenone (d, e, f) for 1 h followed by washing. Mitochondria (in red) are shown in (a) and (d). BODIPY fluorescence (green) is shown in (b) and (e). Merged images are in (c) and (f).
Discussion
Inspired by the therapeutic potential of GA we sought to study its sub-cellular localization and evaluate its effect on cellular organelles toward the goal of identifying its direct molecular targets. We found that this natural product induces mitochondrial structural damage in HeLa cells within 30 min of treatment and prior to activation of caspase-3. During the first hour of incubation, other cellular organelles, such as the ER, the endosomes, and the Golgi apparatus are not structurally affected. We also found that during this time, the cells maintain their ability to undergo endocytosis and to traffic proteins, indicating that GA does not directly affect these normal cell functions. Studies with a radio-labeled analogue of GA indicated that the cellular uptake of GA is independent of transferrin internalization, suggesting that this molecule enters the cell via other endocytotic processes or by passive diffusion.[25] In addition, we showed that GA induces mitochondrial depolarization leading to activation of caspase-3 and caspase-9. More importantly, competition experiments with GA-Bodipy, an equipotent fluorescent analogue of the natural product, demonstrated clearly that GA binds to mitochondria. Collectively, our results indicate that GA induces the mitochondrial pathway of apoptosis through a direct target in the mitochondria. This idea is further supported by our recently reported finding that cluvenone, a simplified analogue of GA, also targets the mitochondria and competes with GA. In addition we found that cluvenone induces ROS formation and depolarizes mitochondria resulting in cytochrome C release and activation of caspase-9.[6d]
Mitochondria are central to cell life and death and have been recently recognized as integrators of a plethora of intrinsic and extrinsic apoptotic pathways.[26] Specifically, alterations to the mitochondria structure and/or function are intimately linked to apoptosis.[27] However, the mere observation that several chemotherapeutic agents induce mitochondrial alterations does not distinguish between two fundamentally different apoptosis pathways: the direct action of this molecule on mitochondria (intrinsic pathway) or the indirect perturbation of mitochondrial function through the activation of proapoptotic signal transduction pathways (extrinsic pathway).[28] In fact, most conventional chemotherapeutic agents affect mitochondria in an indirect fashion by upregulating endogenous apoptosis-induction processes. However, such processes are often compromised in certain cancers leading to chemoresistance thereby effectively limiting the therapeutic benefit of several drugs. One possible strategy to enforce apoptosis may be to trigger downstream events of apoptosis by administering agents that act directly on mitochondria and thus induce apoptosis via the intrinsic pathway and irrespectively of any upstream signaling mechanisms. These agents, often referred to as mitocans,[29] exert selectivity against cancer cells by exploiting the bio-energetic differences between normal and cancer cell mitochondria and, moreover, may prove to be highly useful in killing chemoresistant cancers.[30] For example, many cancer cells have reduced levels of antioxidant enzymes such as the manganese superoxide dismutase (MnSOD) and are subjected to higher levels of oxidative stress than normal cells. 31 Interestingly, an increasing number of recent experimental drugs, including betulinic acid, fenretinide, α-tocopheryl succinate and resveratrol target mitochondria to exert their cytotoxic effects. [32] Furthermore, α-tocopheryl succinate has been found to target complex II of the respiratory chain thus interfering with electron flow and resulting in accumulation of ROS, ultimately, destabilizing mitochondria.[32,33] Along these lines, we propose here that GA represents a new structural class of mitocans setting the foundation for drug discovery efforts focusing in mitochondria related targets.
Conclusion
The documented effect of GA in inducing apoptosis selectively in cancer cells led us to study its subcellular localization. Our studies demonstrate clearly that mitochondria are the main direct target of this compound as other organelles are intact and functional while mitochondria are disrupted. Incubation of cells with GA leads to a rapid mitochondrial membrane depolarization and fragmentation resulting ultimately to apoptosis. Importantly, we also demonstrate that GA binds selectively to mitochondria through competition experiments with a fluorescent analogue. These results are in line with previous observations that GA induces ROS accumulation and collapse of mitochondrial membrane potential resulting in cytochome C release and apoptosis. We have also previously shown that GA and related caged Garcinia xanthones exert selective cytotoxicity against certain cancer cell lines, including chemoresistant cancers.[6a] Combination of these findings, highlight the chemotherapeutic potential of these molecules and provides a possible explanation for their cancer selectivity and efficacy in inducing cancer cell death.
Experimental Section
Material and Methods
Chemical synthesis
Experimental procedures and NMR data for GA, GA-ETA and GA-Bodipy are described in the Supporting Information. The anti-proliferative effect of GA-Bodipy was tested in a 3H-thymidine incorporation assay using HL60/ADR cells as described previously.[6b,34]
Cells, antibodies and reagents
HeLa cells were grown in DMEM supplemented with 10% FBS. BJ cells (human primary fibroblasts) were obtained from Dr. Anne-Laure Prunier (Pasteur Institute) and maintained in EMEM supplemented with 10% FBS. For immunofluorescence and western blot the following antibodies were used: mouse anti-Tom20 (BD Transduction Laboratories, #612278); rabbit anti-cleaved Caspase-9 (Cell Signaling, D315 human spasific); rabbit anti-cleaved Caspase-3 (Cell Signaling, #96645); Calreticulin (Stressgen #SPA-600); rabbit anti-EEA1 (Affinity BioReagents #PA1-063); rabbit anti-Mannosidase II (Chemicon #AB3712); rabbit anti-GFP (AbCam #ab290-50). Transferrin from human serum conjugated with AlexaFluor 488 is from Invitrogen (#T13342). JC-1 (Biotium #30001) was a gift from Dr. Fabrizia Stavru.
Immunofluorescence analysis
HeLa cells plated on 12mm glass coverslips were fixed with 4% formaldehyde, permeabilized with 0.1% TritonX100 in PBS for 5 min, then incubated in blocking buffer (PBS containing 5% fetal goat serum) for 30 min at room temperature. The cells were then incubated for 1 h at room temperature in primary antibody diluted in blocking buffer. The cells were then washed three times with PBS and incubated with secondary antibody, diluted in blocking buffer, for 1 h at room temperature. Alexa fluor 488 goat anti rabbit (1:500) and Alexa Fluor 594 goat anti mouse (1:500) from Molecular Probes were used. Cells were washed three times with PBS containing Hoescht (1:100,000) (H33342, Molecular Probes) to stain DNA. Coverslips were then mounted onto glass slides and visualized using a Zeiss Observer Z1 156 inverted microscope with 63× objective controlled by 157 AxioVision software (Zeiss, Thornwood, NY). The degree of colocalization between mitochondria (red) and GA-bodipy (green) was quantified using the colocalization finder and JaCoP plug-in of ImageJ software (http://rsb.info.nih.gov/ij/). Both Pearson’s and Manders’ overlap coefficients were measured for at least 5 cells per sample.
Immunoblot blot analysis
Cells were lysed with PAGE loading buffer (60 mM Tris, pH 6.8, 5% 2-mercaptoethanol, 2% SDS, 0.01% Bromophenol blue, and 10% glycerol). Proteins in the lysate were separated by SDS–PAGE using a 12% running gel. Proteins were transferred on a nitrocellulose membrane (Western blot) (60 min, 350 mA) that was then kept in blocking buffer (50 mN, Tris, pH 7.5, 150 mM NaCl, 0.05% Tween 20, and 3% BSA) for 1 h. The membrane was incubated for overnight at 4 °C with the primary antibodies diluted in blocking buffer. After washing 3 times with TBS-T buffer, the membranes were incubated for 1 h at room temperature in secondary antibody (anti rabbit IgG HRP-linked, GE Healthcare #NA934V) diluted in blocking buffer. After 3 more washing steps in TBS-T, the reagent for ECL was added (Perkin-Elmer). Kodak Biomax films were used for exposure.
VSVG transport
Cells plated on coverslips at 70% confluency were transfected with VSV-G-GFP (a gift from Dr. Vivek Malhotra) using FuGene 6 (Roche) following the manufacturer’s recommendations. 12h after transfection, the cells were incubated overnight at 40 °C to accumulate VSVG in the ER. GA was added at 2μM for 30 min at 40 °C. The cells were then transferred at 32 °C (in the presence of GA) for 50 min (VSVG at the Golgi) and 180 min (VSVG at the plasma membrane). Cells were fixed after each time point and processed for immunoflorescence. Anti GFP antibody and anti Tom20 antibody were used to label VSV-G-GFP and mitochondria respectively.
Transferrin uptake
Cells were incubated with DMSO or GA (2μM) for 1h at 37 °C. The cells were then transferred on ice for 5 min. Tf-Alexa488 (Invitrogen #T13342) was added (in cold serum free medium) and the cells were kept on ice at 4 °C for 1h to allow Tf to bind to Tf-Receptor at the plasma membrane. The cells were then washed with cold PBS three times. A first group of cells was fixed to verify the ability of Tf to bind TfR. The remaining cells are washed with fresh medium (to remove unbound Tf) and were placed at 37 °C for 5 min and 10 min to allow Tf internalization. After each time point the cells were fixed. Cells were then processed for IF to visualize Tf and mitochondria.
Mitochondrial membrane potential measurement
HeLa cells placed on cover slips were treated with 0.1% DMSO or 2 μM GA for 1 h followed by incubation with 5 μM JC-1 (a gift from Dr. Fabrizia Stavru) for 15 min. The potential sensitive dye, JC-1, exists as aggregates that emit at 590 nm (red fluorescence) when concentrated in mitochondria having intact membrane potential. In depolarized mitochondria JC-1 exists as a monomer and yields green fluorescence (530 nm). Fluorescence was measured using a Zeiss Observer Z1 inverted microscope (excitation at 490 nm).
Binding of 14C-GA-ETA to cells expressing and not expressing TfR
Cells were plated in complete RPMI at 35,000 cells/well/ml in a 24-well plate. Media was removed and wells were blocked with binding buffer (complete RPMI + 0.2 % BSA + 25 mM Hepes) for 2 h at 37 °C. Binding buffer was removed and increasing concentrations up to 20 μM of 14C-GA-ETA were added 1ml/well. Non-specific binding was determined in the presence of excess unlabeled GA-ETA. All conditions were performed in duplicate. Cells were incubated with 14C-GA-ETA for 1 h at 4 °C. Radiolabel was then removed and cells were washed 5X with ice-cold wash buffer (PBS + 0.1% BSA). Cell membranes were then solubilized with solubilization buffer (20 mM Hepes, pH 7.4, +1% Triton X-100, and 10% glycerol) using 0.5 ml/well. Bound 14C-GA-ETA was then determined by counting 0.25 ml aliquots in a scintillation counter. Results are representative of at least 2 determinations.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (CA 133002) is gratefully acknowledged. We thank the National Science Foundation for instrumentation grants CHE9709183 and CHE0741968. We also thank Dr. Anthony Mrse (UCSD NMR Facility) and Dr. Yongxuan Su (UCSD MS Facility).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.For selected references on this topic see: Gustafsson MHG, Bittrich V, Stevens PF. Int J Plant Sci. 2002;163:1045–1054.Sultanbawa MUS. Tetrahedron. 1980;36:1465–1506.Kumar P, Baslas RK. Herba Hungarica. 1980;19:81–91.
- 2.a) Yates P, Karmarkar SS, Rosenthal D, Stout GH, Stout VF. Tetrahedron Lett. 1963;4:1623–1629. [Google Scholar]; b) Anthony A, Desiraju GR. Supramol Chem. 2001;13:11–23. [Google Scholar]; c) Weakley TJR, Cai SX, Zhang HZ, Keana JFW. J Chem Crystallogr. 2001;31:501–505. [Google Scholar]
- 3.For biosynthetic approaches toward the synthesis of caged Garcinia xanthones see: Tisdale EJ, Slobodov I, Theodorakis EA. PNAS. 2004;101:12030–12035. doi: 10.1073/pnas.0401932101.Nicolaou KC, Li J. Angew Chem Int Ed. 2001;40:4264–4268. doi: 10.1002/1521-3773(20011119)40:22<4264::AID-ANIE4264>3.0.CO;2-1.Angew Chem. 2001;113:4394–4398.Tisdale EJ, Chowdhury C, Vong BG, Li H, Theodorakis EA. Org Lett. 2002;4:909–912. doi: 10.1021/ol017278w.Nicolaou KC, Sasmal PK, Xu H, Namoto K, Ritzén A. Angew Chem Int Ed. 2003;42:4225–4229. doi: 10.1002/anie.200351805.Angew Chem. 2003;115:4357–4361.Tisdale EJ, Slobodov I, Theodorakis EA. Org Biomol Chem. 2003;1:4418–4422. doi: 10.1039/b311833a.Tisdale EJ, Vong BG, Li H, Kim SH, Chowdhury C, Theodorakis EA. Tetrahedron. 2003;59:6873–6887.
- 4.For selected recent reviews on the chemistry and biology of caged Garcinia xanthones see: Chantarasriwong O, Batova A, Chavasiri W, Theodorakis EA. Chem Eur J. 2010;16:9944–9962. doi: 10.1002/chem.201000741.Pouli N, Marakos P. Anti-Cancer Agents Med Chem. 2009;9:77–98. doi: 10.2174/187152009787047699.Han Q-B, Xu HX. Curr Med Chem. 2009;16:3775–3796. doi: 10.2174/092986709789104993.
- 5.a) Asano J, Chiba K, Tada M, Yoshii T. Phytochem. 1996;41:815–820. doi: 10.1016/0031-9422(95)00682-6. [DOI] [PubMed] [Google Scholar]; b) Thoison O, Fahy J, Dumontet V, Chiaroni A, Riche C, Tri MV, Sévenet T. J Nat Prod. 2000;63:441–446. doi: 10.1021/np9903088. [DOI] [PubMed] [Google Scholar]; c) Thoison O, Cuong DD, Gramain A, Chiaroni A, Hung NV, Sévenet T. Tetrahedron. 2005;61:8529–8535. [Google Scholar]; d) Zhang HZ, Kasibhatla S, Wang Y, Herich J, Guastella J, Tseng B, Drewe J, Cai SX. Bioorg & Med Chem. 2004;12:309–317. doi: 10.1016/j.bmc.2003.11.013. [DOI] [PubMed] [Google Scholar]; e) Kuemmerle J, Jiang S, Tseng B, Kasibhatla S, Drewe J, Cai SX. Bioorg & Med Chem. 2008;16:4233–4241. doi: 10.1016/j.bmc.2008.02.084. [DOI] [PubMed] [Google Scholar]
- 6.a) Batova A, Lam T, Wascholowski V, Yu AL, Giannis A, Theodorakis EA. Org Biomol Chem. 2007;5:494–500. doi: 10.1039/b612903j. [DOI] [PubMed] [Google Scholar]; b) Chantarasriwong O, Cho WC, Batova A, Chavasiri W, Moore C, Rheingold AL, Theodorakis EA. Org Biomol Chem. 2009;7:4886–4894. doi: 10.1039/b913496d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Batova A, Altomare D, Chantarasriwong O, Ohlsen KL, Creek KE, Lin YC, Messersmith A, Yu AL, Yu J, Theodorakis EA. Mol Cancer Ther. 2010;9:2869–2878. doi: 10.1158/1535-7163.MCT-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Guizzunti G, Theodorakis EA, Yu AL, Zurzolo C, Batova A. Invest New Drugs. 2011 doi: 10.1007/s10637-011-9745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Qiang L, Yang Y, You Q-D, Ma Y-J, Yang L, Nie F-F, Gu H-Y, Zhao L, Lu N, Qi Q, Liu W, Wang X-T, Guo Q-L. Biochem Pharmacol. 2008;75:1083–1092. doi: 10.1016/j.bcp.2007.10.033. [DOI] [PubMed] [Google Scholar]; b) Yang Y, Yang L, You Q-D, Nie F-F, Gu H-Y, Zhao L, Wang X-T, Guo Q-L. Cancer Lett. 2007;256:259–266. doi: 10.1016/j.canlet.2007.06.014. [DOI] [PubMed] [Google Scholar]; c) Guo Q, Qi Q, Gu H, Wu Z. Basic & Clin Pharmacol & Toxicol. 2006;99:178–184. doi: 10.1111/j.1742-7843.2006.pto_485.x. [DOI] [PubMed] [Google Scholar]; d) Zhao L, Zhen C, Wu Z, Hu R, Zhou C, Guo Q. Drug & Chem Toxicol. 2010;33:88–96. doi: 10.3109/01480540903173534. [DOI] [PubMed] [Google Scholar]; e) Hao K, Liu XQ, Wang GJ, Zhao XP. Eur J Drug Metab Pharmacokinet. 2007;32:63–68. doi: 10.1007/BF03190993. [DOI] [PubMed] [Google Scholar]
- 8.Zhou ZT, Wang JW. Chin J New Drugs. 2007;16:79–82. [Google Scholar]
- 9.a) Deorukhkar A, Krishnan S, Gautam S, Aggarwal BB. Expert Opin Investig Drugs. 2007;16:1753–1773. doi: 10.1517/13543784.16.11.1753. [DOI] [PubMed] [Google Scholar]; b) Kim R, Tanabe K, Uchida Y, Emi M, Inoue H, Toge T. Cancer Chemother Pharmacol. 2002;50:343–352. doi: 10.1007/s00280-002-0522-7. [DOI] [PubMed] [Google Scholar]
- 10.For selected reviews on this topic see: Fulda S, Debatin KM. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608.Elmore S. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337.
- 11.a) Slee EA, Adrain C, Martin SJ. J Biol Chem. 2001;276:7320–7326. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]; b) Boatright KM, Salvesen GS. Curr Opin Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 12.a) Acehan D, Jiang X, Morgan DG, Hauser JE, Wang X, Akey CW. Molec Cell. 2002;9:423–432. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]; b) Riedl SJ, Salvesen GS. Nature Rev Mol Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 13.a) Li H, Zhu H, Xu CJ, Yuan J. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]; b) Barnhart BC, Alappat EC, Peter ME. Semin Immunol. 2003;15:185–193. doi: 10.1016/s1044-5323(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 14.a) Wang X, Chen Y, Han Q-b, Chan C-y, Wang H, Liu Z, Cheng CH-k, Yew DT, Lin MCM, He M-l, Xu H-x, Sung JJY, Kung H-f. Proteomics. 2009;9:242–253. doi: 10.1002/pmic.200800155. [DOI] [PubMed] [Google Scholar]; b) Davenport J, Manjarrez JR, Peterson L, Krumm B, Blagg BS, Matts RL. J Nat Prod. 2011;74:1085–1092. doi: 10.1021/np200029q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasibhatla S, Jessen KA, Maliartchouk S, Wang JY, English NM, Drewe J, Qiu L, Archer SP, Ponce AE, Sirisoma N, Jiang S, Zhang H–Z, Gehlsen KR, Cai SX, Green DR, Tsang B. PNAS USA. 2005;102:12095–12100. doi: 10.1073/pnas.0406731102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ortiz-Sánchez E, Daniels TR, Helguera G, Martinez-Maza O, Bonavida B, Penichet ML. Leukemia. 2009;23:59–70. doi: 10.1038/leu.2008.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin Y, Meng L, Hu C, Duan W, Zuo Z, Lin L, Zhang X, Ding J. Mol Cancer Therap. 2007;6:2429–2440. doi: 10.1158/1535-7163.MCT-07-0147. [DOI] [PubMed] [Google Scholar]
- 18.a) Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, Aggarwal BB. Blood. 2007;110:3517–3525. [Google Scholar]; b) Zhu XL, Zhang HM, Lin Y, Chen PS, Min J, Wang ZZ, Xiao W, Chen BA. J Chemother. 2009;21:666–672. doi: 10.1179/joc.2009.21.6.666. [DOI] [PubMed] [Google Scholar]; c) Wang X, Lu N, Yang Q, Gong D, Lin C, Zhang S, Xi M, Gao Y, Wei L, Guo Q, You Q. Eur J Med Chem. 2011;46:1280–1290. doi: 10.1016/j.ejmech.2011.01.051. [DOI] [PubMed] [Google Scholar]
- 19.Yu J, Guo Q-L, You Q-D, Zhao L, Gu H-Y, Yang Y, Zhang H-w, Tan Z, Wang X. Carcinogenesis. 2007;28:632–638. doi: 10.1093/carcin/bgl168. [DOI] [PubMed] [Google Scholar]
- 20.a) Wu ZQ, Guo QL, You QD, Zhao L, Gu HY. Biol Pharm Bull. 2004;27:1769–1774. doi: 10.1248/bpb.27.1769. [DOI] [PubMed] [Google Scholar]; b) Yu J, Guo QL, You QD, Lin S–S, Li Z, Gu H–Y, Zhang H–W, Tan Z, Wang X. Cancer Chemother Pharmacol. 2006;58:434–443. doi: 10.1007/s00280-005-0177-2. [DOI] [PubMed] [Google Scholar]; c) Zhao Q, Yang Y, Yu J, You QD, Zeng S, Gu HY, Lu N, Qi Q, Liu W, Wang XT, Guo QL. Cancer Letters. 2008;262:223–231. doi: 10.1016/j.canlet.2007.12.002. [DOI] [PubMed] [Google Scholar]; d) Guo QL, Lin SS, You QD, Yu J, Zhao L, Qi Q, Liang F, Tan Z, Wang XT. Life Sciences. 2006;78:1238–1245. doi: 10.1016/j.lfs.2005.06.046. [DOI] [PubMed] [Google Scholar]; e) Chen J, Gu H–Y, Lu N, Yang Y, Liu W, Qi Q, Rong J–J, Wang X–T, You Q–D, Guo Q–L. Life Sciences. 2008;83:103–109. doi: 10.1016/j.lfs.2008.05.003. [DOI] [PubMed] [Google Scholar]; f) Wang X, Chen Y, Han Q-b, Chan C-y, Wang H, Liu Z, Cheng CH-k, Yew DT, Lin MCM, He M-l, Xu H-x, Sung JJY, Kung H-f. Proteomics. 2009;9:242–253. doi: 10.1002/pmic.200800155. [DOI] [PubMed] [Google Scholar]
- 21.a) Gu H, Wang X, Rao S, Wang J, Zhao J, Ren FL, Mu R, Yang Y, Qi Q, Liu W, Lu N, Ling H, You Q, Guo Q. Mol Cancer Ther. 2008;7:3298–3305. doi: 10.1158/1535-7163.MCT-08-0212. [DOI] [PubMed] [Google Scholar]; b) Rong JJ, Hu R, Qi Q, Gu HY, Zhao Q, Wang J, Mu R, You QD, Guo QL. Cancer Lett. 2009;284:102–112. doi: 10.1016/j.canlet.2009.04.011. [DOI] [PubMed] [Google Scholar]; c) Gu H-Y, Guo Q-L, You Q-D, Liu W, Qi Q, Zhao L, Yuan S-T, Zhang K. Chin J Nat Med. 2005;3:168–172. [Google Scholar]
- 22.Nie F, Zhang X, Qi Q, Yang L, Yang Y, Liu W, Lu N, Wu Z, You Q, Guo Q. Toxicol. 2009;260:60–67. doi: 10.1016/j.tox.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 23.a) Liu W, Guo QL, You QD, Zhao L, Gu HY, Yuan ST. World J Gastroenterol. 2005;11:3655–3659. doi: 10.3748/wjg.v11.i24.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhai D, Jin C, Shiau C–W, Kitada S, Satterthwait AC, Reed JC. Mol Cancer Ther. 2008;7:1639–1646. doi: 10.1158/1535-7163.MCT-07-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xu X, Liu Y, Wang L, He J, Zhang H, Chen X, Li Y, Yang J, Tao J. Int J Dermatol. 2009;48:186–192. doi: 10.1111/j.1365-4632.2009.03946.x. [DOI] [PubMed] [Google Scholar]
- 24.Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J. J Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGraw TE, Greenfield L, Maxfield FR. J Cell Biol. 1987;105:207–214. doi: 10.1083/jcb.105.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Green DR, Reed JC. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]; b) Mignotte B, Vayssiere JL. Eur J Biochem. 1998;252:1–15. doi: 10.1046/j.1432-1327.1998.2520001.x. [DOI] [PubMed] [Google Scholar]; c) Tait SWG, Green DR. Nature Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 27.a) Fantin VR, Berardi MJ, Scorrano L, Korsmeyer SJ, Leder P. Cancer Cell. 2002;2:29–42. doi: 10.1016/s1535-6108(02)00082-x. [DOI] [PubMed] [Google Scholar]; b) Constantini P, Jacotot E, Decaudin D, Kroemer G. J Natl Cancer Inst. 2002;92:1042–1053. doi: 10.1093/jnci/92.13.1042. [DOI] [PubMed] [Google Scholar]; c) Fantin VR, Leder P. Oncogene. 2006;25:4787–4797. doi: 10.1038/sj.onc.1209599. [DOI] [PubMed] [Google Scholar]
- 28.Armstrong JS. Brit J Pharmacol. 2006;147:239–248. doi: 10.1038/sj.bjp.0706556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a) Ralph SJ, Low P, Dong L, Neuzil J. Recent Pat Anticancer Drug Discov. 2006;1:327–346. doi: 10.2174/157489206778776952. [DOI] [PubMed] [Google Scholar]; b) Neuzil J, Wang XF, Dong LF, Low P, Ralph SJ. FEBS Lett. 2006;580:5125–5129. doi: 10.1016/j.febslet.2006.05.072. [DOI] [PubMed] [Google Scholar]
- 30.a) Ralph SJ, Neuzil J. Mol Nutr Food Res. 2009;53:9–38. doi: 10.1002/mnfr.200800044. [DOI] [PubMed] [Google Scholar]; b) Ralph SJ, Rodriguez-Enriquez S, Neuzil J, Moreno-Sanchez R. Mol Aspects Med. 2010;31:29–59. doi: 10.1016/j.mam.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Borrello S, De Leo ME, Galeotti T. Molec Aspects Med. 1993;14:253–258. doi: 10.1016/0098-2997(93)90012-3. [DOI] [PubMed] [Google Scholar]
- 32.a) Fulda S, Kroemer G. Drug Disc Today. 2009;14:885–890. doi: 10.1016/j.drudis.2009.05.015. [DOI] [PubMed] [Google Scholar]; b) Hail N, Jr, Kim HJ, Lotan R. Apoptosis. 2006;11:1677–1694. doi: 10.1007/s10495-006-9289-3. [DOI] [PubMed] [Google Scholar]; c) Dorrie J, Gerauer H, Wachter Y, Zunino SJ. Cancer Res. 2001;61:4731–4739. [PubMed] [Google Scholar]; d) Neuzil J, Dyason JC, Freeman R, Dong LF, Prochazka L, Wang XF, Scheffer I, Ralph SJ. J Bioenerg Biomembr. 2007;39:65–72. doi: 10.1007/s10863-006-9060-z. [DOI] [PubMed] [Google Scholar]
- 33.a) Lemarie A, Grimm S. Oncogene. 2011;30:3985–4003. doi: 10.1038/onc.2011.167. [DOI] [PubMed] [Google Scholar]; b) Dong L-F, Freeman R, Liu J, Zobalova R, Marin-Hernandez A, Stantic M, Rohlena J, Valis K, Rodriguez-Enriquez S, Butcher B, Goodwin J, Brunk UT, Witting PK, Moreno-Sanchez R, Scheffler IE, Ralph SJ, Neuzil J. Clin Cancer Res. 2009;15:1593–1600. doi: 10.1158/1078-0432.CCR-08-2439. [DOI] [PubMed] [Google Scholar]
- 34.For selected manuscripts on the synthesis of GA and related compounds see: Nicolaou KC, Li J. Angew Chem Int Ed. 2001;40:4264–4268. doi: 10.1002/1521-3773(20011119)40:22<4264::AID-ANIE4264>3.0.CO;2-1.Angew Chem. 2001;113:4394–4398.Tisdale EJ, Slobodov I, Theodorakis EA. PNAS. 2004;101:12030–12035. doi: 10.1073/pnas.0401932101.Tisdale EJ, Vong BG, Li H, Kim SH, Chowdhury C, Theodorakis EA. Tetrahedron. 2003;59:6873–6887.Tisdale EJ, Slobodov I, Theodorakis EA. Org Biomol Chem. 2003;1:4418–4422. doi: 10.1039/b311833a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.