

Abstract
Runx2 is a known master transcription factor for osteoblast differentiation, as well as an essential regulator for chondrocyte maturation. Recently, more and more data has shown that Runx2 regulates hypertrophic chondrocyte-specific type X collagen gene (Col10a1) expression in different species. However, how Runx2 regulation of Col10a1 expression impacts chondrocyte maturation, an essential step of endochondral bone formation, remains unknown. We have recently generated transgenic mice in which flag-tagged Runx2 was driven by a cell-specific Col10a1 control element. Significantly increased level of Runx2 and Col10a1 mRNA transcripts were detected in transgenic mouse limbs at both E17.5 (embryonic day 17.5) and P1 (postnatal day1) stages, suggesting an in vivo correlation of Runx2 and Col10a1 expression. Surprisingly, skeletal staining suggested delayed ossification in both the axial and the appendicular skeleton of transgenic mice from E14.5 until P6. Histological analysis showed elongated hypertrophic zones in transgenic mice, with less von Kossa and TUNEL staining in long bone sections at both E17.5 and P1 stages, suggesting defective mineralization due to delayed chondrocyte maturation or apoptosis. Indeed, we detected increased level of anti-apoptotic genes Bcl-2, Opn, and Sox9 in transgenic mice by real-time RT-PCR. Moreover, immunohistochemistry and Western blotting analysis also suggested increased Sox9 expression in hypertrophic chondrocytes of transgenic mice. Together, our data suggest that targeting Runx2 in hypertrophic chondrocytes upregulates expression of Col10a1 and other marker genes (such as Sox9 etc.). This will change the local matrix environment, delay chondrocyte maturation, reduce apoptosis and matrix mineralization, and eventually, lead to impaired endochondral ossification.
Keywords: Runx2, Col10a1, transgenic mice, chondrocyte maturation, endochondral ossification
INTRODUCTION
Runx2 is an indispensable regulator of osteoblast differentiation during bone formation (Ducy et al., 1997; Ducy et al., 1999). It belongs to the family of runt domain transcription factors that include the hematopoietic Runx1, the osteogenic Runx2, and the neuronal and gastrointestinal Runx3 (Levanon et al., 1994). Runx2 (also known as core binding factor α1, Cbfa1) has two isoforms with different N-terminal sequences (Ogawa et al., 1993; Fujiwara et al., 1999). Both Runx2 isoforms play a pivotal role in maintaining the supply of immature osteoblasts and are required for mesenchymal cell differentiation along the osteoblast lineage in vivo (Ducy et al., 1997; Karsenty et al., 2001; Kanatani et al., 2006). Previous studies have shown that Runx2/Cbfa1 null mice completely lack bone formation and die at birth due to respiratory failure (Komori et al., 1997). Runx2 heterozygotes show skeletal abnormalities that mimic human cleidocranial dysplasia (CCD) which is characterized by delayed closure of the fontanel and hypoplastic clavicles (Otto et al., 1997). These findings demonstrate the essential role of Runx2 in intramembranous bone formation.
Runx2 has also been suggested to be a central regulator of endochondral ossification, especially on chondrocyte maturation or hypertrophy, a critical stage of endochondral bone formation during skeletal development (Karsenty et al., 2001). It is generally accepted that Runx2 is an inducer of chondrocyte hypertrophy, since overexpression of Runx2 in nonhypertrophic chondrocytes induces chondrocyte hypertrophy and endochondral ossification in places where it normally never occurs (Takeda et al., 2001). Runx2 has also been shown to inhibit chondrocyte proliferation and hypertrophy through its expression in the perichondrium. This suggests that Runx2 fulfills antagonistic functions during chondrogenesis (Hinoi et al., 2006). Haploinsufficiency of human RUNX2 is known to cause CCD. Besides the craniofacial and dental abnormalities, CCD is also characterized by short stature and cone-shaped epiphyses which suggest an underlying defect in endochondral ossification (Karagzüel et al., 2010). We have previously reported abnormal endochondral ossification in a fetal case of CCD, possibly due to altered RUNX2 regulation of chondrocyte hypertrophy and down-regulation of its target genes, including type X collagen gene (Zheng et al., 2005).
The type X collagen gene (Col10a1) is specifically expressed in hypertrophic chondrocytes, cells that are eventually surrounded by a calcified extracellular matrix and, favor endochondral ossification (Linsenmayer et al., 1991). Previous studies suggest that type X collagen may facilitate endochondral ossification by regulating matrix mineralization and compartmentalizing matrix components (Shen et al., 2005). Mutations of the human COL10A1 have been associated with an autosomal dominant inherited skeletal disorder, Schmid metaphyseal chondrodysplasia (SMCD) which is characterized by short stature, coxa vara, genu varum and a wide irregular growth plate (Warman et al., 1993; Wallis et al., 1994; McIntosh et al., 1995), suggesting defective long bone development.
The above observations clearly demonstrate that both Runx2 and Col10a1 genes play important roles upon chondrocyte maturation during endochondral bone formation. Meanwhile, interaction between Runx2 and the Col10a1 proximal or core promoters in different species has previously been described extensively (Dourado et al., 1998; Zheng et al., 2003; Simoes et al., 2006; Higashikawa et al., 2009). We recently reported that a 150-bp (-4296 to -4147 bp) murine Col10a1 cis-enhancer element is sufficient to direct hypertrophic chondrocyte-specific reporter expression in vivo (Zheng et al., 2009). More recently, we have demonstrated that two tandem-repeat Runx2 binding sites located within the 3’-end (-4187 to -4176 bp, TGTGGG-TGTGGC) of this Col10a1 cis-enhancer are required for tissue-specific Col10a1 promoter activity (Li et al., 2011). These findings together suggest that Runx2 may contribute to type X collagen gene regulation via different mechanisms. Altered Runx2 and Col10a1 expression may affect downstream target genes, change the local matrix environment, and therefore, impact the process of chondrocyte maturation during endochondral bone formation.
Here we report Runx2 gain-of- function studies by targeting Runx2 overexpression in hypertrophic chondrocytes using the cell-specific Col10a1 regulatory elements that we recently defined (Zheng et al., 2009). Our results demonstrate that these Col10a1-Runx2 transgenic mice show upregulated level of Runx2 and Col10a1 mRNA transcripts, but delayed chondrocyte maturation and ossification at both embryonic and early postnatal skeletal developmental stages. Further expression analysis revealed altered chondrogenic and apoptotic related gene expression, including Bcl-2, Opn, and especially, Sox9, which functionally dominates over Runx2 (Zhou et al., 2006; Cheng A, Genever PG, 2010) and, thereby, leads to delayed endochondral ossification.
MATERIALS AND METHODS
Generation of Col10α1-Runx2 transgenic mice
Transgenic mice were generated in which flag-tagged Runx2 cDNA was driven by the cell-specific Col10a1 regulatory elements that have been described previously (Zheng et al., 2009). Specifically, the Col10a1 regulatory elements contain four copies of the 288-bp Col10a1 distal promoter (-4296 to -4009 bp) followed by a Col10a1 basal promoter (-220 to +45 bp) as illustrated (Fig. 1A). These combined Col10a1 promoter elements were cloned into the BamHI and Asp718 sites of the pcDNA3.1(-) vector (Invitrogen). The full length Runx2 cDNA in-frame with a 3’-flag fragment (Geoffroy et al., 2002; Merciris et al., 2007) was also cloned into pcDNA3.1 (-) and was placed downstream of a HindIII site. After sequence confirmation, a 3.6-kb DNA fragment containing the whole transgenic cassette, which includes the cell-specific Col10a1 promoter elements, the flag-tagged Runx2 cDNA and the bovine growth hormone polyadenylation signal sequence, was released by NheI and PvuII digestion and purified for DNA microinjection. Generation of transgenic mice was conducted at the University of Illinois at Chicago (UIC) Transgenic Production Service core facility. Purified DNA construct was injected into pronuclei of FVBN/J mouse zygotes and transplanted into pseudopregnant ICR mice. Transgenic founders were identified by PCR genotyping using Runx2 and flag-specific primers (5’-CTTCCCAAAGCCAGAGTGGAC-3’ and 5’-TGTCGTCATCGTCTTTGTAGC-3’). The animal studies were approved by the animal care and oversight committees at University of Illinois at Chicago and Rush University Medical Center.
FIG.1. Generation of Runx2 transgenic mice using cell-specific Col10a1-control element.
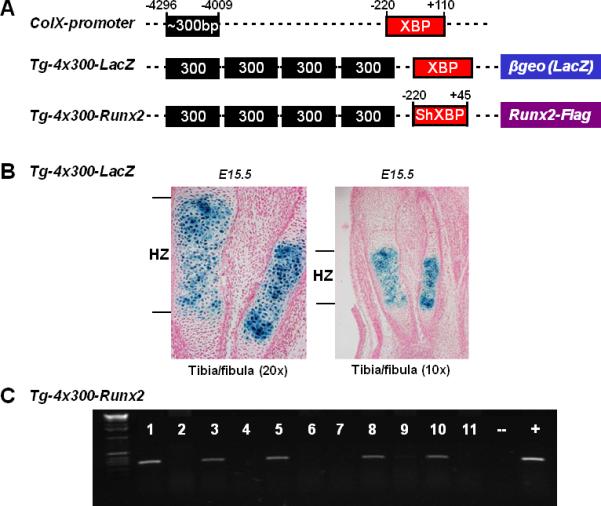
(A). Positions of the ~300-bp (-4296 to -4009 bp) hypertrophic chondrocyte-specific Col10a1 distal promoter and its 330-bp basal promoter (-220 to +110 bp) elements (top) that have been used to generate transgenic reporter (LacZ) construct were as illustrated (middle) (Zheng et al., 2009). Same Col10a1 distal promoter and a shorter Col10a1 basal promoter (-220 to +45 bp) elements were used to generate Runx2-expressing (with a flag-tag) transgenic construct. The 3’-sequence of this shorter basal promoter ends at exon I allowing transgene expression without additional splicing acceptor site. XBP: Col10a1 basal promoter; ShXBP: Shorter Col10a1 basal promoter. (B). Paraffin embedded sections of tibia and fibula from X-gal-stained Tg-4x300 transgenic mouse embryos at E15.5 were counterstained with nuclear fast red. Blue staining indicating reporter activity was exclusively throughout the hypertrophic zone (left panel). No surrounding tissues showed reporter expression (right panel, lower magnification). Tg: transgenic mice, HZ: hypertrophic zone of growth plate. (C). PCR genotyping using Runx2 and Flag fragment specific primers indicated that we have successfully generated five transgenic founders (lanes 1, 3, 5, 8, and 10).
Real-time quantitative reverse-transcription-polymerase chain reaction (qRT-PCR)
Total RNAs were isolated from hind limbs or micro-dissected hypertrophic chondrocyte-enriched femoral growth plates using Trizol reagents (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA) was used to synthesize first-strand cDNA from the purified RNA. qRT-PCR was performed to examine gene expression using the MyiQ Single Color Real-Time PCR Detection System and SYBR Green real-time PCR master mix (Bio-Rad, Hercules, CA). Sequence of the primer pairs were designed for relevant marker genes as well as the internal control Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) gene (Table 1). Relative gene-transcript level was automatically analyzed by the manufacturer provided MyiQ Optical System Software and normalized with endogenous Gapdh.
Table 1.
Primers designed for real-time PCR
| Name | RefSeqID | Sense Primer (5’-3’) | Antisense Primer (5’-3’) | Amplicon (bp) |
|---|---|---|---|---|
| Gapdh | NM_008084 | ACCCAGAAGACTGTGGATGG | CACATTGGGGGTAGGAACAC | 171 |
| Runx2 | NM_001145920 | ACCCAGCCACCTTTACCTAC | TATGGAGTGCTGCTGGTCTG | 150 |
| Col10α1 | NM_009925 | GCAGCATTACGACCCAAGATC | TCTGTGAGCTCCATGATTGC | 201 |
| Sox9 | NM_011448 | TTCATGAAGATGACCGACGA | ATGCACACGGGGAACTTATC | 200 |
| Bax | NM_007527 | TGCAGAGGATGATTGCTGAC | GATCAGCTCGGGCACTTTAG | 173 |
| Bcl-2 | NM_009741 | CTGGCATCTTCTCCTTCCAG | GACGGTAGCGACGAGAGAAG | 183 |
| Opn | NM_009263 | TGCACCCAGATCCTATAGCC | CTCCATCGTCATCATCATCG | 186 |
Gapdh: glyceraldehydes-3-phosphate dehydrogenase; Runx2: runt-related transcription factor 2; Col10α1: type X collagen, alpha 1; Sox9: SRY-box containing gene 9; Bax: BCL-2-associated X protein; Bcl-2: B-cell leukemia/lymphoma 2; Opn: osteopontin.
Skeletal preparation (alcian blue and alizarin red staining)
Mouse skeletons from later fetal, newborn, and early post-natal developmental stages were prepared and stained with alcian blue (for cartilaginous matrices) and alizarin red (for mineralized cartilaginous and bony matrices) according to published protocols with modification (Ovchinnikov D, 2009). Briefly, embryos from E14.5 to E18.5 and mice at the age of P1, P3 and P6 were eviscerated and fixed in 95% ethanol for at least 24 hours and transferred to staining solution consisting of 0.03% alcian blue 8GX (Sigma, St. Louis, MO) in 80% ethanol and 20% acetic acid. After 24 hours, specimens were rinsed with 95% ethanol, 95% ethanol/2% KOH (v/v=1:1) and counterstained overnight with 0.03% Alizarin Red S (Sigma, St. Louis, MO) in 1% KOH and water. Clearing of the samples was conducted by placing them in 1% KOH/20% glycerol for two days or longer. Specimens were transferred into glycerol/95% ethanol (v/v=1:1) followed by 80% and finally stored at 100% glycerol. For each of the developmental stages, at least three lines of transgenic mice were stained and analyzed.
Histological analysis
For histological analysis, mouse limbs at E17.5 or P1 stages were collected and fixed in 10% formalin and stored in 70% ethanol. These mouse limbs were then subjected to dehydration, paraffin embedding, and sectioning. Comparable slides from both transgenic and wild-type littermates were selected for standard Hematoxylin &Eosin staining. Safranin O/Fast Green staining, von Kossa staining, and TUNEL (Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling) assay were also conducted using standard protocols with modifications (Bonewald et al., 2003; Reno et al., 2006). Briefly, for Safranin O/Fast Green staining, after de-paraffin with xylene and gradient ethanol treatment, slides were stained in Fast Green solution (0.1%) for 2 minutes followed by Safranin O (0.1%) staining for 4 minutes. For von Kossa staining, xylene and ethanol treated slides were incubated with 5% silver nitrate and exposed to an ultraviolet light for 30-60 minutes. After rinse with distilled water, slides were treated with 5% sodium thiosulfate for 2-3 minutes and counterstained with 0.1% nuclear fast red. For TUNEL assay, Apoptosis detection was performed on whole hind limb sections using TACS® 2 TdT-DAB In Situ Apoptosis Detection Kit (Trevigen, Inc. Gaithersburg, MD) according to manufacturer's instruction. At least 30 longitudinal (sagittal) sections (5-μm-thick) of the limb growth plate from both TG and WT littermates were observed under microscope (Nikon Eclipse 80i, Nikon Instruments Inc., Melville, NY USA) and analyzed using the Qcapture Suite software (version, 2.95.0, Quantitative Imaging Corp., USA).
Immunohistochemistry (IHC) analysis
Comparable hind limb sections from new born TG and WT mice were subjected to immunohistochemistry analysis using following primary antibodies: anti-Runx2 (C-19, sc-8566, Santa Cruz, CA), anti-Sox9 (H-90, sc-20095, Santa Cruz, CA), and anti-Flag (#2368, Cell Signaling, Danvers, MA). The procedures were based on manufacture suggested protocol with modifications. Briefly, after de-paraffin and rehydration, the selected TG or WT limb sections underwent a series of pretreatments before incubation with the primary antibodies (4°C, overnight). The pretreatments include hot sodium citrate buffer incubation (0.01 M, pH 6.0, 95°C, 10 min) to retrieve antigen, hydrogen peroxide treatment (3% H2O2 in 100% ethanol, 5 min) to quench the endogenous peroxidase, and blocking with 30% normal horse serum (30 min). The concentrations used for the primary antibodies were at 1:50 (anti-Runx2), 1:300 (anti-Sox9) and 1:500 (anti-Flag) dilutions respectively. Non-immune mouse IgG was used as a negative control. After washing with the 1xTBST (Tris Buffered Saline with 0.1% Tween-20), tissue sections were further incubated with biotinylated secondary antibody (anti-rabbit IgG, Santa Cruz, CA). Detection was using the reagents, and protocol as instructed in the ABC kit (Elite PK-6200 Universal, VECTOR laboratories, Burlingame, CA). Slides were counterstained with nuclear fast red (Poly Scientific R&D Corp., NY) before microscopic analysis as described above.
Primary chondrocytes culture and protein extracts preparation
Isolation and culture of primary chondrocytes was based on published protocol with modifications (Gosset et al., 2008). Briefly, femoral heads, femoral condyles and tibial plateau from new born TG or WT mouse hind limbs (left) were collected and rinsed in 1 × PBS twice. Fresh tissues were placed into serum-free (SF) DMEM medium (1% penicillin/streptomycin from Life Technologies, Grand Island, NY; 2% L-glutamine from Sigma, St. Louis, MO) containing Collagenase (3mg/ml, Roche, Germany) and incubated for 45 minutes at 37°C with 5%CO2. Tissues were then transferred into new plates and subjected to overnight culture with SF-DMEM medium containing lower concentration of Collagenase (0.5mg/ml). After digestion, cells were collected from these tissues and cultured for additional 72 hours in DMEM medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. These cells were harvested and used for gene expression analysis by real-time RT-PCR and Western blotting approaches. Cytoplasmic and nuclear protein extracts from above primary chondrocytes as well as from newborn mouse hind limb (right) tissues were prepared using similar protocols as previously described (Martin et al., 2004). Briefly, cell pellets of primary chondrocytes or limb tissues from new born mice were homogenized in 1 X hypotonic buffer with 1:1000 1M DTT and detergent. Cytoplasmic protein was collected from the supernatants after centrifuge for 10 minutes at 850 × g at 4°C. The pellets were then suspended in 500μl 1 × hypotonic buffer and kept on ice for 15 minutes. 25μl of detergent was added into samples before spin down at 14000 × g at 4°C for 10 seconds. The supernatants were combined together with previous cytoplasmic proteins. The remaining pellets were re-suspended with 50μl complete lysis buffer containing 10% 10mM DTT and 1% protease inhibitor cocktail and kept on ice for 30 minutes with shaking on a rocking platform (150rpm). Nuclear extracts were collected from the supernatants after centrifuge at 14000 × g for 10 minutes at 4°C. Proteins were measured using spectrophotometer (GeneQuant1300, GE Healthcare, Piscaraway, NJ).
Western blotting assay
130μg or 20μl of proteins were used for western blotting assay. Specifically, after boiling for 5 minutes, the proteins were loaded on 10%SDS-PAGE (sodium dodecylsulfate polyacrylamide gel electrophoresis) and then transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA). The membranes were blocked in 5% defatted milk and then incubated with different primary antibodies (Mouse anti-Runx2 1:200; Rabbit anti-Sox9 1:200 and Goat anti-Lamin B 1:2000) at 4°C overnight. Horseradish peroxidase (HRP) labeled secondary antibodies were added for 1 hour at room temperature. All antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa cruz, CA). Pierce ECL chemiluminescence Kit (Thermo, Rockford, IL) was used for detecting membrane signals. The target band density was subjected to densitometry analysis using Image J (NIH, Bethesda, MD) software. Lamin B was used as an internal control.
Statistical analysis
Values are presented as means ± SEM. One-way ANOVA was used to assess the significance across experimental groups. Student t-tests were used to compare mean differences between groups. p < 0.05 was considered statistically significant. SSPS v16.0 (Chicago, IL) and GraphPad Prism 5.0 (La Jolla, CA) were used for graph and statistics analysis.
RESULTS
Establishment of Col10a1-Runx2 transgenic mouse lines
To study the in vivo correlation of Runx2 with murine Col10a1 expression and chondrocyte maturation, we performed Runx2 gain-of- function studies by targeting Runx2 overexpression in hypertrophic chondrocytes. We have generated a transgenic construct in which flag-tagged Runx2 cDNA was driven by the cell-specific Col10a1 regulatory elements (Fig. 1A, Tg-4x300-Runx2). We have previously shown that four copies of the 300 bp Col10a1 distal promoter and its basal promoter were able to direct hypertrophic chondrocyte-specific reporter (LacZ) expression in transgenic mice at the P1 stage (Fig. 1A, Tg-4x300-LacZ and Zheng et al., 2009). Further analysis of transgenic mouse embryos at the E15.5 stage confirms that X-gal blue staining is exclusively throughout the hypertrophic zone, but not in adjacent resting or proliferative chondrocytes, nor in surrounding muscles or connective tissues (Fig. 1B). We used a shorter Col10a1 basal promoter which ends at exon I to avoid the additional splicing acceptor site that is required for the Tg-4x300-LacZ reporter construct (Fig. 1A and Zheng et al., 2009). PCR genotyping using Runx2 and Flag specific primers showed that we successfully generated five Col10a1-Runx2 transgenic founders (Fig. 1C).
Elevated Runx2 and Col10α1 expression in Col10α1-Runx2 transgenic mice
To determine if the transgene (flag-tagged Runx2) undergoes germline transmission and expresses in offsprings of these transgenic founders, we have examined Flag-tag expression on TG mouse limb sections by immunohistochemistry analysis using Flag antibody or no antibody control. As illustrated in Fig. 2A, brown staining was primarily restricted to the pre-hypertrophic and hypertrophic zone when Flag antibody was used, suggesting successful target expression of transgene (Flag-tag) by the cell-specific Col10a1 regulatory elements. We also examined Runx2 and its known target Col10a1 expression in these Col10a1-Runx2 transgenic mice. Total RNAs from hind limbs of three transgenic mouse lines at embryonic day 17.5 were prepared. At least nine littermates from each litter size were used for quantitative real-time RT-PCR and for subsequent statistical analysis. Data present here are from a representative transgenic mouse line. The results show that relative Runx2 mRNA was significantly increased (2.11-fold, p = 0.02; Fig. 2B) in transgenic (TG) mice compared to their wild-type (WT) littermates. Meanwhile, Col10a1 transcripts were examined in these E17.5 limb tissues and showed an approximate three-fold (2.89-fold, p = 0.05; Fig. 2B) up-regulation in TG embryos. Expression analysis of Runx2 and Col10a1 genes at the P1 stage showed that both Runx2 (2.25-fold, p = 0.02) and Col10α1 (3.11-fold, p = 0.009) mRNA transcripts were significantly up-regulated in TG mice (Fig. 2C, see also Table 2). We also micro-dissected the hypertrophic zones from mouse femoral growth plates and performed expression analysis. Similar up-regulation of both Runx2 (1.94-fold, p = 0.043) and Col10a1 (2.59-fold, p = 0.031) expression was observed in TG compared to WT littermates (Fig. 2D). Our results together suggest that Runx2 is up-regulated in Col10a1-Runx2 transgenic mice and that up-regulation of Runx2 is associated with increased level of Col10a1 expression in vivo.
FIG.2. Runx2 and Col10α1 mRNA is upregulated in transgenic mice at E17.5 and P1 stages.
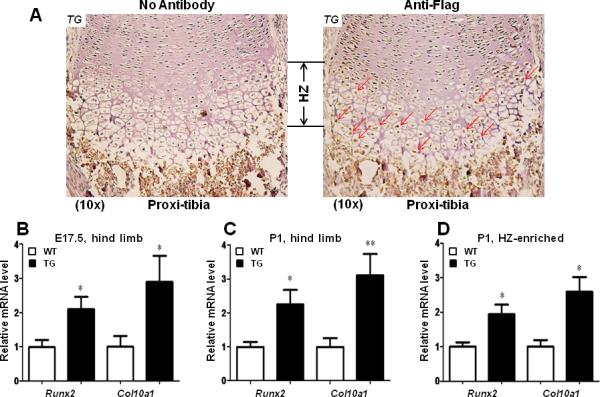
(A). Immunohistochemistry analysis was conducted on TG mouse hind limb sections with or without anti-Flag antibody. Brown staining showing Flag expression was primarily restricted to the pre-hypertrophic and hypertrophic chondrocytes of proximal tibia (right panel, red arrows). Left panel shows no antibody control. TG: Transgenic. HZ: hypertrophic zone. (B). Relative mRNA transcripts of Col10a1 and Runx2 were examined using total RNAs prepared from whole hind limbs at E17.5 stage. Runx2 showed two-fold upregulation in transgenic (TG) mice compared to their wild-type (WT) littermates (WT vs. TG: 1.00 ± 0.20 vs. 2.10 ± 0.36, n = 6, t = 2.69, df = 10, p = 0.02). Meanwhile, Col10a1 expression was also increased in TG mice (WT vs. TG: 1.00 ± 0.32 vs. 2.89 ± 0.77, n = 5, t = 2.27, df = 8, p = 0.05). (C). As illustrated, both Runx2 and Col10a1 showed similar increased level of mRNA transcripts in TG mice at P1 stage (Runx2, WT vs. TG: 1.00 ± 0.15 vs. 2.25 ± 0.43, n = 6, t = 2.78, df = 10, p = 0.02; Col10α1, WT vs. TG: 1.00 ± 0.26 vs. 3.11 ± 0.63, n = 5-6, t = 3.30, df = 9, p = 0.009). (D). Runx2 and Col10a1 also showed upregulation in transgenic mice using total RNAs prepared from hypertrophic zone-enriched tissues at P1 stag (Runx2, WT vs. TG: 1.00 ± 0.13 vs. 1.94 ± 0.28, n = 4-8, t = 2.04, df = 10, p = 0.043; Col10α1, WT vs. TG: 1.00 ± 0.19 vs. 2.59 ± 0.43, n = 4-8, t = 2.50, df = 10, p = 0.031). TG: transgenic; WT: wild-type littermates. Bars denote means ± SE for Runx2 and Col10α1 expression. “*”: p < 0.05; “**”: p < 0.01.
Table 2.
Fold change of chondrogenic/apoptotic marker gene expression
| Gene Name | E17.5 | P1 | ||||
|---|---|---|---|---|---|---|
| Wt | Tg | p | Wt | Tg | p | |
| Runx2 | 1.01 ± 0.20 | 2.04 ± 0.36 | 0.0275* | 1.00 ± 0.15 | 2.25 ± 0.43 | 0.0195* |
| Col10α1 | 1.00 ± 0.32 | 2.89 ± 0.77 | 0.0500* | 1.00 ± 0.26 | 3.11 ± 0.63 | 0.0092** |
| Sox9 | 1.00 ± 0.24 | 2.30 ± 0.45 | 0.0297* | 1.00 ± 0.27 | 17.88 ± 7.51 | 0.0484* |
| Bax | 1.00 ± 0.20 | 1.42 ± 0.22 | 0.1837 | 1.00 ± 0.09 | 1.18 ± 0.13 | 0.2892 |
| Bcl-2 | 1.00 ± 0.23 | 2.00 ± 0.24 | 0.0159* | 1.00 ± 0.23 | 2.30 ± 0.21 | 0.0020** |
| Opn | 1.00 ± 0.13 | 1.93 ± 0.26 | 0.0097** | 1.00 ± 0.10 | 1.69 ± 0.28 | 0.0361* |
p < 0.05
p < 0.01
Delayed ossification in Col10a1-Runx2 transgenic mice
The transgenic mice are grossly normal, fertile with normal life span. However, we have performed detailed skeletal phenotypic analysis of the mice from E14.5 until P6 using alcian blue and alizarin red staining. Here, we show the skeletal staining pattern of representative E17.5 (Fig. 3A) and P1 stages (Fig. 3B) from both TG and WT littermates. Overall, skeletal preparation suggests that the Col10a1-Runx2 TG mice show similar size as their WT littermates at either the E17.5 (Fig. 3A-A1) or P1 stages (Fig. 3B-B1). However, as illustrated in Fig. 3A2, the sutures and fontanels were wider in E17.5 TG mice compared to their littermate controls, whereas at the P1 stage, a much wider sagittal and coronal sutures and posterior fontanel were observed in TG mice, suggesting delayed craniofacial bone formation. Meanwhile, less intense alizarin red staining was observed in E17.5 TG mouse limb digits (Fig. 3A-A4, A5, metatarsal and phalanges, arrows), while significantly reduced alizarin red staining was observed in the xiphoid (Fig. 3B-B3), limbs (calceneus and digits, Fig. 3B-B4, B5), and tails (caudal vertebrae, Fig. 3B-B6) of the TG mice at the P1 stage. Similar results were also observed at stages P3 and P6 of the TG mice (data not shown). Data were collected from at least three Col10a1-Runx2 transgenic mouse lines for each observation. These results suggest that Runx2 over expression in hypertrophic chondrocytes gradually but significantly delays skeletogenesis of both the axial and the appendicular skeletons.
FIG.3. Delayed ossification in Col10α1-Runx2 transgenic mice.
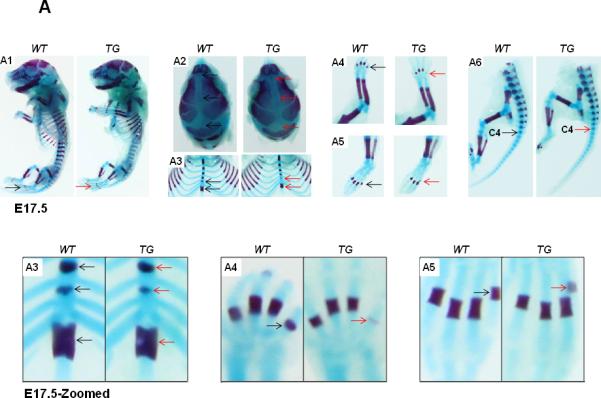
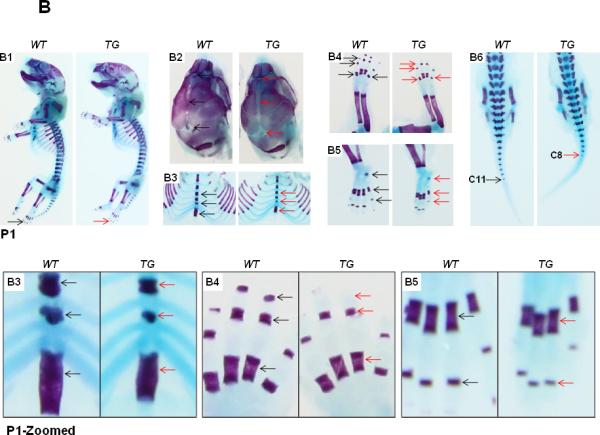
(A). Alcian blue and alizarin red staining of mouse embryos at E17.5 stage showed slightly delayed ossification (less alizarin red staining) in TG mice compared to littermate controls in following skeletal tissues. A1: Whole skeletal staining, no ossification signs were observed in the phalanges in both TG and WT embryos (black and red arrows). A2: Skull, sagittal suture and front/posterior fontanels are wider in TG embryos (red arrows). A3: Rib and thoracic vertebrae, less alizarin red can be observed in vertebrae and xiphoid (red arrows). A4/A5: Fore and hind limbs, obvious alizarin red staining can be seen in the last metatarsal bone of WT embryos while TG embryos can barely see the red staining (red arrows). A6: Tails, alizarin red staining showed no difference in the 4th caudal vertebrae of both WT and TG mouse embryos (black and red arrows). Bottom panel shows zoomed-in pictures of A3, A4, and A5. (B). Less ossification signals in the same panel of skeletons at P1 stage. B1: Whole skeletal staining, detectable alizarin red staining was observed in the phalanges in both TG and WT P1 mice (black and red arrows). B2: Skull, sagittal suture is wider and front/posterior fontanels are more open in TG mice (red arrows) compared to WT ones (black arrows). B3: Rib and thoracic vertebrae, less alizarin red signaling can be observed in vertebrae and the xiphoid is shorter in TG mice (red arrows). B4/B5: Fore and hind limbs, less alizarin red staining show in the metatarsal/phalange bones and calceneus of the TG mice (red arrows) compared to WT controls (black arrows). B6: Tails, alizarin red staining can be seen until the 8th caudal vertebrae of TG mice (red arrow) compared to WT littermates (up to 11th caudal vertebrae, black arrow). Bottom panel shows zoomed-in pictures of B3, B4, and B5.
Delayed chondrocyte maturation in Col10a1-Runx2 transgenic mice
We have performed histological analysis of the growth plate of mouse embryos (E17.5) and new born mice (P1). Hematoxylin & eosin staining of sagittal sections from proximal tibia in E17.5 TG mice showed an elongated hypertrophic zone with increased layers of slightly disorganized hypertrophic chondrocytes compared to their WT littermates (Fig.4A). Similar results were also observed in sagittal sections of the proximal tibia in TG mice at the P1 stage using Safranin O/Fast Green staining (Fig.4B). Histological analysis of the growth plate of the proximal ulna suggests that the increased layers of hypertrophic chondrocytes seen in TG mice are confined within the zone of terminal chondrocyte maturation where bone replacement normally occurs in the WT littermates (Fig. 4C, yellow rectangles). These results demonstrate the delayed chondrocyte maturation in these Col10a1-Runx2 transgenic mice. To determine how the delayed chondrocyte maturation impacts local matrix mineralization, we have performed von Kossa staining on mouse hind limb sections at P1 stage. Less von Kossa staining (black dots) was shown in the hypertrophic zone of TG mice compared to WT littermate controls (Fig. 4D). This result suggests impaired mineralization in Col10a1-Runx2 transgenic mice, possibly due to delayed chondrocyte maturation during endochondral bone formation.
FIG.4. Histology analysis of mouse growth plates.
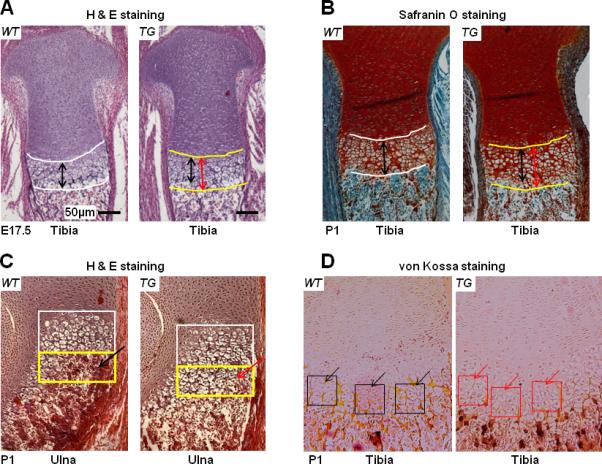
(A). H&E staining of sagittal sections of proximal tibia suggested longer hypertrophic zone in E17.5 TG mouse embryos (red double arrows) compared to WT littermates (black double arrows). Bars represent 50 μm. (B). Safranin O/Fast Green staining of P1 proximal tibia sections also indicated that TG mice (red double arrows) have longer hypertrophic zone than littermate controls (black arrows). (C). More layers of hypertrophic chondrocytes can be seen in sagittal sections of proximal ulna in TG mice (yellow rectangle, red arrow) at P1 stage compared to littermate controls (yellow rectangle, black arrow). (D). von Kossa staining was performed to quantify matrix mineralization on proximal tibia of TG or WT mice at P1 stage. Less von Kossa staining (black dots) suggesting decreased mineralization was shown in the hypertrophic zone of TG mice (red square and arrows) compared to WT littermate controls (black square and arrows).
Increased chondrogenic/apoptotic marker gene expression in Col10a1-Runx2 transgenic mice
To study the potential mechanism of delayed chondrocyte maturation and ossification in these Col10a1-Runx2 transgenic mice, we have performed qRT-PCR to examine the expression level of Bax and Bcl-2 using total RNAs prepared from hind limbs at E17.5 and P1 stages. The results showed that Bcl-2 (B-cell leukemia/lymphoma 2), the anti-apoptosis marker (Tsujimoto et al., 1998), was significantly increased in TG mice at both E17.5 (2-fold, p = 0.016, Fig. 5A) and P1 stages (2.3-fold, p = 0.002; Fig. 5B). Meanwhile, Bax (BCL2-associated X protein), the pro-apoptotic marker (Tsujimoto et al., 1998), did not show significant changes in TG mice compared to their WT littermates at either E17.5 (1.43-fold, p = 0.185; Fig. 5A) or P1 stages (1.18-fold, p = 0.295; Fig. 5B). We have also performed expression analysis of Osteopontin (Opn), a gene that has been implicated with multiple functions in bone remodeling and cell apoptosis (Standal et al., 2004). The result shows that Opn was up-regulated in TG mice at both E17.5 (1.93-fold, p=0.0097, Fig. 5A) and P1 (1.69-fold, p=0.0361, Fig. 5B) stages. Surprisingly, we also detected significant upregulation of Sox9, a gene known to inhibit chondrocyte maturation/apoptosis and osteoblast differentiation (Zhou et al., 2006; Ikegami et al., 2011), in TG mice at both E17.5 (2.30-fold, p=0.0297, Fig. 5A) and P1 (17.88-fold, p=0.0484, Fig. 5B, see also Table 2) stages. These results together suggest a potential correlation of altered chondrogenic apoptosis or differentiation with the delayed chondrocyte maturation and ossification observed in the Col10a1-Runx2 TG mice.
FIG.5. Differential expression of chondrogenic and apoptotic marker genes.
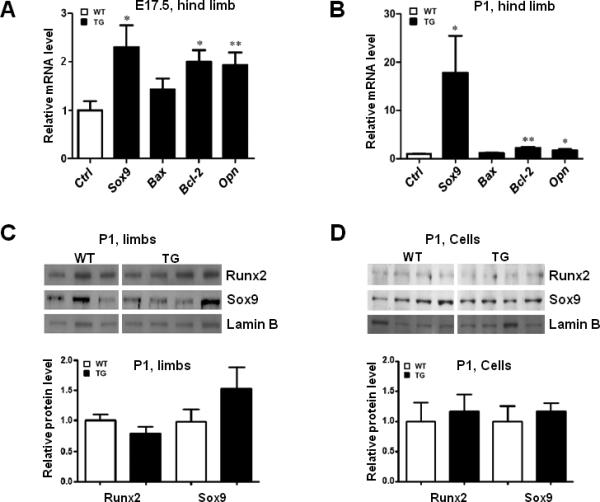
(A). qRT-PCR was performed to examine following chondrogenic and apoptotic marker genes using total RNAs prepared from mouse hind limbs at E17.5 stage. As illustrated, Sox9 (2.30-fold, p=0.0297) is significantly upregulated in TG mice compared to their WT littermates. Meanwhile Bcl-2 (2-fold, p = 0.016), and Opn (1.93-fold, p = 0.0097) were also significantly upregulated in TG mice compared to littermate controls, while Bax (1.42 fold, p=0.1837) is slightly upregulated but without statistical significance. (B). Relevant marker genes were also examined at the P1 stage. As illustrated, Bcl-2 (2.3-fold, p = 0.002), Opn (1.69-fold, p = 0.0361), and Sox9 (17.88-fold, p=0.0484) were also significantly increased in TG mice compared to WT littermates. Similar to the result of E17.5, Bax (1.18 fold, p=0.2892) did not show significant changes between TG and WT controls. Bars denote means ± SE for marker gene expression. TG: transgenic; WT: wild-type. “*”: p<0.05; “**”: p<0.01. (C). Western blotting assay was performed using protein extracts prepared from TG or WT mouse hind limbs at the P1 stage and Runx2 or Sox9 antibodies. Both Runx2 and Sox9 are highly expressed in TG or WT mouse limbs (top). Anti-Lamin B antibody was used as an internal control. Densitometry analysis showed that Sox9 increased ~50%, while Runx2 was slightly decreased in TG mouse limbs (bottom), but not statistically significant. (D). Western blotting was also conducted using protein extracts from primary cultured chondrocytes of TG or WT mice at P1 stage and the antibodies as mentioned above. The results showed that Runx2 is weakly expressed, while Sox9 is highly expressed in all the primary chondrocytes examined (top). Densitometry analysis suggested that both runx2 and Sox9 are slightly increased in TG groups but without statistical significance (bottom).
Runx2 and Sox9 expression in Col10α1-Runx2 transgenic mouse limbs and cells
As described above, we detected increased level of Runx2 and Sox9 mRNA transcripts in TG mouse limbs at E17.5 and P1 stages (Fig. 5A, 5B). To determine the protein level of these genes, we have prepared protein extracts from new born mouse hind limbs (left) of two transgenic mouse lines at P1 stage and performed Western blotting assay using anti-Runx2 and anti-Sox9 antibodies. Anti-Lamin B antibody was used as an internal control. As illustrated in Fig. 5C (top), both Runx2 and Sox9 are highly expressed in TG or WT mouse limbs. The signal density of individual TG or WT sample blotted with anti-Runx2 or anti-Sox9 was calculated and normalized to the sample blotted with anti-Lamin B. The results showed that Sox9 increased ~50%, while Runx2 was slightly decreased in TG mouse limbs (Fig. 5C, bottom). However, no statistically significant difference of Runx2 or Sox9 expression was detected between these TG and WT groups. We have also isolated and cultured primary chondrocytes from TG or WT mouse hind limbs (right). Similar Western blotting assay was conducted using protein extracts prepared from these cells and the antibodies as mentioned above. As expected, Runx2 is weakly expressed, while Sox9 is highly expressed in all the primary chondrocytes examined (Fig. 5D, top). Densitometry analysis suggested that both Runx2 and Sox9 are slightly increased in TG groups, though not statistically significant (Fig. 5D, bottom). These results suggested that targeting Runx2 in hypertrophic chondrocytes may not necessarily increase the whole protein level of marker genes that have increased mRNA transcripts, possibly due to the tissues or cell populations selected and the approaches used for corresponding analysis.
Runx2 and Sox9 expression in hypertrophic chondrocytes and their effects
To determine local expression of Runx2, we have performed IHC analysis on comparable TG and WT mouse limb sections using Runx2 antibody. As expected, Runx2 is highly expressed in pre-hypertrophic chondrocytes and hypertrophic chondrocytes of wild-type mice (Fig. 6A, WT). Surprisingly, Runx2 expression is obviously decreased in these regions of TG mice (Fig. 6A, TG). Meanwhile, we have also performed similar IHC analysis using Sox9 antibody. High level of Sox9 expression was detected in proliferating chondrocytes of both TG and WT mice (data not shown). Sox9 decreased significantly in pre-hypertrophic chondrocytes, while it is barely detectable in hypertrophic chondrocytes of WT mice (Fig. 6B, WT). However, we detected obvious immune-staining signal in terminal hypertrophic chondrocytes, suggesting ectopic expression of Sox9 in TG mice (Fig. 6B, TG). To determine if the altered Runx2 and Sox9 expression impacts chondrocyte apoptosis, we performed TUNEL staining on comparable sections of TG and WT mouse limbs. While the majority of the apoptotic cells were localized in the zone of chondrocyte hypertrophy, much less apoptotic cells were observed in TG mice (Fig. 6C, TG) compared to the littermate controls (Fig. 6C, WT).
FIG.6. Runx2, Sox9 expression and apoptosis assay in mouse growth plates.
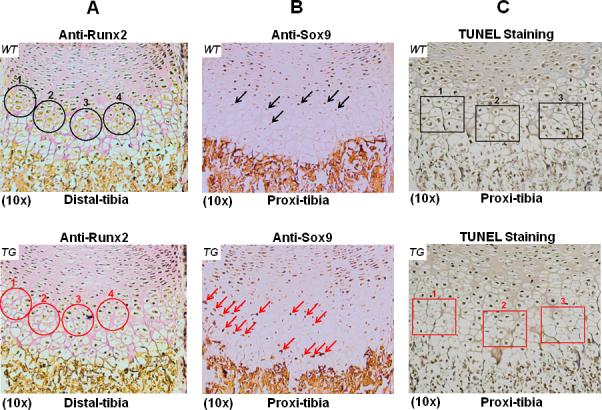
(A). IHC assay was performed on distal tibia sections of TG or WT littermates at P1 stage with anti-Runx2 antibody. Intense brown staining showing Runx2 expression was primarily restricted to pre-hypertrophic and hypertrophic chondrocytes of WT controls (top, black circles). Much less brown staining indicating decreased Runx2 expression was observed in comparable tibia sections of TG mice (red circles, bottom). (B). IHC assay on proximal tibia sections of WT controls with anti-Sox9 antibody showed that Sox9 is weakly expressed in pre-hypertrophic chondrocytes, while it is barely detectable in hypertrophic chondrocytes (top, black arrows). IHC assay on comparable TG tibia sections showed more intense immune-staining signal in pre-hypertrophic and hypertrophic chondrocytes, suggesting ectopic expression of Sox9 in TG mice (bottom, red arrows). (C). Apoptosis was determined by TUNEL staining on comparable proximal tibia sections of TG and WT littermates. The majority of the apoptotic cells were localized in the hypertrophic zone of WT control (top, black rectangles). Less apoptotic cells were observed in comparable tibia section of TG mice (bottom, red rectangles).
DISCUSSION
For the past decade, substantial progress has been made in delineating transcription factors that control both bone development and bone remodeling. Among these, Runx2, a Runt domain family member, has been extensively studied and demonstrated to be a master transcription factor for osteoblast differentiation both in vitro and in vivo (Komori et al., 1997; Otto et al., 1997). Meanwhile, there is increasing data which supports that Runx2 plays an essential role in chondrocyte maturation during endochondral ossification (Inada et al., 1999). However, the mechanism by which Runx2 regulates chondrocyte maturation and impacts skeletal development or disease progression is still not fully understood. One potential pathway could be that altered Col10a1 and Runx2 expression in hypertrophic chondrocytes may change the local matrix or molecular environment and contribute to the corresponding skeletal phenotypic consequence.
There are several lines of evidence which support the correlation of Runx2 and Col10a1 expression with skeletal consequences. Runx2 null mice lack bone formation while Runx2 heterozygotes mimic a human skeletal disorder (Komori et al., 1997; Otto et al., 1997). We have previously detected decreased Col10a1 expression in Runx2 heterozygotes, whereas Col10a1 was barely detectable in Runx2-null mice (Inada et al., 1999; Zheng et al., 2003). In a mouse model of osteoarthritis, Runx2 heterozygotes show decreased cartilage destruction and osteophyte formation, along with reduced Col10a1 and Mmp-13 expression (Kamekura et al., 2006). The human craniofacial dysplasia, which is caused by RUNX2 haploinsufficiency, also shows altered chondrocyte hypertrophy and down-regulation of COL10A1 (Zheng et al., 2005). These observations suggest that abnormal Runx2 and Col10a1 expression is associated with skeletal defects or skeletal diseases.
In our study, we have generated Col10a1-Runx2 transgenic mice using the 300-bp cell-specific Col10a1 control element. This 300-bp Col10a1 distal promoter contains the major cis-enhancer that has been shown to drive reporter (LacZ) expression exclusively in hypertrophic chondrocytes in transgenic studies (Fig. 1B, Zheng et al., 2009). Consequently, we detected transgene (Flag-tagged Runx2) expression primarily within the pre-hypertrophic and hypertrophic region (Fig. 2A). We also detected increased Runx2 and Col10a1 mRNA transcripts in the limb tissues, especially in hypertrophic chondrocyte-enriched skeletal tissues of the TG mice (Fig. 2B, 2C, and 2D). These results suggest that local overexpression of Runx2 upregulates Col10a1 expression in vivo. Interestingly, skeletal preparations suggest delayed ossification in the axial and appendicular skeleton of TG mice, especially in long bones compared to their WT littermates (Fig. 3). An elongated hypertrophic zone and decreased von Kossa and TUNEL staining was also detected in TG mice by histological analysis (Fig. 4, Fig. 6C). These data suggest that enhanced Runx2 and Col10a1 expression has an impact on the skeletal consequence, possibly through altering the status of chondrocyte maturation, apoptosis and local matrix mineralization.
Runx2 has been extensively studied and is known to positively regulate chondrocyte maturation and bone formation (Takeda et al., 2001; Inada et al., 1999; Kim et al., 1999). However, phenotypic analysis of our Col10a1-Runx2 TG mice demonstrated delayed ossification and chondrocyte maturation. This is intriguing and reminds us of two recent independent Sox9 transgenic studies which show skeletal phenotypes similar to our Col10a1-Runx2 TG mice. Kim et al reported that Sox9 overexpression in chondrocytes suppressed chondrocyte hypertrophy and subsequent ossification while misexpression of Sox9 in hypertrophic chondrocytes delayed terminal chondrocyte differentiation (Kim et al., 2010). This finding was confirmed by another group which performed transgenic studies by misexpressing Sox9 in hypertrophic chondrocytes using a BAC-Col10a1 promoter (Hattori et al., 2010). These Col10a1-Sox9 transgenic mice showed defective endochondral bone formation due to significantly delayed vascular invasion along with decreased expression of some Runx2 target genes (Hattori et al., 2010). We therefore examined Sox9, a known negative regulator of endochondral ossification, in our transgenic mice. Surprisingly, Sox9 mRNA transcript was significantly increased at E17.5 and was even more upregulated at the P1stage (Fig. 5A, 5B, and Table 2) by qRT-PCR analysis. We also detected increased level of Sox9 expression in primary cultured chondrocytes and limb tissues of transgenic mice by Western blotting assay, although the increase is not statistically significant (Fig. 5C, 5D). This is possibly due to the fact that, the Col10a1 promoter elements primarily function at the stage of pre-hypertrophic and hypertrophic chondrocytes. These chondrocytes only occupy small portions of the whole limbs that include both osteoblasts and chondrocytes at different stages of maturation. It's also possible that the approach of Western blotting assay may not be sensitive enough to detect the subtle difference of protein expression level between TG and WT groups. However, increased Sox9 expression was detected in terminal hypertrophic chondrocytes of transgenic mouse limb sections by IHC analysis (Fig. 6B). Meanwhile, IHC analysis detected decreased Runx2 expression in pre-hypertrophic and hypertrophic chondrocytes (Fig. 6A), possibly due to increased local expression of Sox9. These results may partially explain the phenotypic findings in our Col10a1-Runx2 TG mice. Previous studies have demonstrated that Sox9 is a master transcription factor both for chondrogenesis and osteogenesis (de Crombrugghe et al., 2001). Moreover, Sox9 has recently been shown to direct intracellular degradation of Runx2 and inhibit the transactivity of Runx2 (Cheng A, Genever PG, 2010), while Sox9 has been demonstrated to dominate over Runx2 function during endochondral ossification (Zhou et al., 2006).
We also observed delayed craniofacial bone and suture development in our transgenic mice at late embryonic and early postnatal stages (Fig. 3 and data not shown). Similar mechanism of impaired endochondral bone formation as described above may apply to these craniofacial skeletal consequences. Flat bones, such as craniofacial, clavicle, scapula bones, are known to develop through an intramembranous pathway. However, cartilage intermediate, which includes hypertrophic chondrocyte-like cells and expresses type X collagen, may also appear in some of the flat bones. Supporting this is the specific reporter (LacZ) expression that we have observed in hypertrophic chondrocyte-like cells within the craniofacial and scapula bones of our Tg-4x300-LacZ transgenic mice (Zheng et al., 2009, and data not shown).
Multiple Runx2 transgenic mice have previously been generated by several research groups for Runx2 gain-of-function studies. Transgenic mice overexpressing Runx2 in osteoblasts using the collagen type I gene (Col1a1) promoter developed severe osteopenia due to increased bone resorption and decreased mineralization (Geoffroy et al., 2002), whereas overexpression of Runx2 under the control of the chondrocyte-specific type II collagen gene (Col2α1) promoter/enhancer resulted in acceleration of endochondral ossification due to precocious chondrocyte maturation (Takeda et al., 2001; Ueta et al., 2001). In our Col10a1-Runx2 transgenic mice, besides delayed endochondral ossification, we also observed a longer hypertrophic zone with increased layers of hypertrophic chondrocytes in the growth plates of long bones at both E17.5 and P1 stages (Fig. 4A, 4B). The increased layers of hypertrophic chondrocytes are within the chondro-osseous junctions, suggesting decreased chondrocyte apoptosis or impaired bone mineralization (Fig. 4C). Indeed, real-time RT-PCR analysis showed that both Bcl-2 and Opn are upregulated in these Col10a1-Runx2 TG mice (Fig. 5A, 5B, and Table 2). Bcl-2 (B-cell leukemia/lymphoma 2) is an anti-apoptosis marker (Tsujimoto et al., 1998). Opn is also an important anti-apoptotic factor that is involved in tumorigenesis and bone remodeling (Standal et al., 2004). As to Sox9's function on chondrocyte survival, Dr. Ikegami et al. has recently reported observation of chondrocyte apoptosis by Sox9 inactivation, suggesting its anti-apoptotic role (Ikegami et al., 2011). Therefore, increased Bcl-2, Opn, and Sox9 expression may result in decreased apoptosis of hypertrophic chondrocytes and thus further impair chondrocyte terminal differentiation and subsequent bone replacement that is essential for endochondral ossification.
In summary, we have successfully generated Runx2 transgenic mice using hypertrophic chondrocyte-specific Col10a1 regulatory elements. These Col10a1-Runx2 transgenic mice have increased level of Runx2 and Col10a1 transcripts in limb tissues. No significant change of Runx2 protein level in whole limbs was detected. However, due to the property of the Col10a1 promoter activity, targeting Runx2 expression in hypertrophic chondrocytes may change the local matrix or molecular environment which is critical for subsequent blood vessel invasion and mineralization. This matrix environment change may trigger expression of a series of chondrogenic and apoptotic related genes, such as Bcl-2, Opn and, Sox9 (Fig. 7). These gene products, especially, Sox9, may interact with or surpass Runx2's function, and thereby lead to impaired chondrocyte maturation, apoptosis, matrix mineralization, and thus, defective endochondral ossification with skeletal phenotypic consequences (Tsujimoto et al., 1998; Standal et al., 2004; Zhou et al., 2006; Wai et al., 2006; Ho et al., 2009).
FIG.7. Potential mechanisms of Runx2-Col10a1 effects on endochondral bone formation.
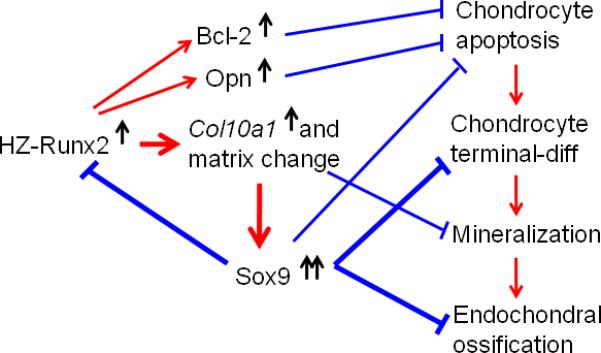
The putative impacts on bone formation in this Col10a1-Runx2 mouse model are proposed: selectively targeting Runx2 overexpression in hypertrophic chondrocytes results in upregulated Col10a1 expression; this will cause local matrix environment change and the subsequent matrix mineralization. The increased Sox9 expression in hypertrophic chondrocytes possibly mediates Runx2 degradation or dominates over Runx2 and other genes’ function, delays chondrocyte maturation and apoptosis, subsequently affects the process of blood vessel invasion, bone mineralization and eventually, leads to impaired endochondral ossification. Meanwhile, upregulation of Bcl-2 and Opn may also delay chondrocyte apoptosis or terminal chondrocyte maturation and therefore, an expanded hypertrophic zone. Red arrows: promote chondrocyte maturation or bone formation; Blue arrows: inhibitory effect.
ACKNOWLEDGEMENTS
We are grateful to Dr. Kotaro Sena, and Mr. David Karwo for technical help on histology analysis of the transgenic mice at the Rush Histology Core. The Col10a1-Runx2 transgenic mice were generated within the Transgenic Production Service core facility (directed by Dr. Roberta Franks) at the University of Illinois at Chicago (UIC). This work was supported by the National Institutes of Health grant (R03 DE16041, NIH/NIDCR, Q.Z.), the Arthritis Foundation (Q.Z.), the 2008 Rush pilot project (Q.Z.) and the Bear Necessities Pediatric Cancer Foundation (Q.Z.).
Footnotes
DISCLOSURES: All authors state that they have no conflicts of interest.
REFERENCES
- Bonewald LF, Harris SE, Rosser J, Dallas MR, Dallas SL, Camacho NP, Boyan B, Boskey A. von Kossa staining alone is not sufficient to confirm that mineralization in vitro represents bone formation. Calcif Tissue Int. 2003 May;72(5):537–47. doi: 10.1007/s00223-002-1057-y. 2003. Epub 2003 May 6. [DOI] [PubMed] [Google Scholar]
- Cheng A, Genever PG. SOX9 determines RUNX2 transactivity by directing intracellular degradation. J Bone Miner Res. 2010;25:2680–2689. doi: 10.1002/jbmr.174. [DOI] [PubMed] [Google Scholar]
- de Crombrugghe B, Lefebvre V, Nakashima K. Regulatory mechanisms in the pathways of cartilage and bone formation. Curr Opin Cell Biol. 2001;13:721–727. doi: 10.1016/s0955-0674(00)00276-3. [DOI] [PubMed] [Google Scholar]
- Dourado G, LuValle P. Proximal DNA elements mediate repressor activity conferred by the distal portion of the chicken collagen X promoter. J Cell Biochem. 1998;70:507–516. doi: 10.1002/(sici)1097-4644(19980915)70:4<507::aid-jcb7>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, Amling M, Karsenty G. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999;13:1025–1036. doi: 10.1101/gad.13.8.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Tagashira S, Harada H, Ogawa S, Katsumata T, Nakatsuka M, Komori T, Takada H. Isolation and characterization of the distal promoter region of mouse Cbfa1. Biochim Biophys Acta. 1999;1446:265–272. doi: 10.1016/s0167-4781(99)00113-x. [DOI] [PubMed] [Google Scholar]
- Geoffroy V, Kneissel M, Fournier B, Boyde A, Matthias P. High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Mol Cell Biol. 2002;22:6222–6233. doi: 10.1128/MCB.22.17.6222-6233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3(8):1253–60. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- Hattori T, Muller C, Gebhard S, Bauer E, Pausch F, Schlund B, Bosl MR, Hess A, Surmann-Schmitt C, von der Mark H, de Crombrugghe B, von der Mark K. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 2010;137:901–911. doi: 10.1242/dev.045203. [DOI] [PubMed] [Google Scholar]
- Higashikawa A, Saito T, Ikeda T, Kamekura S, Kawamura N, Kan A, Oshima Y, Ohba S, Ogata N, Takeshita K, Nakamura K, Chung UI, Kawaguchi H. Identification of the core element responsive to runt-related transcription factor 2 in the promoter of human type X collagen gene. Arthritis Rheum. 2009;60:166–178. doi: 10.1002/art.24243. [DOI] [PubMed] [Google Scholar]
- Hinoi E, Bialek P, Chen YT, Rached MT, Groner Y, Behringer RR, Ornitz DM, Karsenty G. Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. Genes Dev. 2006;20:2937–2942. doi: 10.1101/gad.1482906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WP, Chan WP, Hsieh MS, Chen RM. Runx2-mediated bcl-2 gene expression contributes to nitric oxide protection against hydrogen peroxide-induced osteoblast apoptosis. J Cell Biochem. 2009;108:1084–1093. doi: 10.1002/jcb.22338. [DOI] [PubMed] [Google Scholar]
- Ikegami D, Akiyama H, Suzuki A, Nakamura T, Nakano T, Yoshikawa H, Tsumaki N. Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development. 2011;138:1507–1519. doi: 10.1242/dev.057802. [DOI] [PubMed] [Google Scholar]
- Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kitamura Y, Kishimoto T, Komori T. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. 1999;214:279–290. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, Komori T, Sato S, Takeda S, Karsenty G, Nakamura K, Chung UI, Kawaguchi H. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;54:2462–2470. doi: 10.1002/art.22041. [DOI] [PubMed] [Google Scholar]
- Kanatani N, Fujita T, Fukuyama R, Liu W, Yoshida CA, Moriishi T, Yamana K, Miyazaki T, Toyosawa S, Komori T. Cbf beta regulates Runx2 function isoform-dependently in postnatal bone development. Dev Biol. 2006;296:48–61. doi: 10.1016/j.ydbio.2006.03.039. [DOI] [PubMed] [Google Scholar]
- Karagüzel G, Aktürk FA, Okur E, Gümele HR, Gedik Y, Okten A. Cleidocranial dysplasia: a case report. J Clin Res Pediatr Endocrinol. 2010;2(3):134–6. doi: 10.4274/jcrpe.v2i3.134. Epub 2010 Aug 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G. Minireview: transcriptional control of osteoblast differentiation. Endocrinology. 2001;142:2731–2733. doi: 10.1210/endo.142.7.8306. [DOI] [PubMed] [Google Scholar]
- Kim IS, Otto F, Zabel B, Mundlos S. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 1999;80:159–170. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- Kim Y, Murao H, Yamamoto K, Deng JM, Behringer RR, Nakamura T, Akiyama H. Generation of transgenic mice for conditional overexpression of Sox9. J Bone Miner Metab. 2011;29:123–129. doi: 10.1007/s00774-010-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Levanon D, Negreanu V, Bernstein Y, Bar-Am I, Avivi L, Groner Y. AML1, AML2, and AML3, the human members of the runt domain gene-family: cDNA structure, expression, and chromosomal localization. Genomics. 1994;23:425–432. doi: 10.1006/geno.1994.1519. [DOI] [PubMed] [Google Scholar]
- Li F, Lu Y, Ding M, Napierala D, Abbassi S, Chen Y, Duan X, Wang S, Lee B, Zheng Q. Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. J Bone Miner Res. 2011 Sep 1;26:2899–2910. doi: 10.1002/jbmr.504. 2011. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenmayer TF, Chen QA, Gibney E, Gordon MK, Marchant JK, Mayne R, Schmid TM. Collagen types IX and X in the developing chick tibiotarsus: analyses of mRNAs and proteins. Development. 1991;111:191–196. doi: 10.1242/dev.111.1.191. [DOI] [PubMed] [Google Scholar]
- Martin G, Andriamanalijaona R, Grässel S, Dreier R, Mathy-Hartert M, Bogdanowicz P, Boumédiene K, Henrotin Y, Bruckner P, Pujol JP. Effect of hypoxia and reoxygenation on gene expression and response to interleukin-1 in cultured articular chondrocytes. Arthritis Rheum. 2004 Nov;50(11):3549–60. doi: 10.1002/art.20596. [DOI] [PubMed] [Google Scholar]
- McIntosh I, Abbott MH, Francomano CA. Concentration of mutations causing Schmid metaphyseal chondrodysplasia in the C-terminal noncollagenous domain of type X collagen. Hum Mutat. 1995;5:121–125. doi: 10.1002/humu.1380050204. [DOI] [PubMed] [Google Scholar]
- Merciris D, Marty C, Collet C, de Vernejoul MC, Geoffroy V. Overexpression of the transcriptional factor Runx2 in osteoblasts abolishes the anabolic effect of parathyroid hormone in vivo. Am J Pathol. 2007;170:1676–1685. doi: 10.2353/ajpath.2007.061069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proc Natl Acad Sci USA. 1993;90:6859–6863. doi: 10.1073/pnas.90.14.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov D. Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harb Protoc. 2009;(3) doi: 10.1101/pdb.prot5170. pdb.prot5170. [DOI] [PubMed] [Google Scholar]
- Reno PL, McBurney DL, Lovejoy CO, Horton WE., Jr Ossification of the mouse metatarsal: differentiation and proliferation in the presence/absence of a defined growth plate. Anat Rec A Discov Mol Cell Evol Biol. 2006 Jan;288(1):104–18. doi: 10.1002/ar.a.20268. [DOI] [PubMed] [Google Scholar]
- Shen G. The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthod Craniofac Res. 2005;8:11–17. doi: 10.1111/j.1601-6343.2004.00308.x. [DOI] [PubMed] [Google Scholar]
- Simões B, Conceição N, Viegas CS, Pinto JP, Gavaia PJ, Hurst LD, Kelsh RN, Cancela ML. Identification of a promoter element within the zebrafish colXalpha1 gene responsive to runx2 isoforms Osf2/Cbfa1 and til-1 but not to pebp2alphaA2. Calcif Tissue Int. 2006;79:230–244. doi: 10.1007/s00223-006-0111-6. [DOI] [PubMed] [Google Scholar]
- Standal T, Borset M, Sundan A. Role of osteopontin in adhesion, migration, cell survival and bone remodeling. Exp Oncol. 2004;26:179–184. [PubMed] [Google Scholar]
- Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- Ueta C, Iwamoto M, Kanatani N, Yoshida C, Liu Y, Enomoto-Iwamoto M, Ohmori T, Enomoto H, Nakata K, Takada K, Kurisu K, Komori T. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol. 2001;153:87–100. doi: 10.1083/jcb.153.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai P, Mi Z, Gao C, Guo H, Marroquin C, Kuo P. Ets-1 and runx2 regulate transcription of a metastatic gene, osteopontin, in murine colorectal cancer cells. J Biol Chem. 2006;281:18973–18982. doi: 10.1074/jbc.M511962200. [DOI] [PubMed] [Google Scholar]
- Wallis GA, Rash B, Sweetman WA, Thomas JT, Super M, Evans G, Grant ME, Boot-Handford RP. Amino acid substitutions of conserved residues in the carboxyl-terminal domain of the alpha 1(X) chain of type X collagen occur in two unrelated families with metaphyseal chondrodysplasia type Schmid. Am J Hum Genet. 1994;54:169–178. 1994. [PMC free article] [PubMed] [Google Scholar]
- Warman ML, Abbott M, Apte SS, Hefferon T, McIntosh I, Cohn DH, Hecht JT, Olsen BR, Francomano CA. A type X collagen mutation causes Schmid metaphyseal chondrodysplasia. Nat Genet. 1993;5:79–82. doi: 10.1038/ng0993-79. [DOI] [PubMed] [Google Scholar]
- Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X, Lee B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol. 2003;162:833–842. doi: 10.1083/jcb.200211089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Sebald E, Zhou G, Chen Y, Wilcox W, Lee B, Krakow D. Dysregulation of chondrogenesis in human cleidocranial dysplasia. Am J Hum Genet. 2005;77:305–312. doi: 10.1086/432261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Keller B, Zhou G, Napierala D, Chen Y, Zabel B, Parker AE, Lee B. Localization of the cis-enhancer element for mouse type X collagen expression in hypertrophic chondrocytes in vivo. J Bone Miner Res. 2009;24:1022–1032. doi: 10.1359/JBMR.081249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci USA. 2006;103:19004–19009. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]