

Abstract
Mitochondria sustain damage with aging, and the resulting mitochondrial dysfunction has been implicated in a number of diseases including parkinson disease. We recently demonstrated that the E3 ubiquitin ligase Parkin, which is linked to recessive forms of parkinsonism, causes a dramatic increase in mitophagy and a change in mitochondrial distribution, following its translocation from the cytosol to mitochondria. Investigating how Parkin induces these changes may offer insight into the mechanisms that lead to the sequestration and elimination of damaged mitochondria. We report that following Parkin's translocation from the cytosol to mitochondria, Parkin (but not a pathogenic mutant) promotes the K63-linked polyubiquitination of mitochondrial substrate(s) and recruits the ubiquitin- and LC3-binding protein, p62/SQSTM1, to mitochondria. After its recruitment, p62/SQSTM1 mediates the aggregation of dysfunctional mitochondria through polymerization via its PB1 domain, in a manner analogous to its aggregation of polyubiquitinated proteins. Surprisingly and in contrast to what has been recently reported for ubiquitin-induced pexophagy and xenophagy, p62 appears to be dispensable for mitophagy. Similarly, mitochondrial-anchored ubiquitin is sufficient to recruit p62 and promote mitochondrial clustering, but does not promote mitophagy. Although VDAC1 (but not VDAC2) is ubiquitinated following mitochondrial depolarization, we find VDAC1 cannot fully account for the mitochondrial K63-linked ubiquitin immunoreactivity observed following depolarization, as it is also observed in VDAC1/3−/− mouse embryonic fibroblasts. Additionally, we find VDAC1 and VDAC3 are dispensable for the recruitment of p62, mitochondrial clustering and mitophagy. These results demonstrate that mitochondria are aggregated by p62, following its recruitment by Parkin in a VDAC1-independent manner. They also suggest that proteins other than p62 are likely required for mitophagy downstream of Parkin substrates other than VDAC1.
Key words: sequestration, PINK1, Parkinson disease, porin
Introduction
Mitochondria produce energy for the cell but are also a substantial source of reactive oxygen species (ROS). As mitochondrial damage accumulates with age, particularly in post-mitotic cells with high metabolic demands and/or high oxidation stress, mitochondria can become impaired thereby decreasing energy output, increasing ROS production and diminishing calcium-buffering capacity.1 Increased ROS production and decreased energy production may contribute to the aging phenotypes in animal models,2,3 and mitochondrial dysfunction has been associated with both inherited mitochondrial diseases and common diseases of aging, such as Parkinson disease.1 To help curb the accumulation of dysfunctional mitochondria with aging, the cell has adopted several mechanisms for controlling mitochondrial quality, including proteolytic degradation of damaged proteins, cycles of mitochondrial fission and fusion and the selective elimination of dysfunctional mitochondria by autophagy (hereafter referred to as mitophagy).4 In addition, sequestration of dysfunctional mitochondria has been observed in a mouse model of Parkinson disease,5 suggesting that confining damaged mitochondria in a minimal area within the cell may represent another adaptive mechanism to mitochondrial damage.
The PINK1/Parkin pathway, loss of which causes familial forms of parkinsonism, appears to be an important sensor of mitochondrial dysfunction and a mediator of mitochondrial quality control processes.6 PINK1, a serine/threonine kinase, recruits the E3 ubiquitin ligase Parkin from the cytosol to mitochondria,7–10 upon selective accumulation on the outer mitochondrial membrane of mitochondria with decreased inner membrane potential.9 Following recruitment to depolarized mitochondria, Parkin induces their elimination by mitophagy.11 Loss of either PINK1 or Parkin function leads to dysfunctional mitochondria in Drosophila, mice and human cells, consistent with the hypothesis that loss of PINK1/Parkin leads to failure of a mitochondrial quality control pathway.6 The mechanism by which Parkin induces the selective autophagy of dysfunctional mitochondria, following its recruitment, however, is unclear. Recently VDAC1 ubiquitination and SQSTM1/p62 (hereafter referred to as p62) have been implicated in the process of mitophagy downstream of Parkin translocation.7 We find that following its recruitment Parkin induces the K63-linked polyubiquitination of mitochondrial substrate(s) and recruits the ubiquitin- and LC3-binding adaptor protein p62. p62 mediates aggregation of mitochondria through polymerization via its PB1 domain, in a manner analogous to the aggregation of misfolded proteins.12 Interestingly, knockout or siRNA depletion of p62 does not inhibit Parkin-induced mitophagy, suggesting that, unlike what has been recently reported for pexophagy13 and xenophagy,14 p62 is not required for Parkin-induced mitophagy. In addition, while we confirm that VDAC1 is ubiquitinated, as was recently reported,7 and find that VDAC2 is not ubiquitinated following mitochondrial uncoupling in the presence of Parkin, VDAC1 ubiquitination does not account for the K63-linked polyubiquitin immunoreactivity observed following Parkin recruitment to depolarized mitochondria, as it is also observed in VDAC1/3−/− mouse embryonic fibroblasts (MEFs). Likewise, we find that VDAC1 and VDAC3 are dispensable for Parkin-induced p62 recruitment, mitochondrial clustering and mitophagy in MEFs. These findings demonstrate that p62 mediates the sequestration of impaired mitochondria in a VDAC1-independent manner. Additionally, they suggest that p62-independent mechanisms downstream of ubiquitination may mediate at least some forms of mitophagy in mammalian cells, and that ubiquitination of mitochondrial substrates other than VDAC1 and VDAC3 can mediate Parkin-induced mitophagy.
Results
Parkin, an E3 ubiquitin ligase, is recruited from the cytosol to mitochondria within one hour following loss of the inner mitochondrial membrane potential and over the subsequent 24–48 hours Parkin-bound mitochondria become eliminated by mitophagy.11 Shortly following its recruitment, Parkin induces the aggregation and perinuclear localization of mitochondria (Fig. 1A, top left part).11,15 HeLa cells not expressing Parkin (data not shown) or expressing Parkin mutated in the RING1 domain (R275W), previously found to translocate to mitochondria but not induce mitophagy,7,9 do not display mitochondrial aggregation (Fig. 1A, top right part). To develop a quantitative measure of relative mitochondrial aggregation we exploited the fact that for a given area occupied by mitochondria in the cell, mitochondria that are compacted into a limited area of the cell will have a shorter cumulative circumference than mitochondria that are dispersed throughout the cell. We refer to this measure as the compaction index with 1 being maximally compact (i.e., a circle) and 0 being minimally compact (i.e., with maximum perimeter for a given area) (Fig. 1B). Using this measure we verified that on average depolarized mitochondria from HeLa cells expressing wild-type EYFP-Parkin are more compact than depolarized mitochondria from HeLa cells expressing EYFP-Parkin R275W (Fig. 1C). Mitochondrial clustering seems to be delayed as opposed to absent with EYFP-Parkin R275W, as mitochondrial clustering is clearly seen after 24 hr treatment with CCCP (data not shown).7 These results suggest that wild-type Parkin promotes mitochondrial aggregation better than the R275W Parkin mutant.
Figure 1.

Wild-type Parkin, but not a pathogenic mutant, promotes mitochondrial aggregation. (A) HeLa cells co-transfected with DsRed-Mito and EYFP-Parkin or EYFP-Parkin R275W were treated with 10 µM CCCP for 3 hrs and immunostained for the mitochondrial protein Tom20. (B) Eight circles of equal area have different compaction indexes when dispersed and when aggregated, as measured by ImageJ. The compaction index is ratio of the shortest perimeter possible for an object with the same area as the object of interest divided by the actual perimeter of the object of interest (see Materials and Methods for details). (C) A midplane image for EYFP-Parkin or EYFP-Parkin R275W expressing cells and neighboring untransfected cells treated as in (A) were obtained, the area and perimeter of mitochondria within the cell were measured using ImageJ, and the compaction index was calculated. ≥8 cells per experiment were measured for each condition in at least two independent experiments. Scale bars in images = 10 µm.
To assess Parkin-mediated ubiquitination of proteins associated with mitochondria by immunostaining we used an antibody (FK1) that specifically recognizes polyubiquitinated proteins but not free ubiquitin or monoubiquitinated proteins.16 In HeLa cells expressing EYFP-Parkin treated with vehicle (DMSO) very little ubiquitinated protein can be detected localized with mitochondria (Fig. S1). Following depolarization with CCCP for 1 hr, however, a clear polyubiquitination signal is seen in cells expressing EYFP-Parkin but not in adjacent untransfected cells, which lack endogenous Parkin (Fig. 2A and B). We assessed the kinetics of mitochondrial ubiquitination relative to Parkin recruitment by live cell imaging. In single cells, mCherry-Parkin and EGFP-Ubiquitin consistently accumulated on mitochondria within the same 10 minute window following mitochondrial depolarization (Fig. S2A and B).
Figure 2.

Parkin K63-polyubiquitinates proteins on depolarized mitochondria by confocal imaging. (A) HeLa cells transfected with wild-type EC FP-Parkin (green) or the translocation-competent mutant ECFP-Parkin R275W (green) were treated with CCCP for 1 hr and immunostained for polyubiquitinated protein (red) and Tom20 (white), a mitochondrial marker. Both wild-type Parkin and Parkin R275W co-localize with Tom20. However, polyubiquitinated protein co-localized with depolarized mitochondria only in cells expressing wild-type Parkin. (B) Polyubiquitinated protein fluorescent intensity co-localizing with mitochondria was measured for cells expressing wild-type Parkin or Parkin R275W (WT [+] or R275W [+], respectively) and for untransfected control cells (WT [−] or R275W [−], respectively) from the same fields. ≥8 cells per experiment were measured for each condition in at least two independent experiments. (C) HeLa cells transfected and treated as in (A) were immunostained for K48 polyubiquitinated protein (red) and Tom20 (white). (D) Cells treated as in (C) were analyzed as in (B). (E) HeLa cells transfected and treated as in (A) were immunostained for K63 polyubiquitinated protein (red) and Tom20 (white). (F) Cells treated and immunostained as in (E) were analyzed as described in (B). Scale bars in images = 10 µm.
Although the Parkinson disease-linked mutant of Parkin, R275W, does not exhibit decreased ubiquitination activity in vitro,17 it is defective in mediating mitophagy of uncoupled mitochondria.7,9 Therefore, we tested whether the clumping and mitophagy deficiencies might result from the inability of the R275W mutant to polyubiquitinate mitochondrial substrate(s) in vivo. Consistent with this hypothesis, we found that in cells with similar levels of wild-type and R275W Parkin on depolarized mitochondria, the R275W mutant exhibits a relative deficiency in mitochondrial polyubiquitination (Fig. 2A and B). This association also supports the hypothesis that the mitochondrial clumping and mitophagy induced by Parkin are a consequence of polyubiquitination of mitochondrial substrates.
The effect of polyubiquitination on a protein depends, in part, on how individual ubiquitin monomers are linked in the polyubiquitin chain. While polyubiquitinated proteins with linkages other than K63 (the most common of which is K48) are usually targeted for degradation by the proteasome, K63-linked chains are thought to serve a signaling function and have been implicated in DNA repair, activation of NF-KappaB signaling, sorting of endocytosed receptors to lysosomes, as well as the aggregation and autophagic degradation of misfolded proteins.18,19 As previous reports suggest that Parkin is able to induce K48 and K63 polyubiquitination,20 we used recently developed antibodies, Apu2 and Apu3, which are specific for K48 and K63 linkages, respectively,21 to assess which linkages are predominant following recruitment of Parkin to depolarized mitochondria. We found little K48 polyubiquitination signal could be detected co-localizing with depolarizing mitochondria in HeLa cells expressing ECFP-Parkin (Fig. 2C and D), even though a known K48 polyubiquitinated substrate, SOD1 G93A,22 was readily detected with the same immunostaining procedure (Fig. S3). In contrast, a clear K63-linked polyubiquitin signal colocalized with depolarized mitochondria in the presence of ECFP-Parkin but not in untransfected HeLa cells or Parkin expressing HeLa cells not treated with CCCP (Fig. 2E and F, data not shown). Significantly less K63-linked polyubiquitination was seen co-localizing with depolarized mitochondria in HeLa cells expressing Parkin R275W, consistent with the hypothesis that the R275W mutant is deficient in the polyubiquitination of mitochondrial substrate(s) in vivo (Fig. 2E and F). The relative signals observed for K48- and K63-linked polyubiquitination are unlikely to reflect their relative affinities for native polyubiquitin chains, as the antibody against K48, Apu2, has greater affinity for K48 diubiquitin than the antibody against K63, Apu3, has for K63 diubiquitin in vitro.21 Parkin has been shown to ubiquitinate an N-terminal tag following treatment with CCCP.10,23 To assess if ubiquitination of Parkin's N-terminal EYFP tag can account for the K63 immunoreactivity following Parkin recruitment, we tested the ability to untagged Parkin, which is not robustly ubiquitinated under these conditions,10 to induce K63-linked mitochondrial polyubiquitination by immunocytochemistry. We found untagged Parkin induces K63-linked polyubiquitination to a similar extent as EYFP-Parkin, suggesting that a substrate other than Parkin is K63 polyubiquitinated following the recruitment of Parkin to depolarized mitochondria (Fig. S4A). Together these results suggest that Parkin induces K63-linked polyubiquitination of mitochondrial protein(s) following its recruitment to depolarized mitochondria and supports the hypothesis that at least some mitochondrial substrates polyubiquitinated by Parkin are regulated in a proteasome-independent manner. However, we cannot rule out that Parkin may also induce mono-ubiquitination or polyubiquitination of mitochondrial protein(s) with other linkages such as K27.
p62, which can directly bind polyubiquitin chains and LC3-II, a protein integral to autophagosomes, has been implicated as an adaptor for the aggregation and autophagy of misfolded ubiquitinated proteins.12,24,25 More recently, it has also been implicated in the delivery of ubiquitinated intracellular bacteria,14 experimentally ubiquitinated peroxisomes,13 and Parkin-labeled mitochondria to lysosomes by autophagy.7
Given its role as a ubiquitinbinding adaptor protein, we assessed whether p62 is recruited to mitochondria following Parkin translocation. While endogenous p62 forms aggregates in HeLa cells, consistent with previous reports, p62 did not co-localize with mitochondria in untransfected HeLa cells, which have little or no endogenous Parkin, under depolarizing or basal conditions (Fig. 3A and B). However, in HeLa cells expressing EYFP-Parkin and treated with CCCP for 1 hr (but not the vehicle DMSO), endogenous p62 is readily recruited to mitochondria (Fig. 3A, B and S4B), as recently reported.7 In addition, live-cell imaging experiments demonstrate that, similar to EGFP-ubiquitin, translocation of EGFP-p62 to mitochondria occurs within the same 10-minute window as mCherry-Parkin translocation following CCCP treatment (Fig. S5). In contrast, Parkin R275W, which is deficient in the promotion of in vivo polyubiquitination, did not induce p62 recruitment to depolarized mitochondria. This is consistent with the hypothesis that ubiquitination of mitochondrial substrate(s) is required for p62 recruitment (Fig. 3A and B).
Figure 3.

Ubiquitin-binding adaptor p62 is recruited to depolarized mitochondria in cells expressing Parkin. (A) HeLa cells transfected with wild-type EYFP-Parkin (green) or EYFP-Parkin R275W (green) were treated with DMSO or CCCP for 1 hr and immunostained for p62 (red) and Tom20 (white). (B) Pearson correlation coefficients for p62 intensity vs. Tom20 intensity were measured for cells expressing wild-type Parkin [WT (+)] or Parkin R275W [R275W (+)] and for untransfected control cells from the same fields [WT (−) and R275W (−), respectively]. ≥8 cells per experiment were measured for each condition in at least two independent experiments. (C) Cells transfected as in (A) were treated with CCCP for 1 hr and immunostained for p62. Scale bars in images = 10 µm.
Parkin contains an N-terminal ubiquitin-like (UBL) domain. Thus, it is possible that Parkin recruits p62 through a direct interaction between the UBL domain of Parkin and the ubiquitin-associated (UBA) domain of p62. To determine whether Parkin's UBL is necessary for p62 recruitment, we assessed p62 recruitment to mitochondria in HeLa cells expressing EYFP-Parkin ΔUBL. Although Parkin ΔUBL is not recruited to mitochondria as robustly as wild-type Parkin, p62 co-localized with mitochondrial Parkin ΔUBL, in cells in which Parkin ΔUBL translocation was observed (Fig. S6). This is consistent with the hypothesis that p62 is recruited by substrate(s) on mitochondria that becomes ubiquitinated following recruitment of Parkin rather than by Parkin directly. That Parkin R275W fails to induce p62 localization to mitochondria, despite having an intact UBL domain, also suggests that p62 is recruited following ubiquitination of mitochondrial substrates and not by Parkin's UBL domain.
While endogenous p62 was recruited to some depolarized mitochondria in all HeLa cells expressing Parkin, p62 appeared to be most abundant on clumps of mitochondria coated with Parkin (Fig. 3C). To further investigate the localization and effect of p62 on mitochondria, we observed EGFP-p62 in single cells by live-cell imaging. Consistent with previous reports, we found the threadlike mitochondria begin to fragment 10–15 minutes following depolarization with CCCP, likely due to cleavage and inactivation of the fusion protein OPA1 with continued activity of the fission protein Drp1.26 In the periphery of the cell, around 25–30 minutes following the addition of CCCP, EGFP-p62 appears to coat the fragmented mitochondria (Fig. 4A, top right parts). When the individual, p62-coated mitochondria come into proximity they appear to stick together, forming aggregates of round, fragmented mitochondria, resembling bunches of grapes (Fig. 4A, top right parts). Likewise, near the nucleus p62 appears to connect mitochondria as they are compressed together into a perinuclear aggregate (Fig. 4A and bottom). Occasionally, groups of mitochondria within these aggregates appear stretched apart as one group is pulled toward the nucleus—presumably by microtubule motors, as such events were not observed in the presence of nocodazole (data not shown). Thin filaments of p62 appear stretched between the aggregates under tension, suggesting a force resisting the separation (Suppl. Movie 1). This is consistent with the notion that in order to separate fragmented mitochondria that appear tethered together by polymerized p62, binding between polymerized p62 molecules (and possibly other molecules binding p62, such as NBR1) must be overcome.12,27
Figure 4.

p62 is necessary for mitochondrial aggregation but not mitophagy. (A) HeLa cell co-transfected with GFP-p62 (green) and untagged Parkin in a 1:4 ratio was labeled with Mitrotracker Red (red) and imaged live following addition of 10 µM CCCP at time point 0. Left image depicts the whole cell at time point 0. Changes in morphology of two clusters of mitochondria are shown in the left (L) and right (R) series of images. (A, top right) the left series depicts the fragmentation of thread-like mitochondria from 0 to 70 minutes following CCCP. p62 is recruited to groups of fragmented mitochondria between 20 and 40 minutes. When groups of fragmented mitochondria that co-localize with p62 (yellow) come into contact they begin to move as a single object (a and b at 50 minutes and ab and c at 70 minutes). (A, bottom right) the right series shows p62 labeled structures forming between perinuclear mitochondria as they are compressed together over time. The white line in first image depicts the border of the nucleus. (B) p62+/+ or p62−/− cells transfected with EYFP-Parkin (green) were treated with 20 µM of CCCP for 24 hrs. While mitochondria formed aggregates in p62+/+ cells, mitochondrial failed to form aggregates in p62−/− cells. (C) Cells treated as in (B) were scored for mitochondrial morphology for ≥50 cells/condition in ≥3 independent experiments. Morphology was considered “normal” if it was similar to untransfected cells in the same field and “few dispersed,” if mitochondria appeared reduced in number but no aggregates were apparent. *indicates p-value <0.05. **indicates p-value <0.01. (D) Midplane images of cells treated as in (B) were analyzed using the compaction index as described in (Fig. 1B and C) for ≥8 cells/condition from ≥2 independent experiments. Scale bars in images = 10 µm.
To further test the hypothesis that p62 may be necessary for the observed aggregation and autophagy of mitochondria following recruitment of Parkin to mitochondria, we assessed changes in mitochondrial aggregation and number in p62+/+ and p62−/− MEFs by confocal microscopy. In p62+/+ MEFs expressing Parkin treated with CCCP for 24 hrs, mitochondria often appeared clumped in aggregates (Fig. 4B and C), similar to depolarized mitochondria in HeLa cells expressing Parkin (Fig. 1A). In contrast, mitochondrial aggregates were rarely observed in p62−/− MEFs (Fig. 4B and C). To quantitatively assess of the relative aggregation of mitochondria in p62+/+ and p62−/− MEFs, we measured the compaction index of mitochondria within each cell type. Consistent with our other observations, mitochondria within p62+/+ MEFs had a higher compaction index than mitochondria within p62−/− MEFs (Fig. 4D). Together these findings demonstrate that endogenous p62 can mediate the aggregation of depolarized mitochondria expressing the E3 ubiquitin ligase, Parkin.
Given the role of p62 in mediating the selective autophagy of misfolded proteins,12 peroxisomes,13 and intracellular bacteria,14 we also assessed whether p62 is necessary for Parkin-induced mitophagy. Surprisingly, p62 appears to be dispensable for Parkin-induced mitophagy, with approximately equal numbers of p62+/+ MEFs and p62−/− MEFs lacking mitochondria following treatment with CCCP for 24 hrs (Fig. 4C). To assess whether p62 may be required for Parkin-induced mitophagy in other cell types or following acute depletion, we transfected HeLa cells with siRNA directed against p62. A substantial reduction in p62 protein levels was observed following transfection of HeLa cells with p62 siRNA but not control siRNA (Fig. 5A). Consistent with p62 being dispensable for Parkin-induced mitophagy, HeLa cells acutely depleted of p62 and transiently expressing EYFP-Parkin eliminated depolarized mitochondria to a similar extent as HeLa cells transfected with control siRNA (Fig. 5B and C). In particular, individual p62 siRNA transfected cells that lacked both the abundant p62 puncta apparent in control cells (indicating successful p62 knockdown at the level of individual cells) and Tom20 immunostaining (indicating robust mitophagy) were observed, demonstrating at the single cell level that acute loss of p62 in HeLa cells does not prevent Parkin-induced mitophagy. Taken together these findings demonstrate that p62 is not necessary for Parkin-induced mitophagy.
Figure 5.

Acute depletion of p62 with RNAi does not inhibit Parkin-induced mitophagy. (A) Lysates from HeLa cells transfected with p62 siRNA or control siRNA for three consecutive days were separated on SDS-PAGE gels and immunoblotted for p62 and VDAC1, which served as a loading control. (B) Cells from (A) plated onto slides and transiently transfected with EYFP-Parkin (green) were treated with CCCP for 24 hrs and immunostained for p62 (red) and the mitochondrial protein Tom20 (white). Scale bar in last image = 10 µm. (C) cells in (B) were scored for the presence of mitochondria.
p62 is believed to mediate the aggregation of misfolded proteins by binding the ubiquitinated protein with its UBA domain and then binding to other p62 molecules through its N-terminal PB1 domain (Fig. 6A).12,24,25 To assess whether a similar mechanism is responsible for Parkin-induced aggregation of depolarized mitochondria, we tested the ability of wild-type and mutants of p62 to rescue the mitochondrial aggregation defect in p62−/− MEFs. We found that whereas wild-type p62 and p62 lacking the LIR domain, responsible for the binding of p62 to LC3, were able to rescue mitochondrial aggregation in p62−/− MEFs, p62 with a mutation in the PB1 domain, D69A, failed to restore mitochondrial aggregation (Fig. 6A and B). These findings strongly suggest that p62 mediates mitochondrial aggregation by polymerizing through its PB1 domain.
Figure 6.

Mitochondrial aggregation requires the PB1 domain, but not the LIR domain, of p62. (A) Schematic depicting the domain structure of p62. The PB1 domain (which contains residue D69) is responsible for p62 polymerization, while the LIR domain binds LC3 on autophagosomes and the UBA domain binds ubiquitin. (B) p62−/− cells co-transfected with unlabeled Parkin and EGFP, EGFP-p62 wild-type, EGFP-p62 D69A or EGFP-p62 ΔLIR were treated with 20 µM of CCCP for 24 hrs. (C) Cells treated as in (B) were scored for the presence of mitochondrial aggregates for ≥50 cells/condition in ≥3 independent experiments. Scale bars in images = 10 µm.
Recent findings suggest that experimental fusion of the ubiquitin mutant G76V to peroxisomal membrane proteins is minimally sufficient to induce their selective autophagy, provided ubiquitin faces the cytosol.13 As Parkin induces both the polyubiquitination of substrate(s) on mitochondria and mitochondrial degradation, we created an inducible heterodimerization system to recruit ubiquitin directly to the mitochondrial outer membrane.9,28 This system allows us to assess whether mitochondrial anchored ubiquitin is minimally sufficient for mitophagy, p62 recruitment and/or mitochondrial clumping. The C-terminal mitochondrial anchoring tails of Bcl-XL (213-233), Bax (EYFP20),29 and Fis1 (92–152) were fused to the FRB domain, while UbG76V-GFP or GFP were fused to tandem FKBP domains. While treatment with the rapamycin analog AP21967 (rapalog) caused at least partial co-localization of UbG76V-EGFP-FKBP with mitochondria following co-transfection with each of the FKBP fused outer mitochondrial membrane anchors, increased expression of ubiquitin on mitochondria did not cause a detectable reduction in mitochondrial mass by confocal microscopy after 24 hrs (Fig. 7A). We investigated the UbG76V-EGFP-FKBP paired with the FRB-Fis1 construct further, as this combination resulted in the most stably expressed ubiquitin on the outer mitochondrial membrane. We found no difference in mitochondria mass in HeLa cells expressing UbG76V-EGFP-FKBP and FRB-Fis1 at 24 hrs or 96 hrs of treatment with the rapalog relative to that of HeLa cells expressing EGFP-FKBP and FRB-Fis1 (Fig. 7A and data not shown). Mitochondrial fission, which occurs following mitochondrial depolarization, is thought to precede and possibly promote mitophagy, during basal mitochondrial turnover.30 To test whether mitochondrial fission or another consequence of depolarization is required in conjunction with stable expression of ubiquitin on the outer mitochondrial membrane for mitophagy, we depolarized HeLa cells (which lack endogenous Parkin) expressing UbG76V-EGFP-FKBP/FRB-Fis1 with CCCP for 24 or 96 hrs in conjunction with rapalog treatment (Fig. 7B and C and data not shown). No mitophagy was apparent in the UbG76V-EGFP-FKBP/FRB-Fis1 expressing cells comparable to untransfected cells or cells expressing EGFP-FKBP/FRB-Fis1 (data not shown).
Figure 7.

Anchoring ubiquitin to outer mitochondrial membrane is sufficient for p62 recruitment and mitochondrial clustering but not mitophagy. (A) HeLa cells, which lack Parkin protein expression, were co-transfected with ubiquitin G76V fused to tandem FKBP12 domains and mitochondrial anchor constructs fused to the FRB domain. The cells were subsequently treated with the rapalog AP21967 for 24 hrs. Rapalog induced the translocation of ubiquitin to mitochondria secondary to heterodimerization of the FKBP and FRB domains in the presence of the rapalog. Cells were immunostained with Tom20 and the cells were scored for the absence of mitochondria. ≥50 cells per condition were counted. (B) HeLa cells co-transfected with EGFP-Ubiquitin G76V-FKBP or FKBP-EGFP and FRB-Fis1 (92–152) were treated with rapalog and CCCP for 24 hrs and immunostained for p62 (red) and the mitochondrial protein Tom20 (white). Scale bar in last image = 10 µm. (C) Cells treated as in (B) were scored for the presence of p62 positive and p62 negative mitochondrial clumps. ≥50 cells per condition were counted.
Interestingly, UbG76V-EGFP-FKBP but not EGFP-FKBP was sufficient to recruit p62 to clumped mitochondria in a substantial proportion of cells co-expressing FRB-Fis1 and treated with rapalog in the presence of CCCP (Fig. 7B and C). These findings suggest that anchoring mono-ubiquitin on the outer mitochondrial membrane is sufficient for p62 recruitment to mitochondria and further suggest that p62 recruited to ubiquitinated mitochondria leads to mitochondrial clumping but not mitophagy. These data are also consistent with the hypothesis that ubiquitination of particular mitochondrial substrate(s) and/or modification of the substrate(s) with a particular polyubiquitin chain (e.g., K63-linked) is required for Parkin-induced mitophagy (but not for p62 recruitment and mitochondrial clumping).
VDAC1 was recently reported to be ubiquitinated by Parkin following Parkin translocation to depolarized mitochondria, suggesting that VDAC1 ubiquitination may mediate mitochondrial phenotypes following Parkin recruitment.7 To verify that VDAC1 is ubiquitinated under depolarizing conditions, we treated HeLa cells stably expressing EYFP-Parkin (HeLaEYFP-Parkin) with CCCP for 0, 2 or 5 hrs. Using anti-porin 31HL (VDAC1) mAB6, higher molecular weight immunoreactive bands (near ∼39 and 48 kDa) were observed following CCCP treatment in addition to a band near VDAC1's predicted molecular weight, 31 kDa (Fig. 8A). These findings are consistent with the appearance of mono- and di-ubiquitinated forms of VDAC1, respectively, following CCCP treatment in the presence of Parkin and confirm previous data obtained with this antibody. The immunoreactivity of the 39 and 48 kDa bands compared to the 31 kDa band suggests that a relatively small proportion of VDAC1 is modified.
Figure 8.

VDAC1 but not VDAC2 is ubiquitinated following mitochondrial uncoupling in the presence of Parkin. (A) Lysates from HeLa cells stably expressing EYFP-Parkin and treated with CCCP for 0, 2 or 5 hrs were separated on 12–22% SDS-PAGE gels and immunoblotted with an antibody against VDAC1 (anti-porin 31HL mAB6) or Tubulin. The predicted positions of unmodified VDAC1 and mono- and di-ubiquitinated forms of VDAC1 are indicated. (B) VDAC1 immunoaffinity purified with mAB6 from HeLa cell lysates following treatment with CCCP for 5 hrs was separated on SDS-PAGE gels and stained with Coomassie blue dye. Approximate positions of five bands cut for protein identification are indicated. (C) Table summarizing peptides identified from bands 1–5. (D) Table depicting relative abundance of three unique VDAC2 peptides and three homologous VDAC1 peptides in bands 1–5. Integrated MS ion intensities for each peptide were normalized to that observed in control band 3. Values greater than 2, indicating a 2-fold increase over control band 3, are highlighted in yellow. (E) Table depicting relative abundance of two unique ubiquitin peptides, TITLE and TLS. Integrated MS ion intensities for each peptide were normalized as in (D). Values greater than 2, indicating a 2-fold increase over control band 3, are highlighted in yellow.
Unexpectedly, while the 39 and 48 kDa bands were observed with mAb6, they were not observed with another commonly used VDAC1 antibody, mAB1, which was generated by the same antigen exposure as Ab6,31 and which exhibits fewer cross-reactive bands compared to mAb6 (Fig. S7). To assess whether the higher bands seen with mAb6 represent ubiquitinated VDAC1 or a related cross-reactive protein, such as VDAC2 or VDAC3, endogenous VDAC1 was immunoaffinity purified from HeLaEYFP-Parkin cells treated with CCCP for 5 hrs with the mAb6 antibody. Four strong Coomassie blue labeled bands were observed on a SDS-PAGE gel, roughly corresponding to the molecular weight of unmodified VDAC1 (bands labeled 1 and 2) and mono- and diubiquitinated VDAC1 (bands labeled 4 and 5, respectively) (Fig. 8B). These bands were excised along with a control gel piece (band 3) for identification by LC-MS/MS. Tryptic peptides from VDAC1 were observed in all bands, but a greater number of peptides were identified by MS/MS from the strong Coomassie stained bands 1, 2, 4 and 5, consistent with these representing distinct VDAC1 species (Fig. 8C and Suppl. Table 1). The VDAC1 peptides observed from the control band likely represents a background of protein spread from the neighboring concentrated bands. Evidence for a background protein spread of the strong light chain immunoglobulin band around 25 kDa into bands 1–5 was also observed by MS/MS analysis (data not shown).
Three tryptic peptides unique to the VDAC2 isoform were identified from band 2 but not from the other bands (Fig. 8B and Suppl. Table 1). This demonstrates that although mAb6 is fairly specific for VDAC1, as reported previously,32 it also binds VDAC2. To assess whether VDAC2 is present in higher molecular weight forms, which may be ubiquitinated, we estimated the relative abundance of the three unique VDAC2 peptides and three homologous VDAC1 peptides in the five bands by examining the MS ion chromatograms for each peptide in each sample (Fig. 8D). While the three VDAC1 peptides were substantially more abundant in high molecular weight bands 4 and 5 than in control band 3, the three VDAC2 peptides were no more abundant in bands 4 and 5 than in control band 3. These findings suggest that VDAC2 is present primarily in band 2 and is not ubiquitinated following uncoupling in cells expressing Parkin. To provide independent evidence that VDAC2 is not ubiquitinated, we immunoblotted HeLa lysates treated with CCCP for 0, 2 or 5 hrs with a polyclonal antibody raised against a VDAC2 peptide. The membrane was then stripped of antibody and reprobed for VDAC1 using the anti-VDAC1 mAb6 antibody. While high molecular weight bands immunoreactive to anti-VDAC1 mAb6 appeared with 2 and 5 hrs CCCP treatment (but not 0 hrs of treatment), as was observed above, no corresponding bands reacted with the anti-VDAC2 antibody, indicating no change in VDAC2 mobility following CCCP treatment. Together these findings suggest that VDAC2 is not ubiquitinated following depolarization in cells overexpressing Parkin.
Although we did not identify site(s) of VDAC1 ubiquitination, two unique ubiquitin peptides, TITLEVEPSDTIENVK and TLSDYNKQK (hereafter, TITLE and TLS), were identified by MS/MS from bands 4 and 5 in this initial experiment (Fig. S8B and Suppl. Table 1). As identification of TLS, which fragmented poorly, did not have a p-value of lower than 0.05 in the first experiment in a MASCOT search, the peptide samples were concentrated and rerun to provide additional evidence of this peptide's identity. In the second technical replicate, the identification for the TLS peptide reached significance for band 4 and a third ubiquitin peptide, ESTLHLVLR (hereafter EST), was identified with significance from bands 4 and 5 (Suppl. Table 1). Prominent precursor to fragment ion transitions observed upon collision induced disassociation for these three peptides (894.47 → 1002.5, 541.28 → 867.4 and 534.31 → 637.4 for TITLE, TLS and EST, respectively) have been reported previously,33 further supporting the correct identification of these three ubiquitin peptides in digests of bands 4 and 5 (Fig. S8A–E). No ubiquitin peptides were found from bands 1 or 2, while one, TLS, was found in control band 3 (most likely reflecting background of protein spread from adjacent band 4). The relative abundance of the ubiquitin peptides in each band was calculated from the MS ion chromatograms from the first technical replicate. Bands 4 and 5 exhibited substantially more abundant ubiquitin peptides than background (control band 3), while the abundance of ubiquitin peptides was similar in bands 1 and 2 and band 3 (Fig. 8E). Consistent with the mass shift observed on SDS-PAGE, these results suggest that the higher molecular weight bands (4 and 5) represent ubiquitinated forms of VDAC1, while the lower bands represent VDAC1 that is not ubiquitinated.
In our mass spectrometry analysis of endogenous VDAC1 no ubiquitin peptides were identified with a diglycine modification that would have suggested the topology of the polyubiquitin chain attached to VDAC1, if VDAC1 is polyubiquitinated. To assess whether ubiquitination of VDAC1 (or VDAC3) could account for the K63 polyubiquitin immunoreactivity associated with depolarized mitochondria following Parkin recruitment, we examined K63 polyubiquitin immunoreactivity in VDAC1/3−/− MEFs (Fig. 9A). Following depolarization of mitochondria with CCCP for 3 hrs, we observed robust K63-linked polyubiquitination of mitochondria in cells expressing EYFP-Parkin but not in untransfected cells, demonstrating that a Parkin substrate other than VDAC1 is K63 polyubiquitinated in a Parkin-dependent manner. We next assessed whether VDAC1 or VDAC3 is necessary for the phenotypes observed following Parkin-dependent ubiquitination of mitochondria. Following treatment with CCCP for 24 hrs, a similar proportion of VDAC1/3+/+ and VDAC1/3−/− MEFs expressing EYFP-Parkin lacked mitochondria, demonstrating that VDAC1 and VDAC3 are dispensable for Parkin-induced mitophagy (Fig. 9B and C). In VDAC1/3−/− MEFs that retained mitochondria, the mitochondria were often in perinuclear clusters and exhibited p62 immunoreactivity, demonstrating that VDAC1 and VDAC3 are also dispensable for Parkin-induced p62 recruitment and mitochondrial clustering (Fig. 9D).
Figure 9.

Mitochondria-associated proteins other than VDAC1 and VDAC3 are K63-polyubiquitinated in a Parkin-dependent manner; and VDAC1 and VDAC3 are dispensable for p62 recruitment to mitochondria, mitochondrial sequestration and Parkin-induced mitophagy. (A) VDAC1/3−/− mouse embryonic fibroblasts (MEFs) transiently expressing EYFP-Parkin (white) and treated with 20 µM CCCP for 3 hrs were immunostained for K63 polyubiquitinated proteins (green) and the mitochondrial protein, cytochrome c (red). (B) VDAC1/3+/+ and VDAC1/3−/− MEFs transiently expressing EYFP-Parkin (green) were treated with CCCP for 24 hrs and immunostained for the mitochondrial protein Tom20 (red). (C) Cells in (B) scored for mitochondrial phenotype. (D) Cells treated as in (B) were immunostained for p62 (green) and Tom20 (red). Scale bar in last image = 10 µm.
As was reported previously, with continued depolarization clumped mitochondria in cells expressing Parkin are eventually gathered in a perinuclear location, likely due to retrograde movement along microtubules.8,11 Because lysosomes and autophagosomes are often concentrated in the perinuclear region and retrograde transport has been shown to promote the degradation of aggregated protein,34–36 it has been suggested that Parkin may promote mitochondrial mitophagy primarily by inducing its retrograde transport to the perinuclear location.8 To test this hypothesis directly, we pretreated HeLa cells expressing EYFP-Parkin with nocodazole, which inhibits microtubule polymerization by binding monomeric beta-tubulin, for three hours prior to depolarization with CCCP. Immunostaining for alpha-tubulin confirmed the absence of microtubules following treatment with nocodazole and the mitochondrial distribution was altered in a manner characteristic of nocodazole treatment (Fig. 10A and data not shown). While treatment with nocodazole blocked the perinuclear concentration of mitochondrial aggregates as was reported previously (data not shown),8 it failed to block Parkin-induced mitophagy (Fig. 10B and C). These findings indicate that Parkin promotes the aggregation and degradation of depolarized mitochondria independently of effects it might have on microtubule-dependent transport.
Figure 10.
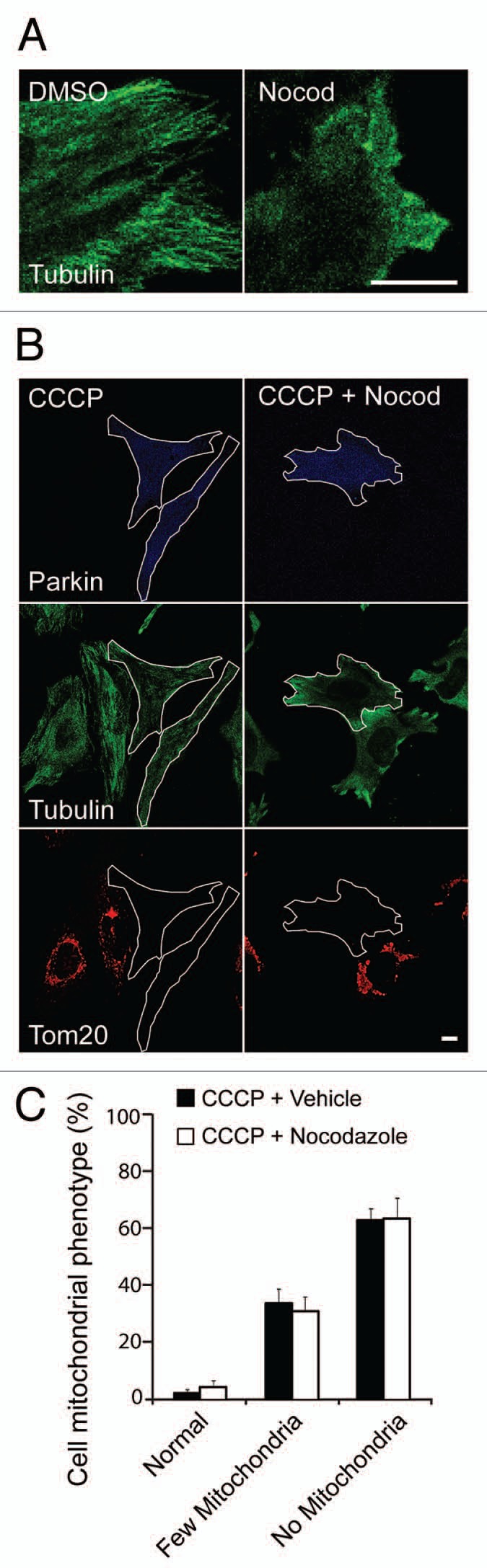
Microtubule network is dispensible for Parkin-induced mitophagy. (A) HeLa cells transfected with EYFP-Parkin were treated with DMSO or 10 µM nocodazole for 3 hrs and then immunostained for α-tubulin (green). Nocodazole treatment led to complete disruption of the microtubule network. (B) HeLa cells transfected with EYFP-Parkin (blue) were pretreated with DMSO or 10 µM nocodazole for 3 hrs and then 10 µM CCCP was added for an additional 24 hrs. Cells were immunostained for Tom20 (red) and α-tubulin (green). (C) Cells treated as in (B) were scored for presence of mitochondria for ≥50 cells/condition in ≥3 independent experiments. Scale bars in images = 10 µm.
Discussion
Mitochondrial damage occurs with aging and when excessive is believed to contribute to common diseases such as Parkinson disease.1 Cells within complex organisms have evolved quality control mechanisms for tempering the accumulation of dysfunctional mitochondria.4 In metazoans, the PINK1/Parkin pathway, loss of which leads to mitochondrial dysfunction in Drosophila and mice and parkinsonism in humans, appears to mediate specific mitochondria quality control pathways.6 PINK1 accumulates selectively on mitochondria with low membrane potential by inhibition of constitutive voltage-dependent proteolysis to recruit Parkin from the cytosol to dysfunctional mitochondria.9,10 Following recruitment, Parkin induces the mitophagy of the dysfunctional mitochondria.11 We have found that following recruitment, Parkin promotes polyubiquitination of mitochondrial substrate(s), which are formed of K63-linked chains with minimal K48 linkages and recruits the ubiquitin- and LC3-binding adaptor protein p62. p62 appears responsible for the aggregation of dysfunctional mitochondria into tight clusters, thereby minimizing the surface area exposed to other cellular components. Mitochondrial clustering by p62 depends on its PB1 domain, which is believed to mediate its polymerization.12 Surprisingly, we find that p62 is not limiting for Parkin-induced mitophagy in fibroblasts or HeLa cells, suggesting that other, possibly parallel, mechanisms must be mediating Parkin-induced mitophagy. Recruitment of p62 to mitochondria labeled with ubiquitin also appears to be insufficient for robust mitophagy, at least in HeLa cells. Additionally, while we confirm that VDAC1 and not VDAC2 is ubiquitinated following depolarization in the presence of Parkin, we find that VDAC1 and VDAC3 do not account for the K63-linked polyubiquitin immunoreactivity of mitochondria following Parkin recruitment and are dispensable for p62 recruitment, mitochondrial clustering and mitophagy.
The sequestration of ubiquitinated mitochondria by p62 appears to be analogous to its clustering of ubiquitinated proteins. By binding ubiquitin with its UBA domain and polymerization through its PB1 domain, p62 mediates the aggregation of misfolded proteins produced by a number of experimental challenges, including puromycin treatment, overexpression of insoluble mutant proteins and inhibition of autophagy, as has been demonstrated in cell culture as well as in Drosophila and mice.12,37,38 In addition, p62 is found with ubiquitin in protein aggregates associated with disease, such as Mallory bodies in alcoholic cirrhosis and, notably, Lewy bodies in Parkinson disease.27,39,40 By minimizing interaction between exposed hydrophobic regions of misfolded proteins and other cellular components, aggregation of misfolded protein may be protective in the context of neurodegenerative disorders—e.g., mutant Huntingtin and alpha-synuclein may be less harmful in larger aggregates compared with small oligomers.41,42 However, in other contexts, such as in autophagy-deficient hepaptocytes, p62-mediated aggregation of misfolded proteins may be detrimental.37
The pathophysiological significance of mitochondrial aggregation is less clear, but interestingly in MitoPark mice, a model of progressive parkinsonism, damaged mitochondria are sequestered in intracellular inclusions that bear some resemblance to Lewy bodies (although they lack alpha-synuclein, a key component of Lewy bodies in sporadic Parkinson disease).5 Parkinsonism in these mice results from conditional knockout of the mitochondrial DNA transcription factor Tfam from tyrosine hydroxylase expressing dopaminergic neurons, experimentally mimicking the electron transport chain dysfunction observed in sporadic Parkinson disease. Reducing the surface of area of impaired mitochondria within the cell by mitochondrial clustering may minimize their uptake of substrates and limit the spread of mitochondrial ROS to other cellular compartments. Additionally, clustering dysfunctional mitochondria would likely prevent their transport to parts of the cell with high-energy requirements, such as neuronal synapses, at the expense of more bioenergetically active mitochondria. Alternatively, as p62 clustering of ubiquitinated substrates has been shown to cause cell death in some cell types in the absence of its degradation by autophagy,37 it is possible that p62 functions as a sensor of defective quality control mitophagy. In this model, p62 would trigger cell death in the face of a deficient removal of mitochondria that have been ubiquitinated by Parkin, in a manner analogous to the p62-dependent cell death triggered by Mallory body-like inclusions in the hepatocytes of Atg7-deficient mice. It will be interesting to learn whether crossing MitoPark mice with p62 null mice and Parkin null mice prevents mitochondrial aggregation and exacerbates or alleviates the MitoPark phenotype.
p62 directly binds the autophagy proteins, LC3 and GABARAP, and is believed to serve as an autophagy receptor for ubiquitinated protein aggregates as well as peroxisomes and intracellular bacteria.12–14 Thus, we were surprised to find mitophagy occurs equally well in cells null for p62 and wild-type cells, suggesting that p62 is not rate limiting for Parkin-induced mitophagy. We also assessed the dependence of Parkin-induced mitophagy on p62 in HeLa cells acutely depleted of p62 by siRNA. Similar to findings in p62 null MEFs, p62 depletion in HeLa cells did not inhibit p62-induced mitophagy. Notably, we observed p62 siRNA transfected HeLa cells that were absent immunostaining for both p62 and Tom20, demonstrating at the single cell level that mitophagy can still proceed in the face of acute p62 depletion. These findings suggest that the p62-independence of Parkin-induced mitophagy is likely not the result of a compensatory mechanism and may be characteristic of many mammalian cell types. These data are in contrast to findings in a recent report published while this manuscript was in preparation suggesting that knockdown of p62 in HeLa cells partially inhibits Parkin-induced mitophagy.7 It is not clear how to account for the discrepancy in these findings, as we appear to have achieved a similar level of p62 protein depletion, using the same p62 siRNAs, in the same cell line, and following a very similar protocol. Nonetheless, Parkin clearly induces mitophagy in single cells with no p62 protein expression, demonstrating that p62 is inessential for Parkin-induced mitophagy at least in some cell types. While this manuscript was in revision, Okatsu et al. reported that whereas p62 is required for mitochondrial clustering it is not essential in MEFs for mitophagy largely consistent with aspects of our study.43
Consistent with the observation that p62 is not required for Parkin-induced mitophagy, knockout of Atg7 but apparently not p62 in hepatocytes leads to the accumulation of dysmorphic mitochondria, suggesting that p62 may not be required for quality control mitophagy in the liver.37,44 In addition, while the phenotype of Drosophila lacking the p62 ortholog is similar to that of Parkin null Drosophila in some respects (both have male sterility), it lacks key features such as muscle degeneration.38,45 This suggests that p62 is unlikely to be the sole downstream mediator of Parkin in Drosophila.
Although at present it is unclear how Parkin induces mitophagy, its ubiquitination activity and ubiqutination of mitochondrial targets appear essential. One possible explanation is the existence of redundant autophagy receptors (e.g., p62 and NBR1) for ubiquitinated mitochondria. Loss of one autophagy receptor, for instance, would not be sufficient to inhibit mitophagy, if a downstream step in the pathway were rate limiting, a possibility that may be addressed by the development of NBR1/p62 double knockout mice. Alternatively, Parkin may induce mitophagy through a different mechanism, such as modification of another autophagy receptor (e.g., Nix).46
Parkin also appears to concentrate depolarized mitochondria within a perinuclear region, in a manner that is likely dependent on retrograde transport of mitochondria along microtubules. As retrograde transport of aggregated misfolded protein appears to enhance delivery to autophagosomes and lysosomes concentrated in this region, particularly in neurons,34–36 it was recently proposed that Parkin's induction of mitophagy might be secondary to its effects on mitochondrial transport.8 This appears not to be the case. In HeLa cells, Parkin is able to eliminate depolarized mitochondria equally well in the presence and absence of a microtubule network. Additionally, Parkin appears to promote the encapsulation of mitochondria by autophagosomes in the periphery of the cell one hour following depolarization, before mitochondria have been concentrated in the perinuclear region.11 It is, however, possible that retrograde mitochondrial transport induced by Parkin may enhance mitochondrial degradation in some cell types, such as neurons. Additionally, the movement of dysfunctional mitochondria, aggregated by p62, into a perinuclear aggresome may help isolate them from other cellular components.
Interestingly, Parkin was recently found to promote retrograde transport of mitochondria in Drosophila follicular tissue, in which loss of Parkin-mediated retrograde movement leads to aggregation of mitochondria. This suggests that in some systems Parkin likely modulates mitochondrial transport independently of its effects on mitophagy and p62-dependent mitochondrial clustering.47
Using antibodies against endogenous K48- and K63-linked polyubiquitin chains, we found that Parkin induces abundant mitochondrial polyubiquitination with a preponderance of K63 linkages relative to K48 linkages. Recently, it was shown that ubiquitin anchored to peroxisomes is sufficient to induce p62 recruitment and pexaphagy.13 Similar to these findings, we observed that anchoring ubiquitin to mitochondria is sufficient to recruit p62 to mitochondria. However, the consequence of p62 recruitment to mitochondria appears to be different than that reported for peroxisomes. While mitochondria tagged with a ubiquitin fusion construct clustered similarly to those ubiquitinated by Parkin, ubiquitin anchored on mitochondria did not appear to induce mitophagy, even when the mitochondria were fragmented by depolarization. These findings suggest that a specific substrate or a polyubiquitin chain with a specific linkage may be required for the induction of mitophagy but not for p62 recruitment and mitochondrial clustering. These findings also support the hypothesis that p62 mediates mitochondrial clustering but not mitophagy. We cannot rule out, however, that mitophagy below our level of detection may have been induced by anchoring ubiquitin to the outer mitochondrial membrane.
Largely in agreement with our finding a predominance of K63-linked polyubiquitin chains following Parkin recruitment to depolarized mitochondria, a recent report, using over-expressed ubiquitin mutants, suggested that K27 and K63 are the predominate linkages in polyubiquitin on mitochondria induced by Parkin.7 The report identified VDAC1 as a likely K27 polyubiquitinated substrate of overexpressed Parkin and tagged Parkin as the primary K63 polyubiquitinated substrate. We confirm that VDAC1 but not VDAC2 is ubiquitinated following depolarization in the presence of Parkin and so is likely a substrate of overexpressed Parkin. Interestingly, a commonly used antibody against VDAC1's acetylated N-terminus failed to detect ubiquitinated VDAC1. The reason for the failure of this antibody to recognize ubiquitinated VDAC1 is unclear but may reflect direct or indirect epitope masking by the attachment of ubiquitin.
We also find that ubiquitination of VDAC1 cannot account for the K63-linked immunoreactivity observed following recruitment of Parkin to depolarized mitochondria, as this immunoreactivity was also observed in MEFs lacking VDAC1 and VDAC3. Similarly, ubiquitination of tagged Parkin is unlikely to account for the K63 polyubiquitin immunoreactivity, as untagged Parkin, which is not detectably ubiquitinated under depolarizing conditions, could also induce the K63-linked polyubiquitination of mitochondria. Thus, it appears that Parkin induces the K63-linked polyubiquitination of substrates other than VDAC1. Additionally, we observed that VDAC1 and VDAC3 are not required for Parkin-induced recruitment of p62, mitochondrial clustering or mitophagy, as these events occur in MEFs lacking VDAC1 and VDAC3. These findings are consistent with the hypothesis that ubiquitination of substrates other than VDAC1 by Parkin are required for p62 recruitment, mitochondrial clustering and mitophagy. Whether VDAC1 is ubiquitinated as a bystander, is functionally redundant with other Parkin substrates, or leads to another functional consequence is unclear. The relatively small pool of VDAC1 that appears to be ubiquitinated despite overexpression of Parkin and relatively long treatment with CCCP seems to support the view that VDAC1 may be ubiquitinated as a bystander or may play a minor and redundant role in the robust mitophagy induced by Parkin under these conditions. In any case, that Parkin appears to ubiquitinate VDAC1 but not VDAC2, which shares 74% sequence identity and 91% sequence similarity with VDAC1, suggests that Parkin does not ubiquitinate mitochondrial proteins indiscriminately and that there are likely contextual cues governing Parkin's choice of substrate. The identification of Parkin ubiquitination sites on VDAC1 and other Parkin substrates may better define what these contextual cues are.
In summary, our findings suggest that p62 is required for the sequestration but not the elimination of depolarized mitochondria following the recruitment of Parkin, in a manner analogous to its aggregation of ubiquitinated proteins. These findings suggest the possibility that sequestration of mitochondria by p62 may play a role in the pathogenesis of Parkinson disease. Additionally, they point to the involvement of autophagy receptors other than p62 and Parkin substrates other than VDAC1 in the selective mitophagy induced by activated Parkin.
Materials and Methods
Cell culture.
p62/SQSTM+/+, p62/SQSTM−/−, VDAC1/3+/+ and VDAC1/3−/− transformed MEFs and HeLa cells stably expressing EYFP-Parkin have been described previously.37,48 ECFP-Parkin, EYFP-Parkin, EYFP-Parkin R275W, EYFP-Parkin ΔUBL, mCherry-Parkin, EGFP-Ub, GFP-p62, SOD1 G93A are in C1 or N1 Clontech vectors and have been described previously.9,11,12,49 EGFP-p62 D69A and EGFP-p62 ΔLIR are in the pDest vectors (Invitrogen) and have been described previously.12 mCherry-p62 was generated by subcloning full length p62 cDNA clone (Open Biosystems) into the mCherry-C1 vector (Clontech). Ubiquitin-G76V-EGFP-FKBP and EGFP-FKBP were created by cloning a PCR fragment containing Ubiquitin-G76V-EGFP (from Addgene deposited by Nico Dantuma) or EGFP into the EcoRI and XbaI sites of the pC4M-F2E vector (ARIAD Pharmaceuticals).50 FRB-Fis1(92–152), FRB-Bcl-XL(213–233) and FRB-Bax(EYFP20) were created by cloning a PCR fragment containing the respective tail anchors, which have been described previously,29 into the BamHI site of the pC4-RHE vector. The rapamycin analog AP21967 was obtained from ARIAD Pharmaceuticals. Cells were transiently transfected using Fugene6 (no. 11815091001 from Roche). In siRNA experiments, HeLa cells in 10 cm dishes were transfected with 30 nM of siGenome pool against p62 (Dharmacon, M-010230-00) or control siRNA (Qiagen, 1027281) for three consecutive days, using Lipofectamine 2000 (Invitrogen, 11668-019). On the morning of the fourth day, cells were disassociated with trypsin for immunoblotting or plating onto chambered coverslides (Nunc, 155383). On the evening of the fourth day, cells were transfected with EYFP-Parkin using Fugene6. Cells were treated with CCCP starting on day five and were fixed for immunostaining and confocal microscopy on day six.
Confocal microscopy.
Confocal microscopy of fixed samples, scoring of Parkin recruitment and Parkin-induced mitophagy and live cell imaging were performed as described previously.11 Measurements of ubiquitinated protein intensity and co-localization between p62 intensity and Tom20 intensity were performed as described previously.9,11 To quantify the compaction index of mitochondria, mid plane images of cells immunostained for Tom20 were obtained. In ImageJ (NIH), the Tom20 channel was converted into binary and the area and perimeter of mitochondria within the cell of interest (selected using the “region of interest” tool) was measured using the “analyze particles” function. The compaction index (which is the perimeter of a circle with the same area as the object of interest divided by the actual perimeter of the object of interest) was calculated from the perimeter (P) and the area (A) using the following formula:
(2π * ((A/π)−2))/P.
Immunocytochemistry.
Cells were fixed and immunostained as described previously.11 The following primary antibodies were used: anti-Tom20 polyclonal (Santa Cruz, sc-11415), anti-cytochrome c monoclonal (BD Biosciences, 556432), anti-alpha-Tubulin monoclonal (Invitrogen, 236-10501), anti-total polyubiquitinated protein (clone FK1; Enzo Life Sciences, PW 8805), anti-K48 polyubiquitinated protein (clone Apu2; Millipore, 05-1307), anti-K48 polyubiquitinated protein (clone Apu2, gift of Vishua Dixit at Genentech), anti-63 polyubiquitinated protein (clone Apu3; Millipore, 05-1308), anti-K63 polyubiquitinated protein (clone Apu2, gift of Vishua Dixit at Genentech), anti-p62 polyclonal guinea pig C-terminus (Progen, GP62-C) and anti-p62 mouse monoclonal (Abcam, ab56416).
Immunoblotting/immunopreciptation.
HeLa cells were lysed in 1% SDS lysate buffer (10 mM Tris/HCl pH 7.4, 150 mM NaCl). DNA was sheared by 5–10 passages through 26 and 30 gauge needles and lysates were cleared by centrifugation (16,000 g). A final concentration of 1x Laemmli loading buffer with 10 mM DTT was added to lysates, they were separated on 12–22% polyacrylamide gradient SDS-PAGE gels, transferred to PVDF membranes (Millipore) and probed with antibodies against VDAC1 (anti-Porin HL Ab3 [referred to as Ab6 in the literature and this paper] and anti-Porin HL Ab1; Calbiochem, 529536 and 529532, respectively), Tubulin (Sigma, T5201), p62 (Abcam, ab56416) or anti-VDAC2 (rabbit polyclonal raised against peptide KKSGKIKSSYKRE corresponding to 120–132 of human VDAC2; Abcam, ab47104) using a standard protocol. For immunoprecipitation of endogenous VDAC1 under denaturing conditions, 5 × 15 cm plates of HeLaEYFP-Parkin treated with 10 µM CCCP for 5 hrs were solubilized in 1% SDS lysis buffer as described above. Then samples were diluted 13-fold in 0.5% NP-40 buffer (10 mM Tris pH 7.5, 150 mM NaCl, 0.1 M EDTA, complete protease tablet [Roche]). Lysates were incubated with 2 µg of VDAC1 antibody per mL of lysate overnight at 4°C. 1 mL of immunocomplex containing lysate was added to 25 µg of Protein A sepharose bead slurry (GE Heathcare, 17-5280-01) and incubated for 3 hrs. The beads were washed twice in NP40 buffer and then eluted in a bead volume of 0.1 M Glycine/HCL pH 2.5. The elutant was neutralized with a final concentration of 0.1 M Tris base and then precipitated with ten volumes of 100% EtOH overnight at −20°C. Sample was resuspended in 1x Laemmli sample buffer with 10 mM DTT, separated on 12–22% SDS-PAGE gels and indicated bands were excised for protein identification. Excised bands were in-gel digested with trypsin (Roche, 11 521 187 001) following a standard protocol.
Mass spectrometry.
Peptides from in-gel digestion diluted in 0.1% formic acid in 5% acetonitrile were loaded onto a nano-column (75 µm ID × 50 mm) with C18 resin and desalted. Peptides were subsequently eluted from the reverse phase column over a 40 minute gradient (5–40% acetonitrile) and analyzed by an LTQ-Orbitrap mass spectrometer (Thermo), using a Top 5 duty cycle (1 MS full scan [400–2,000 amu] and 5 MS/MS scans). For peptide identification, peaks were selected from the resulting MS/MS data using Proteome Discoverer software (Thermo) and compared to the IPI_human database using the MASCOT algorithm (Matrix Science), with oxidation (M), propionamide (C) and diglycine (K) included as variable modifications. For label-free quantification, the peak area was automatically selected from MS ion chromatograms by the ISIC algorithm implemented in Qual Browser (Thermo). When more than one peak was identified the peak retention times were used to assign the correct peak to the ion of interest.
Acknowledgements
We thank C. Smith for help with confocal microscopy, S. Smith for assistance with cell culture, S. Ding, K. Jayawardena for technical assistance with sample preparation and mass spectrometry. We thank J.E. Walker, J. Carroll, the Proteomics Group at the MRC-Mitochondrial Biology Unit, the Youle lab and W. Motley for support and discussions. We thank K. Tanaka and M. Komatsu for transformed p62+/+ and p62−/− MEFs; T. Johansen for EGFP-p62, EGFP-p62 D69A and EGFP-p62 ΔLIR constructs; V. Dixit for the Apu2 and Apu3 antibodies; and B. Turner and J. Atkin for the EGFP-SOD1 G93A construct. We also thank M. Komatsu and T. Johansen for a careful reading of the manuscript and their thoughtful comments.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/13426
Supplementary Material
References
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orr WC, Sohal RS. Extension of life-span by over-expression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263:1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 3.Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci USA. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway: a mitochondrial quality control system? J Bioenerg Biomembr. 2009;41:499–503. doi: 10.1007/s10863-009-9253-3. [DOI] [PubMed] [Google Scholar]
- 7.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 8.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 15.Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujimuro M, Sawada H, Yokosawa H. Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett. 1994;349:173–180. doi: 10.1016/0014-5793(94)00647-4. [DOI] [PubMed] [Google Scholar]
- 17.Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, et al. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- 18.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’/lpage> review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 22.Basso M, Massignan T, Samengo G, Cheroni C, De Biasi S, Salmona M, et al. Insoluble mutant SOD1 is partly oligoubiquitinated in amyotrophic lateral sclerosis mice. J Biol Chem. 2006;281:33325–33335. doi: 10.1074/jbc.M603489200. [DOI] [PubMed] [Google Scholar]
- 23.Matsuda N, Kitami T, Suzuki T, Mizuno Y, Hattori N, Tanaka K. Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J Biol Chem. 2006;281:3204–3209. doi: 10.1074/jbc.M510393200. [DOI] [PubMed] [Google Scholar]
- 24.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 25.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 26.Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 28.Liberles SD, Diver ST, Austin DJ, Schreiber SL. Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proc Natl Acad Sci USA. 1997;94:7825–7830. doi: 10.1073/pnas.94.15.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nechushtan A, Smith CL, Hsu YT, Youle RJ. Conformation of the Bax C-terminus regulates sub-cellular location and cell death. EMBO J. 1999;18:2330–2341. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babel D, Walter G, Gotz H, Thinnes FP, Jurgens L, Konig U, Hilschmann N. Studies on human porin. VI. Production and characterization of eight monoclonal mouse antibodies against the human VDAC “Porin 31HL”/lpage> and their application for histotopological studies in human skeletal muscle. Biol Chem Hoppe Seyler. 1991;372:1027–1034. doi: 10.1515/bchm3.1991.372.2.1027. [DOI] [PubMed] [Google Scholar]
- 32.Winkelbach H, Walter G, Morys-Wortmann C, Paetzold G, Hesse D, Zimmermann B, et al. Studies on human porin. XII. Eight monoclonal mouse anti“porin 31HL”/lpage> antibodies discriminate type 1 and type 2 mammalian porin channels/VDACs in western blotting and enzyme-linked immunosorbent assays. Biochem Med Metab Biol. 1994;52:120–127. doi: 10.1006/bmmb.1994.1042. [DOI] [PubMed] [Google Scholar]
- 33.Kirkpatrick DS, Denison C, Gygi SP. Weighing in on ubiquitin: the expanding role of mass-spectrometry-based proteomics. Nat Cell Biol. 2005;7:750–757. doi: 10.1038/ncb0805-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 35.Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 36.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 37.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 38.Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180:1065–1071. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001;12:2085–2090. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- 40.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–4631. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- 43.Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cox RT, Spradling AC. Clueless, a conserved Drosophila gene required for mitochondrial subcellular localization, interacts genetically with parkin. Dis Model Mech. 2009;2:490–499. doi: 10.1242/dmm.002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turner BJ, Atkin JD, Farg MA, Zang DW, Rembach A, Lopes EC, et al. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J Neurosci. 2005;25:108–117. doi: 10.1523/JNEUROSCI.4253-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.