

Abstract
Ovulation in mammals is gated by a master circadian clock in the suprachiasmatic nucleus (SCN). GnRH neurons represent the converging pathway through which the brain triggers ovulation, but precisely how the SCN times GnRH neurons is unknown. We tested the hypothesis that neurons expressing kisspeptin, a neuropeptide coded by the Kiss1 gene and necessary for the activation of GnRH cells during ovulation, represent a relay station for circadian information that times ovulation. We first show that the circadian increase of Kiss1 expression, as well as the activation of GnRH cells, relies on intact ipsilateral neural input from the SCN. Second, by desynchronizing the dorsomedial (dm) and ventrolateral (vl) subregions of the SCN, we show that a clock residing in the dmSCN acts independently of the light-dark cycle, and the vlSCN, to time Kiss1 expression in the anteroventral periventricular nucleus of the hypothalamus and that this rhythm is always in phase with the LH surge. In addition, we show that although the timing of the LH surge is governed by the dmSCN, its amplitude likely depends on the phase coherence between the vlSCN and dmSCN. Our results suggest that whereas dmSCN neuronal oscillators are sufficient to time the LH surge through input to kisspeptin cells in the anteroventral periventricular nucleus of the hypothalamus, the phase coherence among dmSCN, vlSCN, and extra-SCN oscillators is critical for shaping it. They also suggest that female reproductive disorders associated with nocturnal shift work could emerge from the desynchronization between subregional oscillators within the master circadian clock.
Ovulation is triggered by a surge of LH from the pituitary, which follows a surge of GnRH from medial preoptic area (MPO) neurons. In rats, this surge occurs every 4–5 d, just before the onset of daily locomotor activity. If endogenous estrogen is controlled by removing the ovaries and administering exogenous estradiol (E2), rats show daily afternoon peaks of LH (1). These daily LH peaks are abolished by lesions of the hypothalamic suprachiasmatic nucleus (SCN), which houses the master circadian oscillator (2–4). These and other results have established that the LH surge results from integration of high E2 levels and a circadian signal from the SCN that converge into the hypothalamo-pituitary-gonadal (HPG) axis. In rats, mice, and hamsters, the surge occurs in the evening, 2–3 h before their nocturnal activity onset. In humans, the preovulatory LH surge takes place primarily in the early morning (5, 6), also preceding daily locomotor activity. Disruption of internal circadian timing by shift work or jetlag is associated with reproductive disorders (7–9), suggesting that intact circadian regulation of the HPG axis is critical for regular reproductive cycles and ovulation in women. Despite the central role of the circadian system in ovulation, the output signals from the SCN and the point of convergence between the circadian and ovarian signals remain unknown (10).
In the last 10 yr, kisspeptin has emerged as a key regulator of reproduction in mammals (11). Within the anteroventral periventricular nucleus of the hypothalamus (AVPV), the expression of Kiss1 and the induction of the immediate early gene cFos within Kiss1-expressing cells increase at the time of the LH surge (12–15). Kisspeptin+ AVPV neurons send axonal fibers to GnRH cells, which express the kisspeptin receptor Kissr1, formerly known as G protein-coupled receptor 54 (15–17). The absence of a functional Kissr1 in mice and humans or the Kiss1 gene in mice, leads to hypogonadotropic hypogonadism (18–20), and antisera against kisspeptin blocks the LH surge (15, 21). In summary, kisspeptin is a critical regulator of GnRH and thereby of LH release and ovulation.
We previously demonstrated that Kiss1 expression and cFos expression within Kiss1 cells in the AVPV of ovariectomized E2-primed (OVX+E2) mice increase in a circadian fashion in phase with the LH surge (13). Based on these findings, we hypothesized that Kiss1 neurons represent a key node between the SCN and the GnRH neuronal network in female rats as well. We first recapitulate our findings from mice in female rats. Then, using unilateral SCN lesions, we show that the circadian Kiss1 expression, as well as the downstream activation of GnRH cells, is dependent on intact, ipsilateral SCN projections.
The SCN is a heterogeneous nucleus; neurons within the ventrolateral (vl) and dorsomedial (dm) SCN express different neuropeptides and show different patterns of efferent and afferent projections (22). The role of each subregion cannot be assessed by neuroanatomical lesions mainly because vlSCN efferent fibers go through the dmSCN. Furthermore, knockouts that target gene expression in specific SCN subregions are not available. We have developed a forced desynchrony model that induces the desynchronization of vlSCN neurons from dmSCN neurons in genetically and neurologically intact rats. When rats are exposed to an 11-h light, 11-h dark (LD) cycle (LD22), they develop two locomotor activity rhythms with differing periods: one rhythm is entrained to the 22-h LD cycle and the other rhythm is dissociated from it and shows a period of approximately 25 h. We have previously shown that this approximately 25-h rhythm is not truly free running but still weakly coupled to the vlSCN (23); therefore, we refer to it as the LD-dissociated rhythm. As the two rhythms in the LD22 desynchronized rat come in and out of phase with each other, the animals experience days of alignment, in which both activity phases coincide, and days of misalignment, in which the activity phase of one rhythm ends as the activity phase of the other begins. In males, these rhythmic outputs parallel clock gene expression rhythms within the vlSCN and dmSCN, respectively. Using this animal model, we have shown that the two SCN subregions have different roles in circadian output regulation (23–27). To reveal which of these two subpopulations of SCN neuronal oscillators sends the critical signals timing the LH surge, we first confirmed that the female rat shows similar desynchronization to that observed in the male both at the behavioral and SCN-molecular level. We then show that the timing of Kiss1 expression and the LH surge is associated with the dmSCN activity and not with activity of the light-entrained vlSCN.
Materials and Methods
Animals
Adult female Wistar rats, purchased from Charles River (Wilmington, MA), were used for all the experiments. All experiments were performed according to the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the University of Washington Institutional Animal Care and Use Committee.
Activity cycles and internal desynchronization
Animal activity monitoring and internal desynchronization was carried out as reported previously (26). Briefly, animals were singly housed either under a 12-h light, 12-h dark cycle (LD24 control animals) or under LD22 with 50–150 lux light during the light phase and red light not brighter than 1 lux during the dark phase. LD22 animals were housed under LD22 for 1–2 months. Locomotor activity was recorded using two perpendicular infrared beams crossed through the center of the cage, approximately 2 cm above the bedding, with beam breaks recorded digitally with ClockLab (Actimetrics, Wilmette, IL). Analysis of activity was carried out using the software El Temps (Dr. Antoni Díez-Noguera, University of Barcelona, Barcelona, Spain) and the graphs imported into Adobe Photoshop (San Jose, CA) to prepare the final figures. Only LD22 animals showing two statistically significant rhythms using periodogram analysis were used for any analysis. Assessment of alignment and misalignment during the forced desynchrony protocol was done by eye-fitting, onset-timing lines of the two rhythms found under periodogram analysis (as shown in Fig. 4) by at least two independent researchers. Significantly desynchronized animals were grouped into either the aligned or the misaligned groups for blood and tissue collection, but this did not change their housing or handling, only the time of collection relative to their activity cycles.
Fig. 4.

The circadian timing of the LH surge is coupled to an oscillator within the dorsomedial SCN. A and B, Double-plotted actograms (top) and hourly LH profiles (bottom) of two representative LD22 desynchronized OVX+E2 rats bled during either an aligned day (A) or a misaligned day (B). Arrowhead on left indicates day of ovariectomy for both animals. On actograms, black bars represent locomotor activity, single diagonal lines indicate onset of dmSCN-associated locomotor activity bout with the angle set to match the LD-dissociated period indicated in the periodogram (D), double diagonal bands indicate the time of lights-off. Note that in A the two trend lines cross between the day of bleeding and the preceding day, with the LD-dissociated locomotor activity onset roughly overlapping with the time of lights-off, whereas in B the LD-dissociated locomotor activity onset beings approximately 7 h after lights-off. The day of bleeding is highlighted as in Fig. 3. Thin black lines expand the 24-h time scale on the bleeding day to easily visualize the plasma LH profiles shown below. Squares indicate the time of the LH surge onset both in the actogram and on the LH profile. Black arrows indicate the onset of the LD-dissociated locomotor activity in both actograms and LH plots. X-axes are in clock time, to match the actograms. C, Plasma LH profiles in a representative LD24 animal. The gray circle represents the LH surge onset; black arrow indicates the onset of locomotor activity, as in A and B. C, The x-axis is in Zeitgeber time (ZT) with ZT12 = time of lights-off. *, A high-LH point not considered part of the surge due lack of contiguity (see Materials and Methods). D, Periodograms of animals shown in A (top) and B (bottom).
SCN lesions
Unilateral SCN lesions were carried out in a stereotaxic apparatus under isoflurane anesthesia. The skin over the cranium was sterilized and cut down the middle with a scalpel. Bregma and lamda were visualized and placed on the same horizontal plane. A small hole was drilled in the skull and electrodes (size 00 insect pins coated with nail sealant except for the very tip) lowered at a 10° angle. Coordinates from bregma were −0.9 mm rostrocaudal, 2.25 mm mediolateral, and −9.5 mm dorsoventral. The lesion was made by application of a 1-mA current for 10 sec. The electrode was then removed, the skull sealed with gelfoam, and the skin stapled with wound clips. Surgery was preceded by and followed every 12 h for 2 d by 0.05 mg/kg buprenorphine sc injections to minimize postoperative pain. All SCN-lesioned (SCNX) animals underwent ovariectomy while under anesthesia from the SCNX operation.
Animals with lesions were killed between 1 and 0 h before lights-off. For the purpose of grouping animals as successful or missed lesions, 16-μm brain sections were collected, stained with 4′,6′-diamino-2-phenylindole (DAPI), and the extent of the lesion assessed. Under DAPI staining, the SCN is revealed as a very discrete cluster of nuclei easily recognizable from the surrounding tissue. Lesions were rated successful if the following occurred: 1) the SCN ispilateral to the lesion side was ablated in at least 60% by area of the whole rostrocaudal extent of the nucleus; 2) there was no damage on the contralateral-side SCN throughout its rostrocaudal extent; and (3) the lesion did not extend into either the ipsilateral or contralateral MPO and AVPV. The animals with ipsilateral SCN ablation less than 60% were not included in the study; animals in which the lesions fell outside the SCN but not in the MPO or AVPV were counted as missed lesions.
Ovariectomy, E2 treatment, and catheterization
All ovariectomies were carried out under isoflurane anesthesia, using buprenorphine as indicated above. Internal incisions were closed with biodegradable suture, whereas skin was closed with wound clips. Animals were allowed to heal at least 2 wk. Two days before the animals were killed, E2-treated animals were implanted with 1 cm × 1.57 mm internal diameter, 3.18 mm outer diameter SILASTIC brand capsules (Dow Corning Corp., Midland, MI) filled with 20% E2/80% cholesterol by mass and capped with SILASTIC brand glue. This capsule size and concentration have been shown to yield 1.5–2 times the physiological concentration of E2 during proestrus in rats (2). Capsules were soaked overnight in 70% EtOH before being sc implanted into OVX rats under isoflurane anesthesia. Animals used for LH-profile generation were implanted with SILASTIC brand catheters (Dow Corning) at the same time as the E2 implantation. The jugular vein was exposed and raised from the tissue, nicked but not severed, and SILASTIC brand tubing (Dow Corning) with an internal diameter of 0.51 mm, outer diameter of 0.94 mm inserted 4.3 cm, with its tip in the atrium. The catheter was anchored by suturing, threaded under the skin, extruded through the back of the neck, filled with 20 μl heparin and 120 μg/ml gentamicin saline and plugged until bleeding.
Tissue collection
For LH profiles, hourly blood samples (0.12 ml) were collected through the implanted catheters. Blood volume was replaced with 35 C saline and the catheter refilled with heparinized saline. Bleeding and volume replacement took less than 3 min per animal, from the time of removing the cage from its rack to the time of returning it. Samples were then centrifuged at 4 C, 1500 rpm, for 15 min. Plasma was separated and stored at −80 C for processing by RIA. For circadian sampling of mRNA within brain tissue, brains were collected every 6 h, with predicted peak time selected based on the Kiss1 profile found previously in mice (13). All animals were killed by decapitation. Brains were collected immediately and frozen in −30 C methyl butane; 1 ml of trunk blood was collected into centrifuge tubes with 20 μl heparin (10,000 units/ml) and centrifuged as above. Plasma and brains were then stored at −80 C until processed. All brains were cut into 16-μm sections in a cryostat, mounted on slides, and frozen until histological processing.
Immunolabeling of cFOS and GnRH
Between 18 and 24 slices were stained for cFos and GnRH per animal, spanning the whole rostrocaudal extent of the MPO. Anti-GnRH antibody (FL-92; Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:400 and anti-cFOS antibody (SC-52; Santa Cruz Biotechnology) diluted 1:400 were used as primary antibodies. Secondary antibodies were Alexafluor 594 donkey antirabbit (no. A10042; Invitrogen, Grand Island, NY) and fluorescein isothiocyanate (FITC) donkey antirabbit (no. SC-2090; Santa Cruz Biotechnology). Immunohistochemical labeling was done by fixing tissue in fresh 4% paraformaldehyde in phosphate buffer for 5 min, rinsing 3 × 5 min in PBS and then blocking 30 min in PBS with 5% BSA, 0.5% Triton X-100, 3% normal donkey serum, and 0.3% H2O2. Tissue was then incubated overnight at 4 C with anti-cFOS in a 1:10 dilution of the blocking buffer, rinsed 3 × 5 min with PBS, incubated in FITC antirabbit 1:400 for 2 h, rinsed 3 × 5 min in PBS, reblocked, and stained as before but with anti-GnRH antibody and Alexafluor 594 secondary antibody. The resulting tissue, although using two rabbit primary antibodies, could be scanned for GnRH cells labeled with Alexafluor 594, and the nuclear fill by cFOS could then be checked by assessing FITC. Because GnRH is cytoplasmic and cFOS nuclear, cross-reactivity was not a problem as long as cFOS was labeled first. Slides were coverslipped with Vectashield with DAPI (no. H-1200; Vector Laboratories, Burlingame, CA).
All slides stained for GnRH were scored in random order by two researchers blinded to the condition of the lesion. The GnRH cells on both sides of the brain were counted as with or without cFOS staining under a fluorescent microscope. All the scored slices for each individual were counted and summed, and the percentage of GnRH+ cells that contained a cFOS+ nucleus was quantified on each side of the brain. To assess any side-to-side cFOS expression asymmetry, separate paired Student t tests were done for successful- and missed-lesion animals. Cell counting and lesion assessment was done on a Nikon microphot-FXA fluorescence light microscope (Tokyo, Japan). Photomicrographs were captured using a Leica SP5 confocal microscope (Heidelberg, Germany).
In situ hybridization for Kiss1 mRNA
In situ hybridization (ISH) was carried out as described previously (28). Briefly, a 35S-labeled riboprobe was transcribed from a mouse Kiss1 template (13). After prehybridization washes, slides were incubated overnight at 52 C with 4 × 107 cpm/ml. After posthybridization washes and dehydration in an alcohol series, they were air dried and exposed to autoradiographic films for 4 d. All slides stained for Kiss1 were scored masked to the experimental condition. Kiss1 staining was scored by assigning each slice an area value based on the whole AVPV-stained area above a preset darkness threshold and multiplying that area by the average optical density within that area, normalized to the background optical density in the surrounding tissue. Each animal's score was the mean of the three strongest stained slices. For unilaterally lesioned animals, an average for all the animals for either the ipsilateral or contralateral sides of the lesion was calculated. To assess any side-to-side Kiss1 expression asymmetry, separate paired Student's t tests were done for successful- and missed-lesion animals. All scores are reported as a percentage of the average score of the highest-scoring condition within the groups being compared. Two-way ANOVA was used for experiments with multiple factors existed (treatment and time). Tukey tests were then used for post hoc analysis.
Luteinizing hormone RIA
All LH RIA were run using probe iodinated in-house. Rat LH and antibodies were ordered from Dr. A. F. Parlow (National Hormone and Pituitary Program, Harbor-UCLA Medical Center, Torrance, CA), and iodinated with 125I (no. 016303401; PerkinElmer, Waltham, MA). Plasma from animals was mixed with RIA buffer (0.05 m NaH2PO4, 0.25 m Na2HPO4, 0.025 m EDTA, 0.1% Triton X-100, 0.02% Trasylol protease inhibitor, 0.25% BSA) and incubated with rabbit antirat-LH overnight at 4 C. The next day, 125I-LH was added, mixed, and incubated overnight. Finally, goat antirabbit IgG and normal rabbit serum were added, mixed, and incubated for 4 h. Separation buffer (as RIA buffer but 2.5% BSA, no Triton X-100 or Trasylol) was then added and all tubes were centrifuged. The supernatant was aspirated and discarded and the pellet counted in a γ-counter. All steps were carried out at 4 C. Samples were run in duplicates and serial dilutions of rat LH standard were used to create standard curves. Nonspecific binding was subtracted from all readings and 0-binding controls were used to calculate the final standard curve equation. All samples were assayed in two assays, with three standard curves per assay. The interassay variability was 3.5% and intraassay variabilities were, respectively, 1.9 and 2.2%. The assay was sensitive between 0.5 and 16 ng/ml. Values above these limits were brought to the maximum sensitivity; values below this limit were unchanged.
Rayleigh statistics
To assess the clustering of LH surges, we used a Rayleigh test for circular distribution. Each LH onset time, defined as the first point to exceed 5 times the sd of the lowest six points and to be contiguous with at least two other points also meeting this criterion, was converted into a circular phase with 360° equal to the relevant period (22 h, 24 h, or that animal's LD dissociated period, usually ∼25 h). Rayleigh statistics were then computed for each animal group (LD24, n = 7; LD22, n = 10 on aligned days, n = 9 on misaligned days) and relative to 24 h (for LD24 animals), and to 22 h or approximately 25 h (for LD22 animals). The α-value was always set to 0.05. From the calculated statistics, the Rayleigh data were plotted using quantitative transformation of vectors in Adobe Photoshop.
Results
The master circadian clock is necessary for circadian Kiss1 expression in the AVPV and circadian GnRH cell activation
Mice show E2-dependent Kiss1 expression circadian rhythms in the AVPV (13). We confirmed this result in OVXE2. Expression of Kiss1 mRNA in rats killed every 6 h during the first 24 h after their release into constant darkness showed a rhythm with a peak that coincided with the afternoon LH surge (one way ANOVA for Kiss1 levels, F = 0.023, P = 0.023, n = 4–6 per time point) (Fig. 1). Unilateral lesions of the SCN allowed us to assess the induction of Kiss1 mRNA expression and the activation of GnRH cells without an SCN, compared with the intact SCN side of the same animal. Double-immunohistochemical labeling for GnRH and the cFOS protein at the time of the LH surge in E2-primed females revealed that GnRH cells showed significantly more cFOS-positive nuclei on the intact than on the lesioned side of the brain (n = 7 successful lesions, paired t test, P = 0.004) (Fig. 2). This difference disappeared in animals in which the lesion missed the SCN (n = 9 missed lesions, paired t test, P = 0.08), suggesting that the lack of activation of GnRH cells was due specifically to the loss of the SCN.
Fig. 1.

Circadian Kiss1 expression in the AVPV peaks at the time of the LH surge in OVX+E2 rats. A, Relative Kiss1 mRNA levels (gray line) and plasma LH (dashed line). Sample frequency is one per 6 h. Data represent mean ± sem. B, Representative autoradiographic films of coronal sections at the same AVPV level from animals killed at the indicated times and labeled with an antisense Kiss1 mRNA 35S-tagged riboprobe. One-way ANOVA for Kiss1 levels, F = 0.023, P = 0.023, n = 4–6 per time point; *, Significantly different from circadian time (CT) 0, Tukey post hoc comparison, P = 0.05).
Fig. 2.

Ipsilateral SCN projections gate the daily activation of GnRH cells and the AVPV Kiss1 expression rhythms in OVX+E2 rats. A, Schematic coronal section (modified from Ref. 60) showing the lesion location at two rostrocaudal levels from an animal that received a successful unilateral SCN lesion (shaded). MPO, SCN, and optic chiasm (OX) are indicated. B–G, Representative confocal photomicrograph of GnRH/cFOS double-labeled sections from an animal with a successful lesion. Cells immunostained for GnRH (B and E) cFOS (C and F), and nuclear DAPI stain (D and G) are shown for the contralateral (B–D) or ipsilateral (E–G) side of the lesion. H, The percentage of GnRH cells in the MPO with cFOS+ nuclei is significantly lower on the ipsilateral than on the contralateral side of the lesion in animals with successful unilateral SCN lesions but not in animals in which the unilateral lesion missed the SCN. Bars, Mean ± sem. *, Statistically different (n = 7 successful lesions, paired Student t test, P = 0.004; n = 9 missed lesions, paired Student t test, P = 0.08). I, Schematic coronal section at the AVPV level (modified from Ref. 60). J–L, Representative autoradiographic films of coronal sections at the AVPV level from animals killed at the expected peak time for Kiss1 mRNA expression and labeled with an antisense Kiss1 mRNA 35S-tagged riboprobe in an animal with a successful unilateral SCN lesion and total loss of ipsilateral Kiss1 staining (J), a second animal with a successful unilateral SCN lesion and partial loss of ipsilateral Kiss1 staining (K), and in an animal in which the unilateral lesion missed the SCN (L). M, Kiss1 expression is significantly lower on the ipsilateral than on the contralateral side of the lesion in animals with successful unilateral SCN lesions but not in animals in which the unilateral lesion missed the SCN. Bars, Mean ± sem. *, Statistically different (n = 5 successful lesions, paired Student t test, P = 0.016; n = 7 missed lesions, paired Student t test, P = 0.33).
Similarly, paired t tests of ISH with a 35S-tagged Kiss1 riboprobe revealed a significant reduction of Kiss1 expression on the lesioned side (n = 5 successful lesions, P = 0.016) (Fig. 2). As with GnRH cell activation, this effect disappeared in animals with missed lesions (n = 6, P = 0.33). Kiss1 expression roughly doubled from its circadian trough to its peak (Fig. 1A), as well as from the SCN-lesion side to the contralateral side (Fig. 2M). Taken together, these data suggest that the activation of GnRH cells during the LH surge is associated with increased Kiss1 levels, which in turn depend on circadian signals from the SCN.
Circadian timing of Kiss1 expression and of the LH surge are coupled to an oscillator within the dorsomedial SCN
Because the LH surge in females occurs just before locomotor activity onset, we wondered whether either of the two locomotor rhythms emerging under LD22 desynchrony would predict the LH surge in desynchronized females. The outcome of this experiment would point to which oscillator (vl or dm) of the SCN is responsible for timing ovulation. We first confirmed that OVX females display the same behavioral and neural desynchronization as males (Fig. 3). LD22 led to desynchronization of locomotor activity rhythms in intact females similar to that seen in males, with two statistically significant rhythmic components (22 h and ∼25 h) within each individual. ISH with a 35S-tagged rPer1 riboprobe of OVX females revealed similar anatomical and temporal patterns as previously seen in males (24, 26), with the 22-h and approximately 25-h locomotor activity rhythms associated with clock gene expression within the vlSCN and dmSCN, respectively. After desynchronization was confirmed, ovariectomy and subsequent estrogen implantation did not noticeably perturb desynchronization.
Fig. 3.

Forced desynchronization of locomotor activity rhythms in OVX+E2 rats is associated with rhythmic clock gene expression in the vlSCN and dmSCN. A, Representative actogram of an LD22 desynchronized female; arrowheads represent the specific phases at which other individual animals were killed to process their brain for Per1 ISH (n = 2 per time point). B, A χ2 periodogram analysis detects two statistically significant rhythmic components of 22 h and 25.08 h in the representative animal shown. C and D, Autoradiographic films of SCN Per1 expression in animals killed on the phases indicated by the triangles in A in a typical desynchronized animal (note that the animal whose actogram is shown was not killed for this experiment). Females killed on aligned days (C) show low levels of Per1 mRNA throughout the SCN during the dark phase and high levels during the light phase. During misaligned days the pattern of expression between the two subregions is 180° out of phase: the dmSCN shows daytime levels of Per1 expression during the subjective day (rest) of the approximately 25-h locomotor activity rhythm, whereas the vlSCN shows daytime levels of Per1 expression during the light phase of the 22-h day (subjective night for the approximately 25 h locomotor activity rhythm). For comparison, the Per1 expression pattern in LD22 desynchronized males is shown (modified from Ref. 26) to convey the similarity between the male and female neural bases of circadian desynchrony.
The stability of the forced desynchrony protocol allows for the prediction of the phase relationship between the two locomotor activity rhythms and therefore between the rhythmic clock gene activities of the vlSCN and dmSCN. In days of maximum misalignment, one locomotor activity bout starts as the other ends; in contrast, in days of maximum alignment, both locomotor activity bouts start at the same time. LD24 control animals showed the expected LH surge timing, with levels of LH rising toward the evening and peaking at the time of lights off (Fig. 4C). LD22 desynchronized animals displayed a single LH surge, which was always in phase with the onset of the dmSCN-associated locomotor activity rhythm (Fig. 4, A and B). Thus, on aligned days, the LH surge occurred, on average, at the same phase (relative to the LD cycle) as it did in LD24 controls. On misaligned days, however, the LH surge had the same phase relationship to the onset of dmSCN locomotor activity rhythm but no significant relationship to the LD cycle or the entrained vlSCN-associated locomotor activity rhythm. Raleigh tests for clustering of the phases of LH surge onsets for all animals within each group confirmed a significant clustering of onsets for LD24 controls (onset time = lights-off − 2.71 h; n = 7; P = 0.010) and for all desynchronized animals relative to the dmSCN locomotor activity rhythm, whether they had been bled during aligned (onset time = lights-off − 2.49 h; n = 10; P = 0.0018) or misaligned days (onset time = dmSCN-associated activity onset − 3.35 h; n = 9; P = 0.013). Clustering relative to the vlSCN locomotor activity rhythm was only significant for animals bled during aligned days (onset time = lights-off + 2.07 h, P = 0.007 for aligned; onset time = lights-off + 4.82 h, P = 0.23 for misaligned). One-way ANOVA for phases revealed no differences between LD24 controls (relative to the LD cycle), LD22 aligned animals (relative to the LD cycle), and LD22 misaligned animals (relative to the dmSCN associated locomotor activity onset) (F = 3.422; P = 0.47) (Fig. 5). Taken together, these results indicate that regardless of alignment between the vlSCN and dmSCN neuronal oscillators, the LH surge always occurred in phase with the predicted activity of the dmSCN. Although there is an apparent bifurcation of the clusters on misaligned days relative to the dmSCN-associated rhythm, this probably does not reflect a real biological timing phenomenon. Rather, it likely arises from days in which the misalignment between the 22 h and the approximately 25 h rhythms is not complete (see Fig. 4B).
Fig. 5.

The circadian timing of the LH surge is coupled to an oscillator within the dorsomedial SCN. A, Polar plot and Raleigh test shows significant clustering of LH surge onset in control animals (gray circles, as in Fig. 4.C.) with a mean of 2.71 h before lights-off and locomotor activity onset. Vector direction indicates mean phase with 360° = 24 h, and vector length indicates significance (n = 7, Rayleigh test, P = 0.010; dotted circle indicates a P = 0.05). B and C, Same as for A but for LD22 OVX+E2 animals. Animals bled during misaligned (points as black squares, descriptive vector in black) and aligned days (points as white squares, descriptive vector in white) are represented in polar plots in which each individual animal's LH-surge onset is plotted relative to the period of one of its locomotor activity rhythms set equal to 360°. Thus, 360° corresponds to the period of the dmSCN-associated activity bout (∼25 h) (B) or to the period of the vlSCN-associated activity bout (22 h) (C). Note that the LH surges of animals bled during misaligned days do not significantly cluster relative to the period of the vlSCN-associated activity rhythm (black arrow in C does not reach significance). LD22 Rayleigh tests: B, Days of alignment: n = 10, P = 0.0018, mean phase 2.49 h before locomotor activity onset; days of misalignment: n = 9, P = 0.013, mean phase 3.35 h before locomotor activity onset; C, Days of alignment: n = 10, P = 0.007, mean phase 2.07 h after lights-off and locomotor activity onset; days of misalignment: n = 9, P = 0.23.
The LH surge amplitude, but not duration, was significantly lower in LD24 controls than in LD22 desynchronized animals bled either during aligned or misaligned days (Fig. 6). A two-way ANOVA with group and time as factors yielded a significant effect of group (F = 26.71, P < 0.0001), time (F = 23.02; P < 0.0001), and the interaction (F = 2.51, P < 0.0001). Post hoc Tukey contrasts (P = 0.05) indicated that each group was significantly different from the others (LD24 < LD22 misaligned < LD22 aligned). Although the timing of the LH surge in LD24 animals was as expected, the amplitude of their LH surge was lower than expected and lower than the LH surge drawn from trunk blood (Fig. 1).
Fig. 6.

Circadian desynchronization affects the LH surge amplitude. Means ± sem of LH surges centered at their peak time (0), grouped by phase on the day of bleeding. Each group is significantly different from each other (two way ANOVA, effect of group, F = 26.71, P < 0.0001; time, F = 23.02; P < 0.0001; and the interaction, F = 2.51, P < 0.0001).
Our analysis indicates that the LH surge is coupled to the clock gene activity of the dmSCN. Our hypothesis that the circadian gating of Kiss1 expression is associated with the timing of the LH surge predicts that the peak of Kiss1 expression should occur just before the dmSCN locomotor activity onset in desynchronized rats. We killed LD22 desynchronized OVX+E2 females during misaligned days just before the dmSCN-associated locomotor activity onset or just before the vlSCN-associated locomotor activity onset as well as during aligned days just before the coinciding locomotor activity onsets (n = 4 for each of the groups). We assessed Kiss1 mRNA expression in coronal sections of the AVPV by ISH and found a significant effect of phase (Fig. 7; one way ANOVA, F = 4.25, P = 0.05). Tukey post hoc contrasts (P = 0.05) revealed that whereas the levels of Kiss1 mRNA in misaligned animals before the dmSCN-associated locomotor activity onset did not differ from those of aligned animals, the levels in misaligned animals before the vlSCN-associated locomotor activity onset were significantly lower. A planned comparison between the levels before the vlSCN-associated locomotor activity onset, and the other two groups taken together indicated the expression was lower in misaligned animals killed during the vlSCN-associated locomotor activity onset (P = 0.02). These data indicate that, similarly to the timing of the LH surge, Kiss1 expression is coupled to the clock gene activity of the dmSCN but not to the activity of the light-entrained vlSCN.
Fig. 7.

Circadian Kiss1 expression is coupled to an oscillator within the dorsomedial SCN. A, Kiss1 expression on misaligned days 1 h before dmSCN-associated activity onset, but not 1 h before the vlSCN-associated locomotor activity onset, is similar to that in aligned animals before the locomotor activity onset [one way ANOVA (P = 0.05)]. Bars, Mean ± sem; groups not sharing the same capital letter are statistically significant according to Tukey post hoc comparisons (P = 0.05). *, Significantly different from aligned and misaligned taken together, planned comparison, P = 0.02. B, Representative Kiss1 ISH autoradiographic films from animals killed on aligned days just before locomotor activity onset and on misaligned days just before dmSCN-associated activity onset (dm) or just before the vlSCN-associated activity onset (vl).
Discussion
There has been considerable debate about how the SCN gates the LH surge. Our data provide strong support for the hypothesis that GnRH cells in the MPO are timed by multisynaptic rather than monosynaptic input from the SCN. Our data also support a role for AVPV kisspeptin cells as an SCN relay station that confers circadian timing information to GnRH cells. We show that AVPV Kiss1 circadian expression and GnRH neuronal activation are both dependent on ipsilateral input from the SCN. Furthermore, we show that both the expression of Kiss1 in AVPV neurons and the LH surge are phase locked to a circadian oscillator located within the dmSCN but not to the oscillations within the vlSCN.
Whereas the master regulation of circadian locomotor rhythms relies on diffusible factors released by SCN neurons (29), the circadian release of hormones, including melatonin, glucocorticoids and LH, relies on synaptic connections between specific neurons in the SCN and specific extra-SCN neuronal targets (23, 30, 31). Our results provide evidence for the identity of specific SCN subregional oscillators and their downstream targets critical for the circadian timing of the LH surge.
The dmSCN and vlSCN are heterogeneous not only under our forced desynchrony protocol but also in their pattern of afferent and efferent projections and their expression of specific neurotransmitters (22). Whereas neurons synthesizing arginine-vasopressin (VP) are localized within the dmSCN, neurons synthesizing vasoactive intestinal polypeptide (VIP) reside in the vlSCN. Although SCN neurons send axonal projections to the MPO (32, 33), they likely emerge from vlSCN VIPergic SCN neurons (34). Our findings suggest that this SCN monosynaptic pathway to GnRH cells is likely not responsible for the initiation of the LH surge. Rather, circadian signals emerging from the dmSCN and ovarian estrogen signals likely converge on Kiss1 cells, which in turn signal GnRH cells to induce the surge.
The rat is the only animal model in which the stable desynchronization of the vlSCN and dmSCN can be achieved. The specific role of these SCN subregions cannot be addressed by classic neuroanatomical lesions because of the difficulty to individually target them and because vlSCN efferent fibers course through the dmSCN (35). Thus, the forced desynchronized rat represents a unique model in which the output of each subregion can be dissected out. Although our results in the forced desynchronized OVX+E2 female rat do not demonstrate a causal link between the dmSCN activity and the LH surge, the association between this region's clock gene activity and the timing of the LH surge point to the dmSCN as the site of a critical circadian oscillator timing the LH surge.
Our data suggest a putative role for the vlSCN in modulating the amplitude of the LH surge. Given the mismatch between the amplitude values of circadian profiles (from samples every 6 h under constant darkness in animals released from LD24) and serially bled animals under LD24, it is likely that the LH surge amplitude of this latter group was diminished by the handling schedules, stress of bleeding, or other factors. Importantly, there was a difference in LH-surge amplitude between LD22 aligned and misaligned animals, with maximum LH levels in aligned animals. These two groups were housed in the same chamber, handled identically, and bled simultaneously. This indicates clearly that although the dmSCN may be initiating the surge, the vlSCN may be modulating the surge amplitude through as-yet-undefined alternate pathways. Recent studies have pointed to extra-SCN circadian oscillators within several sites of the HPG axis (see reviews in Refs. 10 and 36). They include neural centers such as GnRH cells themselves, which express circadian clock genes (37), as well as peripheral organs such as the anterior pituitary (38) and the ovary (39). The increased LH surge amplitude in desynchronized aligned animals suggests that stimulation of GnRH cells is taking place when the GnRH network and its downstream pathways to initiate an LH surge are highly sensitized. We speculate that whereas the dmSCN may send the critical signal to initiate the LH surge, the vlSCN may regulate the sensitivity to this signal in downstream targets (Fig. 8). Recent work by Kriegsfeld and colleagues in the Syrian hamster suggest the vlSCN could directly gate the response of GnRH cells (40) and/or disinhibit them by decreased RF amide-related peptide release (41). Finally, other signals, directly released by the SCN or by neurons regulated in turn by SCN efferents, could potentially represent a link between the SCN activity and the activity of the GnRH neuronal network (see ref. 42 for a recent review).
Fig. 8.
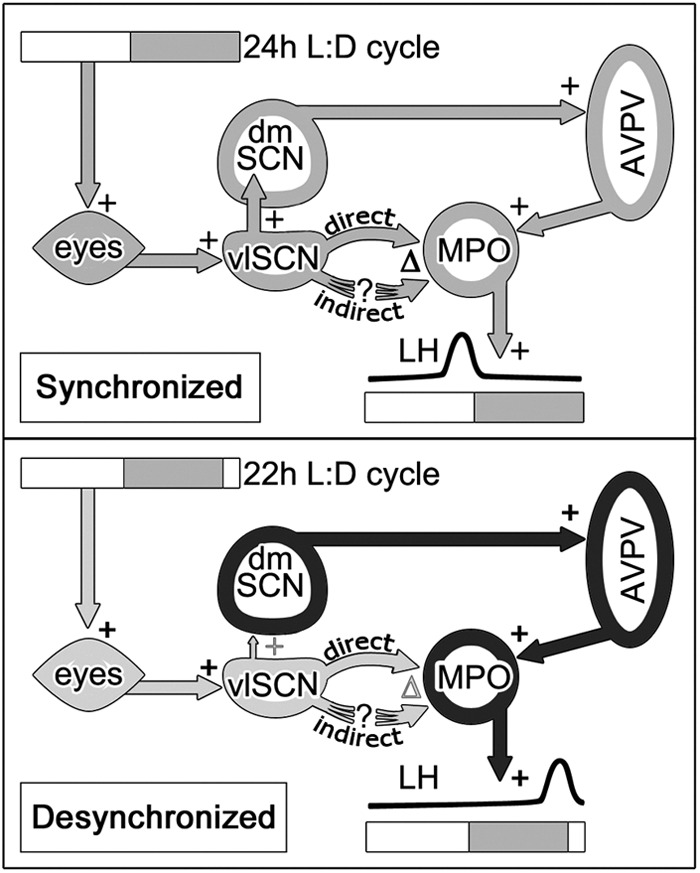
Model of subregional SCN regulation of the LH surge. Top, Synchronized animals receive light input, which entrains the vlSCN. The vlSCN and dmSCN are coupled and send projections to hypothalamic regions involved in timing and shaping the LH surge. Bottom, In desynchronized females, the vlSCN entrains to light signals but the dmSCN is weakly coupled to the vlSCN. Downstream signals are then sent on different phases (respectively indicated in black and light gray), leading to LH surges timed by the dmSCN through AVPV kisspeptin but modulated (via monosynaptic or multisynaptic pathways) by the influence of as-yet-uncertain vlSCN modulators of GnRH cell activity. +, Stimulatory connections; Δ, modulatory connections.
The role of VP and VIP peptides as putative SCN signals triggering the LH surge has been addressed by several laboratories. VP can have strong stimulatory effects on GnRH and LH release (43–45) and induce LH surges in SCNX animals (2). These effects of VP appear to be indirect because VP receptors are barely detectable in GnRH neurons in female rats (46), and although evidence for VP innervation of GnRH cells has been found in the diurnal Nile rat, it is not clear that these fibers are of SCN origin (47). In contrast, vasopressinergic, but not VIPergic, fibers that are likely of SCN origin project to and synapse on kisspeptin cells in the AVPV, kisspeptin cells in this region express the VP receptor V1a, and the expression of this receptor in the AVPV is increased by estrogen in OVX rats (40, 46, 48). These results, together with the association of the timing of Kiss1 expression and the LH surge with clock gene activity in the VP-rich dmSCN that we describe, are consistent with a critical role of SCN VPergic cells in the gating of the LH surge though the regulation of Kiss1-expressing cells.
SCN VIPergic efferent fibers, on the other hand, project directly to GnRH cells (34). The functional relevance of these projections is suggested by the fact that GnRH cells express the VIP receptor, VIP2 (49), and that VIP-innervated GnRH cells show increased cFos expression at the time of the LH surge (50). The specific role of VIP in the regulation of the LH surge is unclear; VIP can either stimulate (51, 52) or inhibit (53–56) the release of GnRH and LH, depending on the conditions. This discrepancy could in part be due to the fact that the response of GnRH cells to the peptide depends both on estrogen levels and the time of the day (57). Our data show that the timing of the LH surge is not guided by the VIP-rich vlSCN but rather that this region may be setting the phase of extra-SCN oscillators that decode a timing signal emerging from the dmSCN (Fig. 8).
Nearly 20 million Americans work on nocturnal shifts (Department of Labor, 2007), as presumably do many more people worldwide. Women working under this disruptive temporal environment are significantly more likely to suffer severe reproductive ailments including irregular menstrual cycles, reduced fertility, miscarriages, and preterm births (7–9). Although the causes for these increased health risks are unknown, a salient feature in humans under nocturnal shift work is the internal desynchronization of circadian rhythms (58, 59). Progress in understanding the adverse health consequences of circadian internal desynchronization has been slow because of the lack of animal models in which desynchronization can be stably induced. Rats are the only rodent in which the vlSCN and dmSCN can be stably desynchronized, and the result of this desynchronization on the regulation of circadian outputs bears remarkable similarities with the regulation of outputs in humans under forced-desynchrony protocols (24, 25). Thus, the forced desynchronized rat offers a unique opportunity to not only dissect out the independent circadian outputs of the vlSCN and dmSCN but also to study the mechanisms underlying adverse physiological consequences of circadian internal desynchrony in a genetically, neurologically, and pharmacologically intact animal. Our results clearly show that circadian internal desynchronization leads to a timing of the LH surge that is at odds with the LD cycle and the behavioral and physiological rhythms that are associated with it. Reproductive ailments in women exposed to temporal challenges such as nocturnal shift work may emerge from a similar misalignment between an LH surge timed by a clock that fails to synchronize to the nocturnal shift and physiological processes that adapt to this abnormal temporal environment.
Acknowledgments
We thank Drs. Robert Steiner and Don Clifton for fruitful discussions about our work and recombinant plasmids and Don Hamlin at Dr. Scott Wilbur's lab for help with iodinations.
This work was supported by National Institutes of Health Grants R01MH075016 and R03 HD061853 (to H.O.d.l.D.); National Science Foundation Doctoral Dissertation Improvement Grants in the Directorate for Biological Sciences 0909716 (to H.O.d.l.D. and B.S.); E.M. was supported by the Mary Gates Research Scholarship for Undergraduates.
Disclosure Summary: The authors declare no conflict of interest.
Footnotes
- AVPV
- Anteroventral periventricular nucleus of the hypothalamus
- DAPI
- 4′,6′-diamino-2-phenylindole
- dm
- dorsomedial
- E2
- estradiol
- FITC
- fluorescein isothiocyanate
- HPG
- hypothalamo-pituitary-gonadal
- ISH
- in situ hybridization
- LD
- light-dark cycle
- LD22
- 11-h light, 11-h dark cycle
- LD24
- 12-h light, 12-h dark cycle
- MPO
- medial preoptic area
- OVX
- ovariectomized
- SCN
- suprachiasmatic nucleus
- SCNX
- SCN-lesioned
- VIP
- vasoactive intestinal polypeptide
- vl
- ventrolateral
- VP
- arginine-vasopressin.
References
- 1. Caligaris L, Astrada JJ, Taleisnik S. 1971. Release of luteinizing hormone induced by estrogen injection into ovariectomized rats. Endocrinology 88:810–815 [DOI] [PubMed] [Google Scholar]
- 2. Palm IF, Van Der Beek EM, Wiegant VM, Buijs RM, Kalsbeek A. 1999. Vasopressin induces a luteinizing hormone surge in ovariectomized, estradiol-treated rats with lesions of the suprachiasmatic nucleus. Neuroscience 93:659–666 [DOI] [PubMed] [Google Scholar]
- 3. Klein DC, Moore RY, Reppert SM. 1991. Suprachiasmatic nucleus: the mind's clock. 1 ed New York: Oxford University Press [Google Scholar]
- 4. Coen CW, MacKinnon PC. 1980. Lesions of the suprachiasmatic nuclei and the serotonin-dependent phasic release of luteinizing hormone in the rat: effects on drinking rhythmicity and on the consequences of preoptic area stimulation. J Endocrinol 84:231–236 [DOI] [PubMed] [Google Scholar]
- 5. Edwards RG. 1981. Test-tube babies, 1981. Nature 293:253–256 [DOI] [PubMed] [Google Scholar]
- 6. Cahill DJ, Wardle PG, Harlow CR, Hull MG. 1998. Onset of the preovulatory luteinizing hormone surge: diurnal timing and critical follicular prerequisites. Fertil Steril 70:56–59 [DOI] [PubMed] [Google Scholar]
- 7. Labyak S, Lava S, Turek F, Zee P. 2002. Effects of shiftwork on sleep and menstrual function in nurses. Health Care Women Int 23:703–714 [DOI] [PubMed] [Google Scholar]
- 8. Schernhammer ES, Kroenke CH, Laden F, Hankinson SE. 2006. Night work and risk of breast cancer. Epidemiology 17:108–111 [DOI] [PubMed] [Google Scholar]
- 9. Knutsson A. 2003. Health disorders of shift workers. Occup Med (Lond) 53:103–108 [DOI] [PubMed] [Google Scholar]
- 10. de la Iglesia HO, Schwartz WJ. 2006. Timely ovulation: Circadian regulation of the female hypothalamo-pituitary-gonadal axis. Endocrinology 147:1148–1153 [DOI] [PubMed] [Google Scholar]
- 11. Popa SM, Clifton DK, Steiner RA. 2008. The role of kisspeptins and GPR54 in the neuroendocrine regulation of reproduction. Annu Rev Physiol 70:213–238 [DOI] [PubMed] [Google Scholar]
- 12. Clarkson J, d'Anglemont de Tassigny X, Moreno AS, Colledge WH, Herbison AE. 2008. Kisspeptin-GPR54 signaling is essential for preovulatory gonadotropin-releasing hormone neuron activation and the luteinizing hormone surge. J Neurosci 28:8691–8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robertson JL, Clifton DK, de la Iglesia HO, Steiner RA, Kauffman AS. 2009. Circadian regulation of Kiss1 neurons: implications for timing the preovulatory gonadotropin-releasing hormone/luteinizing hormone surge. Endocrinology 150:3664–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kauffman AS, Gottsch ML, Roa J, Byquist AC, Crown A, Clifton DK, Hoffman GE, Steiner RA, Tena-Sempere M. 2007. Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology 148:1774–1783 [DOI] [PubMed] [Google Scholar]
- 15. Kinoshita M, Tsukamura H, Adachi S, Matsui H, Uenoyama Y, Iwata K, Yamada S, Inoue K, Ohtaki T, Matsumoto H, Maeda K. 2005. Involvement of central metastin in the regulation of preovulatory luteinizing hormone surge and estrous cyclicity in female rats. Endocrinology 146:4431–4436 [DOI] [PubMed] [Google Scholar]
- 16. Clarkson J, Herbison AE. 2006. Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology 147:5817–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Irwig MS, Fraley GS, Smith JT, Acohido BV, Popa SM, Cunningham MJ, Gottsch ML, Clifton DK, Steiner RA. 2004. Kisspeptin activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology 80:264–272 [DOI] [PubMed] [Google Scholar]
- 18. Lapatto R, Pallais JC, Zhang D, Chan YM, Mahan A, Cerrato F, Le WW, Hoffman GE, Seminara SB. 2007. Kiss1−/− mice exhibit more variable hypogonadism than Gpr54−/− mice. Endocrinology 148:4927–4936 [DOI] [PubMed] [Google Scholar]
- 19. Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MB, Crowley WF, Jr, Aparicio SA, Colledge WH. 2003. The GPR54 gene as a regulator of puberty. N Engl J Med 349:1614–1627 [DOI] [PubMed] [Google Scholar]
- 20. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. 2003. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 100:10972–10976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adachi S, Yamada S, Takatsu Y, Matsui H, Kinoshita M, Takase K, Sugiura H, Ohtaki T, Matsumoto H, Uenoyama Y, Tsukamura H, Inoue K, Maeda K. 2007. Involvement of anteroventral periventricular metastin/kisspeptin neurons in estrogen positive feedback action on luteinizing hormone release in female rats. J Reprod Dev 53:367–378 [DOI] [PubMed] [Google Scholar]
- 22. Moore RY, Speh JC, Leak RK. 2002. Suprachiasmatic nucleus organization. Cell Tissue Res 309:89–98 [DOI] [PubMed] [Google Scholar]
- 23. Schwartz MD, Wotus C, Liu T, Friesen WO, Borjigin J, Oda GA, de la Iglesia HO. 2009. Dissociation of circadian and light inhibition of melatonin release through forced desynchronization in the rat. Proc Natl Acad Sci USA 106:17540–17545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee ML, Swanson BE, de la Iglesia HO. 2009. Circadian timing of REM sleep is coupled to an oscillator within the dorsomedial suprachiasmatic nucleus. Curr Biol 19:848–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cambras T, Weller JR, Angles-Pujoràs M, Lee ML, Christopher A, Díez-Noguera A, Krueger JM, de la Iglesia HO. 2007. Circadian desynchronization of core body temperature and sleep stages in the rat. Proc Natl Acad Sci USA 104:7634–7639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de la Iglesia HO, Cambras T, Schwartz WJ, Díez-Noguera A. 2004. Forced desynchronization of dual circadian oscillators within the rat suprachiasmatic nucleus. Curr Biol 14:796–800 [DOI] [PubMed] [Google Scholar]
- 27. Schwartz WJ. 2009. Circadian rhythms: a tale of two nuclei. Curr Biol 19:R460–R462 [DOI] [PubMed] [Google Scholar]
- 28. de la Iglesia HO. 2007. In situ hybridization of suprachiasmatic nucleus slices. In: Rosato E, ed. Methods in molecular biology. Totowa, NJ: Humana Press; 513–531 [DOI] [PubMed] [Google Scholar]
- 29. Silver R, LeSauter J, Tresco PA, Lehman MN. 1996. A diffusible coupling signal from the transplanted suprachiasmatic nucleus controlling circadian locomotor rhythms. Nature 382:810–813 [DOI] [PubMed] [Google Scholar]
- 30. Meyer-Bernstein EL, Jetton AE, Matsumoto SI, Markuns JF, Lehman MN, Bittman EL. 1999. Effects of suprachiasmatic transplants on circadian rhythms of neuroendocrine function in golden hamsters. Endocrinology 140:207–218 [DOI] [PubMed] [Google Scholar]
- 31. de la Iglesia HO, Meyer J, Schwartz WJ. 2003. Lateralization of circadian pacemaker output: activation of left- and right-sided luteinizing hormone-releasing hormone neurons involves a neural rather than a humoral pathway. J Neurosci 23:7412–7414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van der Beek EM, Horvath TL, Wiegant VM, Van den Hurk R, Buijs RM. 1997. Evidence for a direct neuronal pathway from the suprachiasmatic nucleus to the gonadotropin-releasing hormone system: combined tracing and light and electron microscopic immunocytochemical studies. J Comp Neurol 384:569–579 [DOI] [PubMed] [Google Scholar]
- 33. de la Iglesia HO, Blaustein JD, Bittman EL. 1995. The suprachiasmatic area in the female hamster projects to neurons containing estrogen receptors and GnRH. Neuroreport 6:1715–1722 [DOI] [PubMed] [Google Scholar]
- 34. van der Beek EM, Wiegant VM, van der Donk HA, van den Hurk R, Buijs RM. 1993. Lesions of the suprachiasmatic nucleus indicate the presence of a direct vasoactive intestinal polypeptide-containing projection to gonadotrophin-releasing hormone neurons in the female rat. J Neuroendocrinol 5:137–144 [DOI] [PubMed] [Google Scholar]
- 35. Card JP, Brecha N, Karten HJ, Moore RY. 1981. Immunocytochemical localization of vasoactive intestinal polypeptide-containing cells and processes in the suprachiasmatic nucleus of the rat: light and electron microscopic analysis. J Neurosci 1:1289–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chappell PE. 2005. Clocks and the black box: circadian influences on gonadotropin-releasing hormone secretion. J Neuroendocrinol 17:119–130 [DOI] [PubMed] [Google Scholar]
- 37. Hickok JR, Tischkau SA. 2010. In vivo circadian rhythms in gonadotropin-releasing hormone neurons. Neuroendocrinology 91:110–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS. 2004. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci USA 101:5339–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sellix MT, Menaker M. 2010. Circadian clocks in the ovary. Trends Endocrinol Metab 21:628–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Williams WP, 3rd, Jarjisian SG, Mikkelsen JD, Kriegsfeld LJ. 2011. Circadian control of kisspeptin and a gated GnRH response mediate the preovulatory luteinizing hormone surge. Endocrinology 152:595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gibson EM, Humber SA, Jain S, Williams WP, 3rd, Zhao S, Bentley GE, Tsutsui K, Kriegsfeld LJ. 2008. Alterations in RFamide-related peptide expression are coordinated with the preovulatory luteinizing hormone surge. Endocrinology 149:4958–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Christian CA, Moenter SM. 2010. The neurobiology of preovulatory and estradiol-induced gonadotropin-releasing hormone surges. Endocr Rev 31:544–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palm IF, van der Beek EM, Wiegant VM, Buijs RM, Kalsbeek A. 2001. The stimulatory effect of vasopressin on the luteinizing hormone surge in ovariectomized, estradiol-treated rats is time-dependent. Brain Res 901:109–116 [DOI] [PubMed] [Google Scholar]
- 44. Funabashi T, Shinohara K, Mitsushima D, Kimura F. 2000. Gonadotropin-releasing hormone exhibits circadian rhythm in phase with arginine-vasopressin in co-cultures of the female rat preoptic area and suprachiasmatic nucleus. J Neuroendocrinol 12:521–528 [DOI] [PubMed] [Google Scholar]
- 45. Funabashi T, Aiba S, Sano A, Shinohara K, Kimura F. 1999. Intracerebroventricular injection of arginine-vasopressin V1 receptor antagonist attenuates the surge of luteinizing hormone and prolactin secretion in proestrous rats. Neurosci Lett 260:37–40 [DOI] [PubMed] [Google Scholar]
- 46. Kalamatianos T, Kallóó I, Goubillon ML, Coen CW. 2004. Cellular expression of V1a vasopressin receptor mRNA in the female rat preoptic area: effects of oestrogen. J Neuroendocrinol 16:525–533 [DOI] [PubMed] [Google Scholar]
- 47. Mahoney MM, Smale L. 2005. Arginine vasopressin and vasoactive intestinal polypeptide fibers make appositions with gonadotropin-releasing hormone and estrogen receptor cells in the diurnal rodent Arvicanthis niloticus. Brain Res 1049:156–164 [DOI] [PubMed] [Google Scholar]
- 48. Vida B, Deli L, Hrabovszky E, Kalamatianos T, Caraty A, Coen CW, Liposits Z, Kalló I. 2010. Evidence for suprachiasmatic vasopressin neurones innervating kisspeptin neurones in the rostral periventricular area of the mouse brain: regulation by oestrogen. J Neuroendocrinol 22:1032–1039 [DOI] [PubMed] [Google Scholar]
- 49. Smith MJ, Jiennes L, Wise PM. 2000. Localization of the VIP2 receptor protein on GnRH neurons in the female rat. Endocrinology 141:4317–4320 [DOI] [PubMed] [Google Scholar]
- 50. van der Beek EM, van Oudheusden HJ, Buijs RM, van der Donk HA, van den Hurk R, Wiegant VM. 1994. Preferential induction of c-fos immunoreactivity in vasoactive intestinal polypeptide-innervated gonadotropin-releasing hormone neurons during a steroid-induced luteinizing hormone surge in the female rat. Endocrinology 134:2636–2644 [DOI] [PubMed] [Google Scholar]
- 51. Vijayan E, Samson WK, Said SI, McCann SM. 1979. Vasoactive intestinal peptide: evidence for a hypothalamic site of action to release growth hormone, luteinizing hormone, and prolactin in conscious ovariectomized rats. Endocrinology 104:53–57 [DOI] [PubMed] [Google Scholar]
- 52. Samson WK, Burton KP, Reeves JP, McCann SM. 1981. Vasoactive intestinal peptide stimulates luteinizing hormone-releasing hormone release from median eminence synaptosomes. Regul Pept 2:253–264 [DOI] [PubMed] [Google Scholar]
- 53. Alexander MJ, Clifton DK, Steiner RA. 1985. Vasoactive intestinal polypeptide effects a central inhibition of pulsatile luteinizing hormone secretion in ovariectomized rats. Endocrinology 117:2134–2139 [DOI] [PubMed] [Google Scholar]
- 54. Stobie KM, Weick RF. 1989. Vasoactive intestinal peptide inhibits luteinizing hormone secretion: the inhibition is not mediated by dopamine. Neuroendocrinology 49:597–603 [DOI] [PubMed] [Google Scholar]
- 55. Weick RF, Stobie KM. 1992. Vasoactive intestinal peptide inhibits the steroid-induced LH surge in the ovariectomized rat. J Endocrinol 133:433–437 [DOI] [PubMed] [Google Scholar]
- 56. van der Beek EM, Swarts HJ, Wiegant VM. 1999. Central administration of antiserum to vasoactive intestinal peptide delays and reduces luteinizing hormone and prolactin surges in ovariectomized, estrogen-treated rats. Neuroendocrinology 69:227–237 [DOI] [PubMed] [Google Scholar]
- 57. Christian CA, Moenter SM. 2008. Vasoactive intestinal polypeptide can excite gonadotropin-releasing hormone neurons in a manner dependent on estradiol and gated by time of day. Endocrinology 149:3130–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Haus E, Smolensky M. 2006. Biological clocks and shift work: circadian dysregulation and potential long-term effects. Cancer Causes Control 17:489–500 [DOI] [PubMed] [Google Scholar]
- 59. Sack RL, Blood ML, Lewy AJ. 1992. Melatonin rhythms in night shift workers. Sleep 15:434–441 [DOI] [PubMed] [Google Scholar]
- 60. Paxinos G, Watson C. 1998. The rat brain in stereotaxic coordinates. 4th ed San Diego: Academic Press [Google Scholar]