

Abstract
Delirium is a profound, acute confusional state that leads to long-term cognitive decline. Increased anticholinergic medications and prior dementia, in which basal forebrain cholinergic degeneration is a prominent feature, both predict delirium. Thus, cholinergic hypoactivity is thought to be important in cognitive dysfunction during delirium, and acute systemic inflammation is a major trigger for this dysfunction. Here, we hypothesize that decreased cholinergic function confers increased susceptibility to acute inflammation-induced cognitive deficits. We used the murine-p75-saporin immunotoxin (mu-p75-sap) to induce selective lesions of the basal forebrain cholinergic system in mice, mimicking early dementia-associated cholinergic loss, and superimposed systemic inflammation using low-dose bacterial lipopolysaccharide (LPS). Intracerebroventricular injection of mu-p75-sap produced depletion of cholinergic neurons in the basal forebrain and decreased innervation of the hippocampus, but left performance on hippocampal-dependent reference and working memory tasks relatively intact. However, systemic LPS (100 μg/kg) induced acute and transient working memory deficits in lesioned animals without effect in unlesioned controls. CNS inflammatory responses were similar in normal and lesioned animals and the acetylcholinesterase inhibitor, donepezil (1 mg/kg), protected against the acute cognitive deficits in this cholinergic-dependent task. Thus, cholinergic depletion predisposes to development of acute cognitive deficits upon subsequent systemic inflammatory insult. These data provide a useful model for examining interactions between acute systemic inflammation and chronic cholinergic hypofunction in delirium and have implications for the recent trial of rivastigmine in sepsis-associated delirium.
Introduction
Delirium is a profound and highly prevalent neuropsychiatric syndrome of acute onset that is characterized by disturbances in attention, memory, orientation, and perception. The major risk factors for delirium are aging and dementia (Inouye, 2006), and it is now clear that episodes of delirium predict long-term cognitive decline (Witlox et al., 2010). A lack of biologically relevant animal models with which to study delirium has hampered progress in investigating the mechanisms underlying this condition. Sepsis can induce delirium in an otherwise healthy individual, but much milder inflammation may induce episodes in those with prior susceptibility. We have recently shown that prior neurodegenerative pathology, including synaptic loss and microglial priming, increases the vulnerability of mice to show acute working memory deficits upon systemic challenge with bacterial endotoxin (lipopolysaccharide, LPS; Murray et al., 2012). Since the nature of these deficits are acute and transient, with impairments in attention, recall, and short-term/working memory, this model displays key core features of delirium as defined by DSM-IV (American Psychiatric Association, 1994) and ICD-10 (World Health Organization, 1992) and will be useful in delineating mechanisms.
Additionally, there is clinical evidence that anticholinergic medications can induce delirium (Tune et al., 1981), and cholinergic hypofunction is a cornerstone of the “final common pathway” hypothesis of delirium (Trzepacz, 2000). Since the basal forebrain cholinergic system shows significant degeneration in Alzheimer's disease and dementia (Davies and Maloney, 1976), and these patient groups are among the most susceptible to delirium, we hypothesized that chronic cholinergic hypofunction increases the susceptibility of individuals to acute cognitive deficits induced by systemic inflammation.
Here we have produced and characterized a model of limited selective lesioning of the basal forebrain cholinergic system using the murine-p75-saporin (mu-p75-sap) immunotoxin, a ribosome-inactivating toxin saporin, bound to an antibody directed against the p75 neurotrophin receptor (Berger-Sweeney et al., 2001; Moreau et al., 2008), which is enriched on basal forebrain cholinergic neurons. We subsequently challenged lesioned and control mice systemically with LPS (100 μg/kg), assessed their differential susceptibility to working memory deficits, and examined the inflammatory and cholinergic dependence of these impairments.
Materials and Methods
Animals and stereotaxic surgery.
Female C57BL/6 mice (Harlan) were housed in groups of five at 21°C with a 12:12 h light-dark cycle with food and water ad libitum. Mice were anesthetized intraperitoneally (i.p.) with Avertin (2,2,2-tribromoethanol; Sigma) and positioned in a stereotaxic frame (Kopf Instruments). Two 1 μl injections of mu-p75-sap (Advanced Targeting Systems) at a concentration of either 0.04 or 0.2 μg/μl (total dosage, 0.08 μg or 0.4 μg) were made into the lateral ventricles using a pulled glass micropipette [coordinates (mm) from bregma: anteroposterior, −0.45; lateral, ±1.0; depth, −1.9]. Control animals were injected with sterile PBS. Following injection, the needle remained in place for 5 min to minimize reflux. Animals recovered fully from surgical procedures (20 d) before starting cognitive experiments. All procedures were performed under Republic of Ireland Department of Health and Children license and efforts were made to minimize the suffering and number of animals used.
Intraperitoneal challenges.
For some cognitive and inflammatory experiments, animals were injected intraperitoneally with 100 μg/kg LPS (Salmonella equine abortus L5886, Sigma) in sterile saline. Some animals received donepezil hydrochloride (1 mg/kg, i.p.; Aricept, Pfizer), in endotoxin-free water, 1 h after LPS. The muscarinic antagonist scopolamine hydrobromide (Tocris Bioscience) was injected intraperitoneally at 1 mg/kg in sterile saline, administered 1 h before testing in a cohort of naive C57BL/6 mice. In all cases, control animals were administered sterile saline.
Cognitive testing.
We used the “paddling” Y-maze visuospatial task, as previously described (Cunningham et al., 2009), to investigate hippocampal-dependent reference memory following basal forebrain cholinergic lesioning. Mice were placed in one of two possible start arms in a pseudo-randomized sequence for 10 trials, and the groups were counter-balanced with respect to the location of the exit. In a subset of animals, the task was conducted for 15 trials. A correct trial was defined as entry to the exit arm without entering other arms. Hippocampal-dependent working memory was assessed as previously described in our “paddling” T-maze alternation task (Murray et al., 2012). Each mouse was placed in the start arm of the maze with one arm blocked such that they were forced to make a left (or right) turn to exit the maze, selected in a pseudorandom sequence (equal left and right turns, ≤2 consecutively to same arm). The mouse was held in a holding cage for 25 s (intratrial interval) during which time the door was removed and the exit tube was switched to the alternate arm. The mouse was then replaced in the start arm and could choose either arm. Alternation from its original turn constitutes a correct trial. On choosing incorrectly, the mice were allowed to self-correct to find the correct exit arm. Animals were trained for blocks of 10 trials (20 min intertrial interval) until reaching performance criterion. Most animals reached alternation of >80% but no animals were challenged with LPS or saline unless they showed consecutive days performing at 70% or above, never <70% in the last block of five trials before challenge and no evidence of a side preference.
Histochemistry and immunohistochemistry.
At the end of behavioral experiments (42–50 d) animals were transcardially perfused with heparinized saline for acetylcholinesterase (AChE) histochemistry and additionally with 10% formalin for choline acetyltransferase (ChAT) immunohistochemistry. AChE histochemistry was performed on fresh frozen tissue (20 μm coronal sections) at the level of the hippocampus using the thiocholine method (Paxinos and Watson, 1998). Coronal sections (10 μm) of paraffin wax-embedded tissue were cut on a microtome, at the level of the medial septum. Sections were labeled with goat anti-human ChAT (1/500; Millipore) or goat anti-IBA1 (Abcam), after pretreatment with 1% H2O2/methanol (20 min), microwaving in citrate buffer (pH 6) for 2 × 5 min and preblocking with normal rabbit serum. IBA-1 sections were pretreated with 0.04% pepsin for 20 min before blocking. Thereafter, the ABC method was used as previously described (Cunningham et al., 2005). Both AChE density and cholinergic cell counting was performed using ImageJ software (rsbweb.nih.gov). ChAT-positive cells were identified and counted at three predefined areas moving rostrocaudally through the medial septum of all animal groups (anteroposterior +1.10–1.05, 1.00–0.95, 0.90–0.85).
RNA extraction and quantitative PCR.
Tissue for RNA extraction was precisely scalpel-dissected from coronal sections at the appropriate level after rapid transcardial perfusion with saline. Samples weighed 20–30 mg. Isolation of total RNA, cDNA synthesis, and analysis of transcription using relative quantification after normalization to GAPDH expression, have been performed as previously described (Cunningham et al., 2005). Total RNA was extracted from brain samples using Qiagen RNeasy Plus mini kits (Qiagen) and yields determined by spectrophotometry at 260 nm. For Taqman quantitative PCR, cDNA was generated from total RNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer's instructions. Two hundred nanograms of total RNA were reverse transcribed in a 10 μl reaction volume. One microliter of the RT reaction (equivalent to 20 ng of RNA) was used for PCR. Primer pairs were as follows: CXCL1, forward 5′-CACCCAAACCGAAGTCATAGC-3′ and reverse 5′-AATTTTCTGAACCAA GGGAGCTT-3′; ChAT, forward 5′-CAGCCAATTGGGTCTCTGAATAC-3′ and reverse 5′-GAAGTGCTGCCGAGCAAAG-3′. Other primer pairs were as previously described (Palin et al., 2008).
Statistical analyses.
Behavioral, ChAT-positive cell loss and CNS transcription data were analyzed by two-way ANOVA with Bonferroni post hoc analysis after significant main effects. AChE density measurements were analyzed by two-tailed t test.
Results
Cognitive and neuropathological characterization of saporin lesions
We assessed animals injected with 0.08 μg and 0.4 μg of mu-p75-sap on tests of reference and working memory. PBS and mu-p75-sap-treated animals were tested in the visuo-spatial reference memory Y-maze task (Fig. 1A) for two blocks of five trials. There was a main effect of treatment (F = 3.147, df 2, 76; p < 0.05) by two-way repeated-measures ANOVA and Bonferroni post hoc tests revealed that in the second block of five trials, the 0.4 μg mu-p75-sap group showed significantly fewer correct trials compared with other groups. In a subset of animals (PBS, n = 14; 0.08 μg, n = 20; 0.4 μg, n = 10) tested for 15 trials, there were no differences in performance in the final block of trials, indicating that the higher dose mu-p75-sap-lesioned animals can learn the task but do so at a slower rate than other groups.
Figure 1.

Cognitive and neuropathological consequences of saporin lesions. A, Visuo-spatial reference memory, assessed in Y-maze (10 trials: PBS, n = 24; 0.08 μg, n = 30; 0.4 μg, n = 25; 15 trials: PBS, n = 14, 0.08 μg, n = 20; 0.4 μg, n = 10). Main effects by two-way repeated-measures ANOVA are shown in the text. Significant post hoc differences between 0.4 μg mu-p75-sap and all other groups are denoted by *p < 0.05. B, Working memory performance, assessed by T-maze alternation (PBS, n = 29; 0.08 μg, n = 52; 0.4 μg, n = 32). C–E, Representative photomicrographs of ChAT-positive neuronal depletion in the medial septum/diagonal band in PBS (C), 0.08 μg mu-p75-sap (D), and 0.4 μg mu-p75-sap (E). Scale bar: (in E) C–E, 200 μm. F–H, Histochemical AChE enzyme activity using the thiocholine method in the hippocampus of 0.08 μg (G) and 0.4 μg mu-p75-sap-treated (H) animals compared with nonlesioned controls (F). Scale bar: (in H) F–H, 200 μm. I, Correlation between ChAT-positive cells and Y-maze performance in trial block two. J, Mean ChAT-positive cells, in three regions spanning the medial septum/diagonal band, in 0.08 μg mu-p75-sap (n = 13) and PBS controls (n = 6), 50 d postlesion (*p < 0.05, Bonferroni post hoc after significant two-way ANOVA). K, Histochemical AChE enzyme activity in the hippocampus of 0.08 μg mu-p75-sap (n = 15) and PBS controls (n = 5; t test, **p < 0.01). L, ChAT mRNA expression in the medial septum in 0.08 μg mu-p75-sap (n = 10) animals relative to PBS controls (n = 9; t test, *p < 0.05).
Animals were assessed on learning of a T-maze working memory task for 10 blocks of 10 trials (Fig. 1B). There was no effect of lesion on learning of this task, as determined by two-way repeated-measures ANOVA (F = 1.34, df 2, 990; p = 0.2670), although there was a trend toward slower learning at the higher dose (0.4 μg). Cerebellar deficits have been reported with high doses of this toxin (Berger-Sweeney et al., 2001; Moreau et al., 2008) but we observed no motor coordination impairments at either dose upon formal testing (data not shown).
There was clear evidence for loss of ChAT-positive neurons in 0.08 μg and 0.4 μg mu-p75-sap-treated animals, and this was more severe in the 0.4 μg group (Fig. 1C–E). Similarly, AChE histochemistry in the hippocampus, a major projection area from the basal forebrain (Mesulam et al., 1983), showed robust loss of AChE activity in each of the mu-p75-sap treatment groups, with more severe loss at 0.4 μg (Fig. 1F–H). Linear regression analysis indicated a significant correlation between remaining cholinergic cell density and Y-maze performance (Fig. 1I; r2 = 0.36, p = 0.0084). Since our hypothesis was based upon limited cholinergic deficiency that, in itself, did not produce cognitive impairments, the lower dose (0.08 μg) was used for all further experiments. ChAT-positive neurons were quantified in three areas of the medial septum/diagonal band moving rostrocaudally from bregma (Fig. 1J). Significant ChAT depletion was found in mu-p75-sap-lesioned mice compared with PBS controls. Repeated-measures ANOVA revealed a main effect of lesion (F = 7.61, df 1, 34; p = 0.0134) and of rostrocaudal location (F = 65.56, df 2, 34; p < 0.0001), and Bonferroni post hoc tests showed significant differences between PBS and mu-p75-sap at 1.0–0.95 and 0.9–0.85 mm from bregma (p < 0.05). We assessed for nonselective neuronal loss using NeuN immunolabeling in a standardized area of 0.48 mm2 in the intermediate lateral septum (periventricular, proximal to injection site). There were no significant differences in neuronal counts at this location (324 ± 14 for mu-p75-sap vs 320 ± 14 for PBS).
Histochemical AChE enzyme activity was decreased in the hippocampus of mu-p75-sap-lesioned animals (0.08 μg; Fig. 1K). There was significantly less AChE activity in lesioned animals (t test, p < 0.01), although four lesioned animals showed limited AChE loss. These were subsequently omitted from analysis of cognitive performance. Assessment of ChAT mRNA in the medial septum/diagonal band also revealed significantly less ChAT expression in mu-p75-sap-lesioned animals compared with controls (t test, p < 0.05).
Microglial response to cholinergic lesion
Quantitative PCR for the proinflammatory cytokine TNF-α (Fig. 2A) and scavenger receptor A2 (SRA2; Fig. 2B), showed that there was a statistically significant acute microglial activation at 3 d postinjection of mu-p75-sap (p < 0.001 by Bonferroni post hoc after significant one-way ANOVA) which had completely resolved at 35 d postinjection. Similarly, IBA-1 staining for microglia showed clear evidence of activation in both the medial septum and in the dentate gyrus of the hippocampus at 6 d postlesion (Fig. 2E,F), but not in PBS-injected animals or in mu-p75-sap animals at 40 d postlesion.
Figure 2.
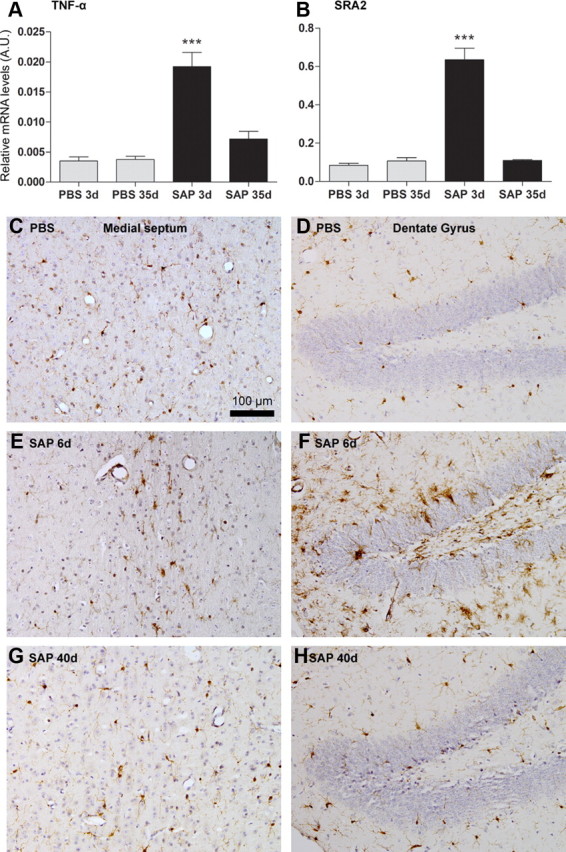
Acute microglial response to cholinergic lesion. A, B, Quantitative PCR data for TNF-α and SRA2 at 3 and 35 d postinjection of PBS or mu-p75-saporin. ***p < 0.001 by Bonferroni post hoc test after a significant one-way ANOVA. Representative micrographs of microglial IBA-1 immunolabeling in the medial septum and dentate gyrus 6 d after PBS injection (C, D) and 6 d (E, F) or 40 d (G, H) after mu-p75-sap (0.08 μg) injection. Scale bar: (in C) C–H, 100 μm.
LPS reveals vulnerability of cholinergic-dependent working memory
Mu-p75-sap-lesioned (0.08 μg) and control animals, trained on T-maze working memory were challenged with saline or LPS at 40 ± 2 d postlesion. Lesioned animals challenged systemically with LPS (100 μg/kg) showed marked working memory deficits (Fig. 3A), peaking 3 h postchallenge and persisting at 7 h. Neither mu-p75-sap alone nor LPS alone was sufficient to induce such deficits. Two-way repeated-measures ANOVA revealed a main effect of treatment (F = 3.93, df 3, 240; p = 0.015) and an interaction between treatment and time (F = 1.89, df 18, 240; p = 0.0173). Bonferroni post hoc tests revealed that mu-p75-sap + LPS animals were impaired with respect to all other groups at 3 h (p < 0.001) and 7 h (p < 0.05). Since this task is scored on correct choices rather than latency it cannot be confounded by motor deficits.
Figure 3.
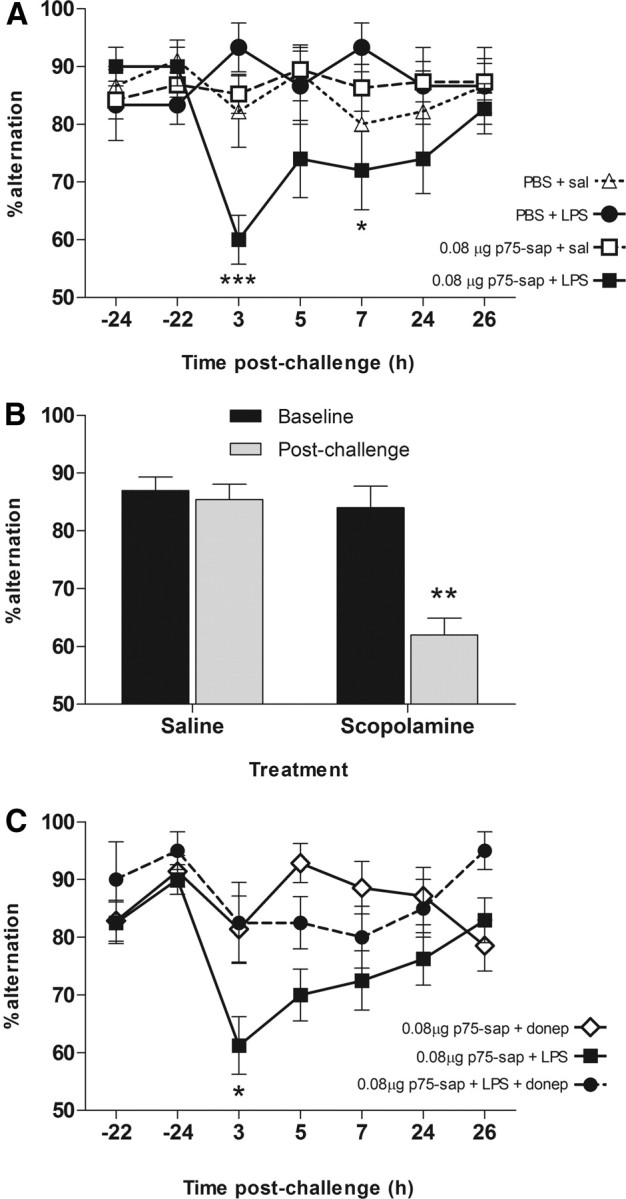
LPS-induced working memory deficits and cholinergic dependence. A, T-maze alternation of PBS or saporin-lesioned animals challenged intraperitoneally with LPS (100 μg/kg) or sterile saline 40 ± 2 d after surgery (n: PBS + sal, 9; PBS + LPS, 6; SAP + sal, 19; SAP + LPS, 10). Bonferroni post hoc tests after significant two-way repeated-measures ANOVA are indicated by ***p < 0.001 and *p < 0.05. B, Cholinergic dependence of T-maze performance was verified by testing 1 h postsystemic challenge with scopolamine hydrobromide (1 mg/kg, i.p., n = 10) or saline (n = 13). Significant interaction between time and treatment by repeated-measures ANOVA is indicated by **p < 0.01. C, Working memory deficits 3 h post-LPS (100 μg/kg) were partially protected against by the acetylcholinesterase inhibitor donepezil (1 mg/kg, i.p.), administered 1 h after LPS. Statistically significant differences by Bonferroni post hoc test after significant main effects by repeated-measures ANOVA are denoted by *p < 0.05 (p75-sap + donepezil n = 14; p75-sap + LPS n = 16; p75-sap + LPS + donepezil n = 8).
To verify cholinergic dependence of this T-maze, we trained naive C57BL/6 mice and administered the muscarinic antagonist scopolamine hydrobromide (1 mg/kg i.p.) or sterile saline. Scopolamine induced a marked impairment in working memory compared with saline controls (Fig. 3B). Repeated-measures ANOVA revealed main effects of block (F = 16.74, df 1, 21; p = 0.0005) and treatment (F = 20.18, df 1, 21; p = 0.0002) and a significant interaction (F = 12.65, df 1, 21; p = 0.0019), demonstrating cholinergic dependence of this task. We then assessed the ability of the acetylcholinesterase inhibitor, donepezil, to block the LPS-induced deficits. Animals were challenged systemically with LPS and received donepezil (1 mg/kg i.p.) or vehicle 1 h later. Donepezil afforded significant, though incomplete, protection against the LPS-induced working memory deficits without effect in normal animals (Fig. 3C). Repeated-measures ANOVA analysis of the LPS-treated groups revealed significant main effects of treatment (F = 12.17, df 1, 132; p = 0.0021). Bonferroni post hoc tests revealed a significant difference between mu-p75-sap + LPS and mu-p75-sap + LPS + donepezil at 3 h post-LPS challenge (p < 0.05). Thus, acetylcholinesterase inhibition partially protects against the LPS-induced deficits in animals with existing cholinergic hypofunction.
Microglia are not primed after cholinergic depletion
We examined the CNS transcription of a number of cytokine mRNA species in the medial septum and hippocampus 3 h after systemic challenge with LPS (100 μg/kg) to assess for microglial priming in lesioned animals (Fig. 4). Systemic LPS induces robust upregulation of the proinflammatory genes IL-1β, TNFα, IL-6, and CXCL1 in the medial septum/diagonal band and hippocampus. Since CNS detection of cytokine protein is unreliable after 100 μg/kg LPS, we also assessed the secondary response genes PTX-3 and TNFAIP2 as indicators of IL-1β and TNF-α action. Two-way ANOVA revealed a main effect of LPS in all cases (F ≥ 8.17, df 1, 15; p < 0.0027). However, there was no effect of lesion in the case of any of these genes. Thus, 40 d after these limited cholinergic lesions, systemic LPS induces CNS inflammation, but microglia are not primed to respond more robustly to this inflammatory challenge.
Figure 4.

CNS inflammatory response to systemic LPS in lesioned animals. Expression of proinflammatory genes IL-1β, TNFα, IL-6, CXCL1, and downstream genes PTX-3 and TNFAIP2 in septum and hippocampus 3 h after LPS (100 μg/kg, i.p.) or saline (40 d postlesion). n = 4 for PBS + saline and n = 5 for other groups. Main effects of LPS (two-way ANOVA) are described in main text.
Discussion
In the current study we have shown that moderate basal forebrain cholinergic hypofunction leaves animals susceptible to systemic inflammation-induced acute working memory deficits. Systemic inflammation alone produced no deficits and, consistent with prior reports of selective cholinergic lesioning (Wrenn et al., 1999), limited cholinergic hypofunction alone was also insufficient. These data demonstrate an important interaction between prior cholinergic pathology and subsequent inflammatory insults, and offer a novel animal model with obvious relevance for delirium during dementia.
We had previously shown, in an animal model of progressive neurodegenerative disease, that microglia are primed to produce exaggerated CNS responses to systemic inflammation (Cunningham et al., 2005), and that these animals are now more susceptible to working memory deficits (Murray et al., 2012). The current study also demonstrates LPS-induced deficits only in those animals with prior neurodegeneration, although in this case, the degeneration is cholinergic. We have shown evidence of acute inflammation postlesioning that, given the considerably lower saporin dosing, is proportionally more subtle than in prior studies (Hunter et al., 2004). There is no evidence that the microglia of the basal forebrain or hippocampus are primed at 35 d, suggesting that although primed microglia may increase the likelihood of inflammation-induced cognitive impairment, this exaggerated inflammatory response is not essential. Therefore, loss of cholinergic modulation becomes the likely predisposing factor. In previous studies we have shown that increased susceptibility is associated with significant loss of presynaptic terminals (Murray et al., 2012), and here we show that it is associated with the loss of cholinergic input to the hippocampus and can be blocked with cholinesterase inhibition. Centrally administered IL-1β can impair memory in a cholinesterase-sensitive manner and can directly diminish acetylcholine outflow (Rada et al., 1991; Matsumoto et al., 2001; Taepavarapruk and Song, 2010). Our data show that systemic inflammation is sufficient to induce acute memory dysfunction in a cholinergic-dependent manner only in the context of existing cholinergic neurodegeneration.
Cholinergic neuromodulation is important in normal cognitive function and loss of this can lead to cognitive and attentional deficits (McGaughy et al., 2000). Anticholinergic medications can induce delirium (Tune et al., 1981), contributing to the consensus that cholinergic hypofunction is a key feature (Trzepacz, 2000) and cholinergic inhibition has been used to mimic delirium in rats (Trzepacz, 1996). The cholinesterase inhibitor rivastigmine was recently tested as a treatment for intensive care unit (ICU) delirium (van Eijk et al., 2010) based both on the known neuromodulatory role of acetylcholine and on its suppression of peripheral macrophage function via vagal stimulation of α7-nicotinic receptors (Tracey, 2007). Agonism at this receptor on microglia can downregulate microglial activation (De Simone et al., 2005) but endogenous acetylcholine has not been shown to inhibit CNS microglia in a context relevant to sepsis. Data in the current study suggest that at least with ∼20% depletion of hippocampal cholinergic innervation, microglial responses to systemic inflammation are not accentuated. Whether more severe hypocholinergia leads to more robust LPS responses remains to be established.
We have shown that a pre-existing cholinergic deficit results in a vulnerability to the cognitive effects of systemic inflammation. The demonstration that acetylcholinesterase inhibition is protective, but only partially, in this paradigm has implications for the reconciliation of inflammatory and cholinergic hypotheses of delirium. The rivastigmine trial was stopped prematurely due to a lack of efficacy and increased mortality and we would hypothesize that procholinergic strategies in sepsis, where individuals lack “hypocholinergic predisposition,” may be less effective than in those on a background of progressive dementia. Dementia is the major risk factor for delirium, and extensive degeneration of the basal forebrain cholinergic neurons is observed in AD (Davies and Maloney, 1976). Even early in disease there is clear evidence of decreased cholinergic innervation of the frontal cortex (Ikonomovic et al., 2007). Consistent with previous studies with mu-p75-sap (Moreau et al., 2008), the current lesion produced limited neuronal loss in the nucleus basalis and thus, the effects observed here likely rely on dysfunction in the septohippocampal pathway (Mesulam et al., 1983) rather than in the frontal cortex. Further studies with mu-p75-sap injections directly into the nucleus basalis will be necessary to specifically investigate hypocholinergia in the frontal cortex. Nonetheless, we have previously shown that as neurodegenerative pathology becomes more global, so too does the breadth of CNS changes induced by systemic inflammation (Cunningham et al., 2009); thus, while the current model predicts only a subset of symptoms based on its deliberately limited extent, it provides an excellent paradigm in which to interrogate the interaction between systemic inflammation and cholinergic function. We propose that this novel animal model can contribute significantly to our understanding of delirium pathophysiology, which is currently rather limited in both ICU and geriatric settings.
Footnotes
This work was supported by The Wellcome Trust. R.H.F. and A.G. were supported by postgraduate awards from Trinity College Dublin and the Health Research Board, respectively. We thank Anne-Marie O'Connor for technical assistance and Prof. Shane O'Mara for providing donepezil.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Ed 4. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- Berger-Sweeney J, Stearns NA, Murg SL, Floerke-Nashner LR, Lappi DA, Baxter MG. Selective immunolesions of cholinergic neurons in mice: effects on neuroanatomy, neurochemistry, and behavior. J Neurosci. 2001;21:8164–8173. doi: 10.1523/JNEUROSCI.21-20-08164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, Rawlins JN, Perry VH. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65:304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- De Simone R, Ajmone-Cat MA, Carnevale D, Minghetti L. Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflammation. 2005;2:4. doi: 10.1186/1742-2094-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CL, Quintero EM, Gilstrap L, Bhat NR, Granholm AC. Minocycline protects basal forebrain cholinergic neurons from mu p75-saporin immunotoxic lesioning. Eur J Neurosci. 2004;19:3305–3316. doi: 10.1111/j.0953-816X.2004.03439.x. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Isanski BA, Wuu J, Mufson EJ, DeKosky ST. Superior frontal cortex cholinergic axon density in mild cognitive impairment and early Alzheimer disease. Arch Neurol. 2007;64:1312–1317. doi: 10.1001/archneur.64.9.1312. [DOI] [PubMed] [Google Scholar]
- Inouye SK. Delirium in older persons. N Engl J Med. 2006;354:1157–1165. doi: 10.1056/NEJMra052321. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Yoshida M, Watanabe S, Yamamoto T. Involvement of cholinergic and glutamatergic functions in working memory impairment induced by interleukin-1beta in rats. Eur J Pharmacol. 2001;430:283–288. doi: 10.1016/s0014-2999(01)01374-7. [DOI] [PubMed] [Google Scholar]
- McGaughy J, Everitt BJ, Robbins TW, Sarter M. The role of cortical cholinergic afferent projections in cognition: impact of new selective immunotoxins. Behav Brain Res. 2000;115:251–263. doi: 10.1016/s0166-4328(00)00262-x. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6) Neuroscience. 1983;10:1185–1201. doi: 10.1016/0306-4522(83)90108-2. [DOI] [PubMed] [Google Scholar]
- Moreau PH, Cosquer B, Jeltsch H, Cassel JC, Mathis C. Neuroanatomical and behavioral effects of a novel version of the cholinergic immunotoxin mu p75-saporin in mice. Hippocampus. 2008;18:610–622. doi: 10.1002/hipo.20422. [DOI] [PubMed] [Google Scholar]
- Murray C, Sanderson DJ, Barkus C, Deacon RM, Rawlins JN, Bannerman DM, Cunningham C. Systemic inflammation induces acute working memory deficits in the primed brain: relevance for delirium. Neurobiol Aging. 2012;33:603–616.e3. doi: 10.1016/j.neurobiolaging.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palin K, Cunningham C, Forse P, Perry VH, Platt N. Systemic inflammation switches the inflammatory cytokine profile in CNS Wallerian degeneration. Neurobiol Dis. 2008;30:19–29. doi: 10.1016/j.nbd.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Rada P, Mark GP, Vitek MP, Mangano RM, Blume AJ, Beer B, Hoebel BG. Interleukin-1 beta decreases acetylcholine measured by microdialysis in the hippocampus of freely moving rats. Brain Res. 1991;550:287–290. doi: 10.1016/0006-8993(91)91330-4. [DOI] [PubMed] [Google Scholar]
- Taepavarapruk P, Song C. Reductions of acetylcholine release and nerve growth factor expression are correlated with memory impairment induced by interleukin-1beta administrations: effects of omega-3 fatty acid EPA treatment. J Neurochem. 2010;112:1054–1064. doi: 10.1111/j.1471-4159.2009.06524.x. [DOI] [PubMed] [Google Scholar]
- Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzepacz PT. Anticholinergic model for delirium. Semin Clin Neuropsychiatry. 1996;1:294–303. doi: 10.1053/SCNP00100294. [DOI] [PubMed] [Google Scholar]
- Trzepacz PT. Is there a final common neural pathway in delirium? Focus on acetylcholine and dopamine. Semin Clin Neuropsychiatry. 2000;5:132–148. doi: 10.153/SCNP00500132. [DOI] [PubMed] [Google Scholar]
- Tune LE, Damlouji NF, Holland A, Gardner TJ, Folstein MF, Coyle JT. Association of postoperative delirium with raised serum levels of anticholinergic drugs. Lancet. 1981;2:651–653. doi: 10.1016/s0140-6736(81)90994-6. [DOI] [PubMed] [Google Scholar]
- van Eijk MM, Roes KC, Honing ML, Kuiper MA, Karakus A, van der Jagt M, Spronk PE, van Gool WA, van der Mast RC, Kesecioglu J, Slooter AJ. Effect of rivastigmine as an adjunct to usual care with haloperidol on duration of delirium and mortality in critically ill patients: a multicentre, double-blind, placebo-controlled randomised trial. Lancet. 2010;376:1829–1837. doi: 10.1016/S0140-6736(10)61855-7. [DOI] [PubMed] [Google Scholar]
- Witlox J, Eurelings LS, de Jonghe JF, Kalisvaart KJ, Eikelenboom P, van Gool WA. Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta-analysis. JAMA. 2010;304:443–451. doi: 10.1001/jama.2010.1013. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Diagnostic criteria for research. Geneva: World Health Organization; 1992. The ICD-10 classification of mental and behavioural disturbances. [Google Scholar]
- Wrenn CC, Lappi DA, Wiley RG. Threshold relationship between lesion extent of the cholinergic basal forebrain in the rat and working memory impairment in the radial maze. Brain Res. 1999;847:284–298. doi: 10.1016/s0006-8993(99)02099-5. [DOI] [PubMed] [Google Scholar]