

Abstract
A summary of the initial discovery and characterization of the enzyme fatty acid amide hydrolase (FAAH), and the subsequent advancement of an important class of competitive, reversible, potent, and selective inhibitors is presented. Initially explored using substrate-inspired inhibitors bearing electrophilic carbonyls, the examination of α-ketoheterocyle-based inhibitors of FAAH with the benefit of a unique activity-based protein-profiling (ABPP)-based proteome-wide selectivity assay, a powerful in vivo biomarker-based in vivo screen, and subsequent retrospective X-ray cocrystal structures with the enzyme, is summarized. These efforts defined the impact of the central activating heterocycle and its key substituents, provided key simplifications in the C2 acyl side chain and clear interpretations for the unique role and subsequent optimization of the central activating heterocycle, and established the basis for the recent further conformational constraints in the C2 acyl side chain, providing potent, long-acting, orally active FAAH inhibitors.
Keywords: Fatty acid amide hydrolase, FAAH, α-ketoheterocycles, pain, sleep
The characterization of fatty acid amides1 as a fundamental class of endogenous signaling molecules, of which anandamide2 and oleamide3−6 were the early prototypical members, led to the identification of the enzyme fatty acid amide hydrolase (FAAH).7−9 The distribution of FAAH in the central nervous system (CNS)10,11 indicates that the enzyme is localized to degrade signaling fatty acid amides at their site of action, and control the intensity and duration of their effects. FAAH is a member of the amidase signature family of serine hydrolases, and it is the only well-characterized mammalian enzyme in the family that bears an unusual Ser–Ser–Lys catalytic triad. Although FAAH acts on a wide range of amide or ester substrates,7−12 it preferentially hydrolyzes arachidonoyl and oleoyl substrates13 where primary amides are hydrolyzed faster than ethanolamides.13
Recently, FAAH has emerged as an exciting new therapeutic target of clinical interest. Since FAAH inhibition potentiates only an activated signaling pathway thereby increasing the levels of a released signaling molecule, it provides a temporal and spatial pharmacological control not available to classical receptor agonists. Thus, the development of FAAH inhibitors, that raise endogenous fatty acid amide levels only at their released sites of action and sustain their duration of action by blocking their hydrolysis, has emerged as an attractive new approach to pharmacological intervention that avoids the side effects that accompany the blunt force use of more conventional receptor agonists. A series of seminal studies summarized in recent reviews14−17 have detailed the discovery of FAAH as well as its potential to serve as a new therapeutic target for the treatment of a range of disorders including pain, inflammation, and sleep disorders.18−20 Herein, we summarize our discovery and development of α-ketoheterocycle inhibitors of FAAH conducted alongside many of these studies.
Isolation, Structure Determination, and Characterization of Oleamide
In 1994, collaborating groups at Scripps reported the detection of a lipid that progressively appeared in the cerebrospinal fluid (CSF) of sleep-deprived cats and slowly dissipated upon restfulness.5 Given the apparent simplicity of the molecule and the challenges associated with isolating sufficient quantities for unambiguous identification, candidate lipid structures incorporating the established molecular formula (HRMS) were prepared and correlated with the endogenous substance (Figure 1).3−5 By using this approach, the unknown substance was identified as oleamide (1), the primary amide of oleic acid.3,4 In addition to subsequent studies that showed it induces natural or physiological sleep in laboratory animals,5,6,21,22 oleamide was also subsequently found to exhibit cannabinoid-like activity, and potentially behave as an agonist at CB1 (cannabinoid-1) receptors.23,24 The examination of a number of close structural analogues revealed that the sleep-inducing effects are specific for oleamide.4 These studies established oleamide as an endogenous signaling fatty acid amide and provided the second prototypical member of this new and growing class of signaling molecules: fatty acid amides.1 Although less is known about the endogenous synthesis or storage of oleamide25 and key insights into its site(s) of action are still emerging,26−28 the most well understood and extensively studied feature of this new class of signaling molecules is their hydrolysis by the enzyme fatty acid amide hydrolase (FAAH).
Figure 1.
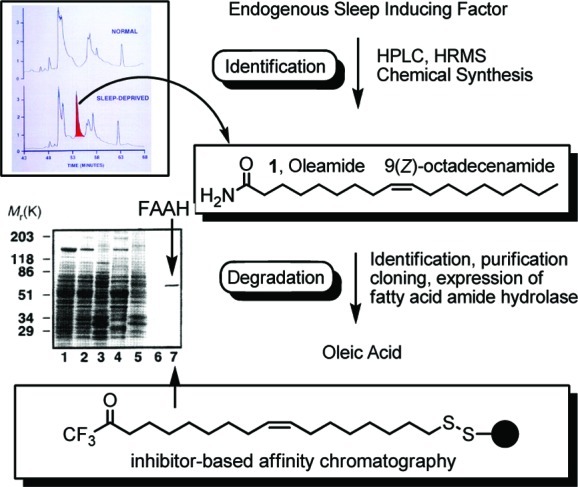
Characterization of endogenous oleamide (1) and FAAH.
Degradation and Regulation of Oleamide: Discovery and Characterization of FAAH
The discovery of oleamide led to the detection4 of enzymatic activity that was responsible for its hydrolysis and inactivation. This enzymatic deactivation of oleamide led to the isolation, purification, sequencing, cloning, expression, and characterization of rat7 and human8 FAAH and its subsequent validation as therapeutic target. The initial purification and characterization of the enzymatic activity that hydrolyzes oleamide was accomplished by inhibitor-bound affinity chromatography (Figure 1).7 The purified rat FAAH was subsequently sequenced, permitting the cloning of the cDNA encoding the enzyme. As mentioned above, the expressed enzyme was found to degrade several fatty acid amides,7 including not only oleamide but also anandamide indicating that the enzyme serves to inactivate the fatty acid amide family of signaling molecules. With the pure enzyme in hand, its substrate scope,13its unusual Ser–Ser–Lys catalytic triad, mechanistic details of fatty acid amide hydrolysis,29−32 and its X-ray structure were established.33
Early Inhibitors: Activated Carbonyl Inhibitors
Shortly following the initial characterization of FAAH, early studies revealed that the endogenous sleep-inducing molecule 2-octyl α-bromoacetoacetate34 is a potent, reversible inhibitor of FAAH (Ki = 0.8 μM) and several related synthetic analogues were established to be effective FAAH inhibitors.35 Similarly, the first series of reversible competitive FAAH inhibitors that were reported also possessed an electrophilic carbonyl within substrate inspired inhibitors, and include the corresponding oleoyl aldehyde, α-ketoamide, α-ketoester, and trifluoromethyl ketone.36 The potency of the inhibitors followed the expected trends characteristic of the electrophilic carbonyls, culminating with the α-ketoesters and trifluoromethyl ketones (Figure 2). The profile of active versus inactive designs explored in this work established FAAH as a candidate serine hydrolase before much was known about the enzyme. An analogous series of derivatives of arachidonic acid and simpler fatty acids was independently examined37 for inhibition of anandamide hydrolase before the two enzymes (oleamide hydrolase and anandamide hydrolase) were recognized as being identical (FAAH).
Figure 2.

Representative initial inhibitors of FAAH.
A more extensive series of trifluoromethyl ketones was subsequently examined and defined structural and conformational properties that contribute to enzyme active site binding and inhibition.38 The lipophilic tail group had significant effects on the enzyme inhibition, and the inhibitors exhibited a now characteristic alkyl chain length parabolic relationship, a feature that has figured heavily in our and other subsequent design efforts. The corresponding methyl ketone was inactive (Ki > 100 μM), illustrating the importance of the electrophilicity of the carbonyl for enzyme inhibition (Figure 3).
Figure 3.

Representative trifluoromethyl ketone inhibitors.
α-Ketoheterocycle Inhibitors
The use of α-ketoheterocycles has emerged as a powerful design concept for the development of inhibitors of serine and cysteine proteases and hydrolases.39 By virtue of possessing electrophilic carbonyls, they form enzyme-stabilized reversible covalent hemiketals or hemithioketals with the enzyme catalytic nucleophile. In our studies and at a time when only a handful of articles on α-ketohetrocycles had been published, a series of representative heterocycles were incorporated into oleyl α-ketoheterocycles.40,41 Based on the observation that incorporation of an additional basic nitrogen into the activating heterocycle correlated with enhanced inhibition, conversion of the traditional benzoxazoles to the oxazolopyridines that contain a fused pyridine afforded a significant increase in potency (>100-fold increase) with N4 incorporation providing the most potent inhibitors (Figure 4). Today, we appreciate that these results not only reflect the enhanced electron-withdrawing properties of oxazolopyridines that increase the electrophilic character of the reactive carbonyl, but they also represents the flexible positioning of a key H-bond acceptor (the pyridyl N), allowing the inhibitors to flip 90° in order to present any of the isomeric pyridyl nitrogens with the opportunity to engage a cytosolic port water hydrogen bond.
Figure 4.

Initial α-ketoheterocycle FAAH inhibitors.
Inhibitors containing the oleoyl cis alkene were more potent than the trans isomers, which in turn were more potent than inhibitors where the alkene was reduced. Systematic variation in the fatty acid saturated side chain of the α-keto-oxazolopyridines showed the greatest potency with C12–C8 chain lengths and exhibited the now characteristic parabolic relationship with chain length (Figure 5). The incorporation of π-unsaturation into the acyl chain increased potency and introduction of a simplifying phenyl ring provided inhibitors with subnanomolar Ki’s, with the most potent inhibitor possessing a Ki of 200 pM. The chain length linking the phenyl group and the oxazolopyridine exhibited an optimal length, indicating that π-unsaturation at the position corresponding to the oleyl alkene might be important40 and such aryl group incorporations are now characteristically found in most FAAH inhibitors.
Figure 5.

Representative modifications in the fatty acid side chain.
Further optimization of the inhibitors,42 simultaneously examining potency and proteome-wide selectivity for FAAH using newly developed ABPP technology,42−46 defined the impact of the C2 acyl chain length separating the oxazole and phenyl ring, and examined the incorporation of an activating 1,3,4-oxadiazole heterocycle.47 This study represented the first use of a proteome-wide selectivity assay conducted alongside traditional efforts to optimize enzyme inhibition potency and led to the expedited discovery of exceptionally potent (Ki < 300 pM) and selective (>100-fold selective) FAAH inhibitors that lacked off target activity and that exhibited in vivo efficacy.47,48 Significantly, the ABPP proteome-wide selectivity assay of all potential competitive enzymes does not require the use of expressed or purified enzymes, no enzyme substrate is needed, no modification of the inhibitors is required, and the relative potency for all competitive enzymes can be quantified, including those that lack known substrates or function.46 An examination43 of oxazole C4 and C5 aryl substituents revealed unique enhancements in potency and selectivity with incorporation of a 2-pyridyl group,49 and the activity of such C5 substituted α-ketooxazoles47 paralleled the relative placement of a heteroatom and their hydrogen bond acceptor properties (Figure 6).
Figure 6.
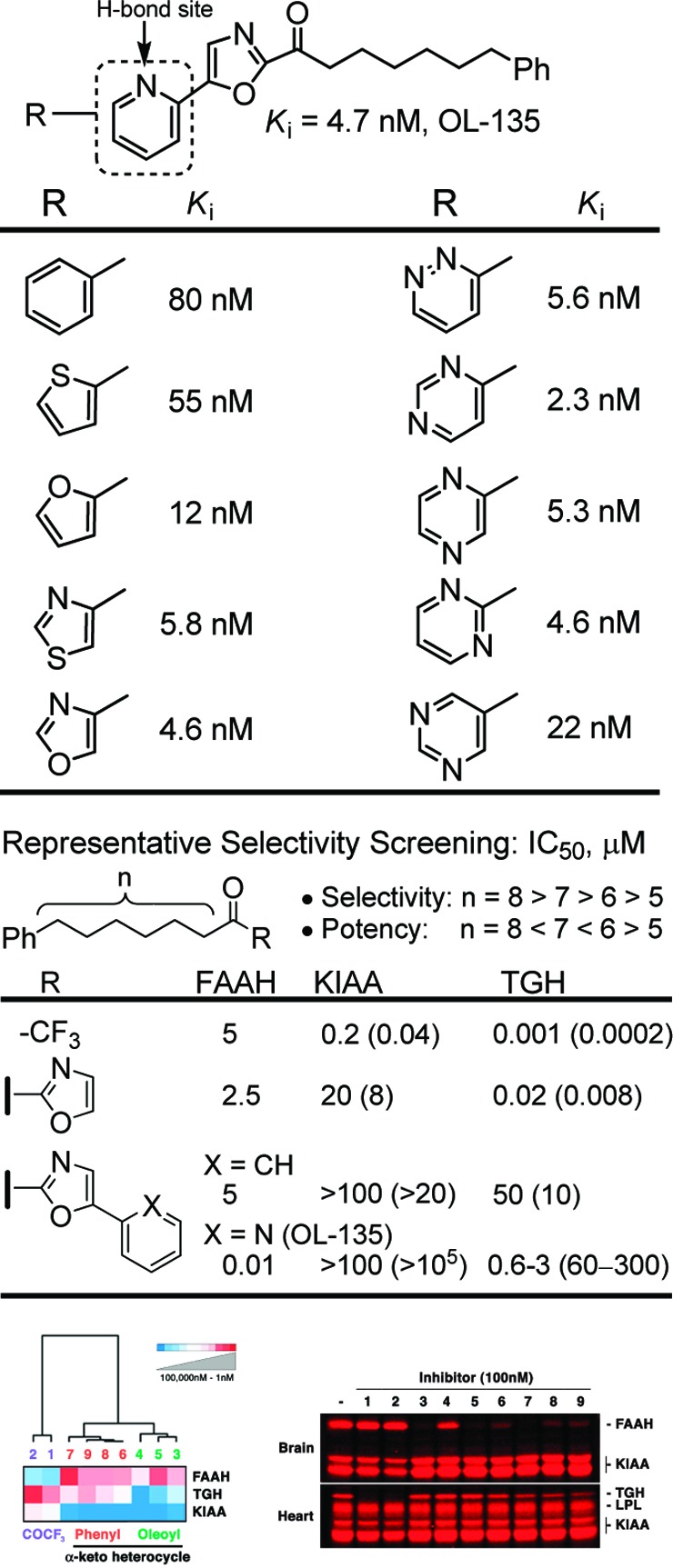
Representative C5 substituted α-ketooxazoles and ABPP selectivity assay results.
An extensive exploration of the α-ketoheterocycle inhibitors of FAAH, providing more than 800 candidates, culminated in one of the most widely recognized lead α-ketoheterocycle compounds disclosed to date (OL-135, Figure 6).47 OL-135 is a potent (Ki = 4.7 nM) and selective (>60–300 fold)42 reversible, competitive FAAH inhibitor that produces analgesia and increases endogenous anandamide levels in vivo.48,50−53 It exhibits analgesic activity in a range of animal models, including the tail flick, hot plate assay, formalin test of noxious pain (first and second phase),48 the mild thermal injury (MIT) model of peripheral pain, the spinal nerve ligation (SNL) and chronic constriction injury (CCI) models of neuropathic pain,50,51 and models of pruritus52 and LPS-induced allodynia,53 with efficacies that match or exceed those of morphine (1–3 mg/kg in MTI/SNL), ibuprofen (100 mg/kg in MTI), or gabapentin (500 mg/kg in SNL) and at doses (10–20 mg/kg, i.p.) that are below many of the commonly used medications. Importantly, OL-135 is not active in FAAH knockout mice, confirming that FAAH is the biological target responsible for the in vivo effects. The compound lacks significant offsite target activity (Cerep profiling), does not bind cannabinoid (CB1 or CB2) or vanilloid (TRP) receptors, and does not significantly inhibit P450 metabolism enzymes or hERG. The in vivo effects in the CCI model of neuropathic pain are blocked by a CB1 or CB2 receptor antagonist and are unaffected by opioid antagonists, consistent with production of increased levels of anandamide at the sites of injury. Moreover, OL-135 has no effect on feeding, mobility, or motor control observed with classical CB1 and CB2 receptor agonists, and it does not produce respiratory depression or the tolerance observed with chronic dosing of opioid agonists.50−53 FAAH inhibition produces efficacious analgesia in each of the pain models examined to date suggesting clinical applications in chronic, neuropathic, and inflammatory pain. This validation of FAAH as a therapeutic target subject to small molecule intervention and the accompanying demonstration that its potentiation of an endogenous signaling molecule that targets a GPCR may avoid the side effects of a more conventional receptor agonist served to motivate many large pharmaceutical and small biotech companies to initiate programs targeting FAAH.17−20
Fundamental α-Ketoheterocycle Substituent Effect for Design of Enzyme Inhibitors
Continued extensive studies on OL-135, examining the substitution of the central oxazole,47,54−56 the C2 acyl side chain,47,56,57 and the central heterocycle,58 were conducted, and each was found to independently impact inhibitor potency or selectivity. These studies demonstrated that incorporation of 2-pyridine at the C5 position of the 2-ketooxazole significantly enhanced both binding affinity and FAAH selectivity by formation of a hydrogen bonded array between the pyridyl nitrogen and Lys142/Thr236 in the active site. They also defined a role for the central activating heterocycle distinct from that observed with serine proteases39 that explains the unique substituent effects observed. The work illustrated the importance of the electrophilic character of the ketone in driving the FAAH inhibition. An exquisite linear correlation between the Hammett σp constant of the α-ketooxazole C5 or C4 substituent and FAAH inhibition was established that is of a magnitude to dominate the behavior of inhibitors (ρ = 3.0–3.4), indicating that a unit increase in σp results in a 1000-fold increase in Ki. This provides an important predictive tool for the rational design of α-ketoheterocycle-based serine hydrolase inhibitors beyond FAAH (Figure 7).49,54,55,57
Figure 7.

Plot of −log Ki versus σp.
Systematic studies examined the effect of substituents found on the C2 acyl side chain phenyl group,56 defined the required hydrophobic character of the C2 acyl side chain,56 identified sites amenable to polar substituent introduction used to modulate solubility and PK properties, and established beneficial conformational constraints in the C2 side chain54,56 (Figure 8). The combination of the optimized C5 oxazole substituents with the optimized C2 acyl side chains provided exceptionally selective and potent FAAH inhibitors.57
Figure 8.

Representative further explorations of the C2 acyl side chain.
Changes in the central heterocycle of OL-135 were also explored and found to significantly influence the inhibitor activity: 1,3,4-oxadiazoles and 1,2,4-oxadiazoles > tetrazoles, the isomeric 1,2,4-oxadiazoles, 1,3,4-thiadiazoles > oxazoles >1,2-diazines and thiazoles >1,3,4-triazoles (Figure 9).58 Several activating heterocycles were found to improve the inhibitor potency relative to an oxazole (e.g., OL-135). In short, the introduction of an additional heteroatom at position 4 (oxazole numbering, N > O > CH) substantially increased inhibitory activity that may be attributed in part to both the increased electron-withdrawing properties of the activating heterocycle and a reduced destabilizing steric interaction59,60 at the active site.
Figure 9.

Key variations of the central activating heterocycle.
Orally Active, Long-Acting, Reversible α-Ketoheterocycle FAAH Inhibitors
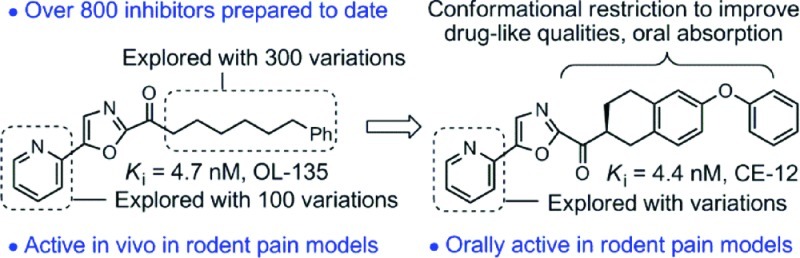
Most recently, introduction of further conformational constraints in the C2 side chain of OL-135 improved on the druglike characteristics of the inhibitors. With such inhibitors, a chiral center is introduced adjacent to the electrophilic carbonyl in which only one of the two enantiomers displayed FAAH inhibition with potencies comparable to OL-135 (Figure 10).61 In vivo characterizations demonstrated that inhibitors in this series raised brain anandamide levels following intraperitoneal (i.p.) or oral (p.o.) administration, displayed an advantageous reversible metabolic ketone/alcohol reduction/reoxidation equilibrium in vivo, and exhibited efficacy in models of thermal hyperalgesia and neuropathic pain.61 Significantly, the inhibitor CE-12 was found to be an orally active, long-acting analgesic attenuating mechanical (>6 h) and cold (>9 h) allodynia for sustained periods (>9 h). These effects mirrored the long-acting effects of CE-12 in raising endogenous levels of anandamide (>10-fold) in the CNS (>9 h) following oral administration (Figure 11). Prior studies demonstrated that >90% inhibition of FAAH is required for sustained elevations in anandamide levels or observation of analgesic effects, and it is especially notable that the duration of action of CE-12 in this class of reversible, competitive α-ketoheterocycles was similar to those reported for the irreversible urea inhibitor PF-3845 and exceed those reported for the irreversible carbamate inhibitor URB597. It is striking how similar CE-12 is in structure to OL-135 even though the exploration of the acyl side chain conformational constraints was not limited to this nearly exact overlay of key features (Figure 11).
Figure 10.

Conformational constraints in the C2 acyl side chain.
Figure 11.

Structural comparison of OL-135 and CE-12 and endogenous brain levels of anandamide (AEA) following oral administration of CE-12 (50 mg/kg p.o., mouse).
X-ray Structures of α-Ketoheterocycle-Based Inhibitors Bound to FAAH
The first X-ray structures of the α-ketoheterocycle-based inhibitors bound to FAAH were disclosed in 2009.62 The cocrystal structures of OL-135 and its isomer with FAAH confirmed that the catalytic Ser241 is covalently bound to the inhibitor electrophilic carbonyl, providing a deprotonated hemiketal mimicking the enzymatic tetrahedral intermediate (Figure 12). Additional cocrystal structures of key α-ketoheterocycles systematically probed the three active site regions central to substrate or inhibitor binding: (1) the conformationally mobile acyl chain-binding pocket and membrane access channel, (2) the active site catalytic residues and surrounding oxyanion hole that covalently binds the α-ketoheterocycle inhibitors, and (3) the cytosolic port and a newly identified anion binding site.63 These structures, including a representative member of the inhibitors containing a conformationally constricted C2 acyl side chain,61 confirmed covalent attachment through nucleophilic addition of Ser241 on the inhibitor electrophilic carbonyl and they captured the catalytic residues in an “in action” state. These studies also revealed an unusual Ser217 OH-π H-bond to the activating heterocycle, and revealed a prominent role that bound water in the cytosolic port plays in stabilizing inhibitor binding. These studies established that the dominant role of the activating heterocycle is its intrinsic electron-withdrawing properties and identified a key role of an ordered cytosolic port water in mediating the stabilizing hydrogen bonding of optimized oxazole substituents. Additionally, an exceptionally potent α-ketoheterocycle inhibitor bound to FAAH in two states was reported, representing covalently and noncovalently bound states of the inhibitor.64 Key to obtaining the structure of the noncovalently bound state of the inhibitor was the use of fluoride ion in the crystallization conditions that binds the oxyanion hole, precluding inhibitor covalent adduct formation. The opportunity to examine the noncovalently bound state of the α-ketoheterocycle inhibitor revealed that they bind in their keto versus gem diol state, and that the hydrophobic C2 side chain phenyl group binding in the acyl chain-binding pocket overrides the inhibitor’s intricate polar interactions in the cytosolic port. The X-ray structures not only confirmed key elements of inhibitor binding discussed, but they provided exquisite insights into the SAR accumulated to date, provided accurate structural templates on which high resolution structure-based design can be confidently conducted, and captured the enzyme structure and its active site catalytic residues bound to a mimic of its tetrahedral intermediate state.
Figure 12.
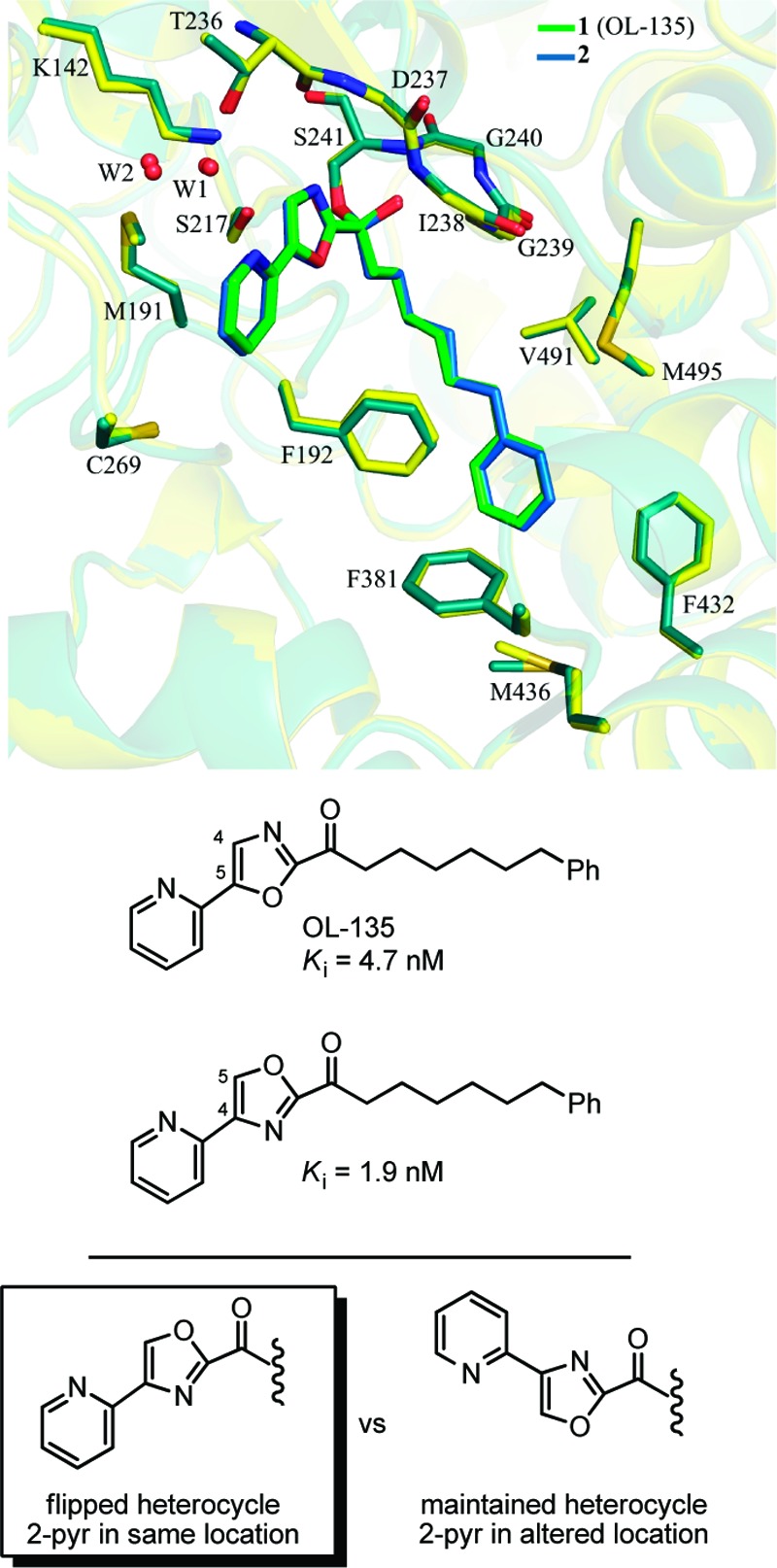
Superimposition of OL-135 (green) and its isomer (blue) bound to FAAH.
Conclusions
A remarkable series of potent, selective, and efficacious α-ketoheterocycle-based inhibitors of the enzyme fatty acid amide hydrolase (FAAH) have been disclosed, which have been utilized to mechanistically and structurally characterize the enzyme, define the mechanism of fatty acid amide hydrolysis, and validate the enzyme as a therapeutic target for the treatment of pain or sleep disorders. Their examination was conducted with the benefit of a unique activity-based protein-profiling (ABPP)-based proteome-wide selectivity screen, a powerful in vivo biomarker-based in vivo screen, and subsequent retrospective X-ray cocrystal structures, culminating in the disclosure of selective, potent, long-acting, orally active FAAH inhibitors. Since FAAH inhibition only potentiates an activated signaling pathway, increasing the endogenous levels of released fatty acid amide signaling molecules at their immediate sites of action, it provides a unique opportunity for the development of treatments for pain and sleep disorders potentially free of the side effects encountered with existing therapies. It is also likely that additional therapeutic applications will emerge from the in vivo examination of FAAH inhibitors in the years ahead with the quality of inhibitors now in hand to establish their pharmacological properties.
Acknowledgments
We wish to especially thank our collaborators with whom it has been and continues to be true pleasure to work with and include Professors R. A. Lerner, B. F. Cravatt, S. J. Henriksen, G. Suizdak, N. B. Gilula, A. H. Lichtman, R. C. Stevens, W. L Jorgensen, and E. J. Bilsky.
We gratefully acknowledge the financial support of the National Institutes of Health (DA015648). K.O. is a Skaggs fellow.
The authors declare no competing financial interest.
Author Contributions
Both authors contributed equally to composing the invited manuscript.
Funding Statement
National Institutes of Health, United States
References
- Ezzili C.; Otrubova K.; Boger D. L. (2010) Fatty acid amide signaling molecules. Bioorg. Med. Chem. Lett. 20, 5959–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane W. A.; Hanus L.; Breuer A.; Pertwee R. G.; Stevenson L. A.; Griffin G.; Gibson D.; Mandelbaum A.; Etinger A.; Mechoulam R. (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258, 1946–1949. [DOI] [PubMed] [Google Scholar]
- Cravatt B. F.; Lerner R. A.; Boger D. L. (1996) Structure determination of an endogenous sleep-inducing lipid, cis-9-octadecenoamide (oleamide): a synthetic approach to the chemical analysis of trace quantities of a natural product. J. Am. Chem. Soc. 118, 580–590. [Google Scholar]
- Cravatt B. F.; Prospero-Garcia O.; Suizdak G.; Gilula N. B.; Henriksen S. J.; Boger D. L.; Lerner R. A. (1995) Chemical characterization of a family of brain lipids that induce sleep. Science 268, 1506–1509. [DOI] [PubMed] [Google Scholar]
- Lerner R. A.; Siuzdak G.; Prospero-Garcia O.; Henriksen S. J.; Boger D. L.; Cravatt B. F. (1994) Cerebrodiene: a brain lipid isolated from sleep-deprived cats. Proc. Natl. Acad. Sci. U.S.A. 91, 9505–9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Henriksen S. J.; Cravatt B. F. (1998) Oleamide: an endogenous sleep-inducing lipid and prototypical member of a new class of biological signaling molecules. Curr. Pharm. Des. 4, 303–314. [PubMed] [Google Scholar]
- Cravatt B. F.; Giang D. K.; Mayfield S. P.; Boger D. L.; Lerner R. A.; Gilula N. B. (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty acid amides. Nature 384, 83–87. [DOI] [PubMed] [Google Scholar]
- Giang D. K.; Cravatt B. F. (1997) Molecular characterization of human and mouse fatty acid amide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 94, 2238–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney M. K.; Cravatt B. F. (2005) Structure and function of fatty acid amide hydrolase. Annu. Rev. Biochem. 74, 411–432. [DOI] [PubMed] [Google Scholar]
- Egertova M.; Cravatt B. F.; Elphick M. R. (2003) Comparative analysis of fatty acid amide hydrolase and CB1 cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience 119, 481–496. [DOI] [PubMed] [Google Scholar]
- Long J. Z.; LaCava M.; Jin X.; Cravatt B. F. (2011) An anatomical and temporal portrait of physiological substrates for fatty acid amide hydrolase. J. Lipid Res. 52, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli M. P.; Cravatt B. F. (2001) Characterization and manipulation of acyl chain selectivity of fatty acid amide hydrolase. Biochemistry 40, 6107–6115. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Fecik R. A.; Patterson J. E.; Miyauchi H.; Patricelli M. P.; Cravatt B. F. (2000) Fatty acid amide hydrolase substrate specificity. Bioorg. Med. Chem. Lett. 10, 2613–2616. [DOI] [PubMed] [Google Scholar]
- Cravatt B. F.; Lichtman A. H. (2003) Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Curr. Opin. Chem. Biol. 7, 469–775. [DOI] [PubMed] [Google Scholar]
- Lambert D. M.; Fowler C. J. (2005) The endocannabinoid system: drug targets, lead compounds, and potential therapeutic applications. J. Med. Chem. 48, 5059–5087. [DOI] [PubMed] [Google Scholar]
- Ahn K.; McKinney M. K.; Cravatt B. F. (2008) Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 108, 1687–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K.; Johnson D. S.; Cravatt B. F. (2009) Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Exp. Opin. Drug Discovery 4, 763–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seierstad M.; Breitenbucher J. G. (2008) Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J. Med. Chem. 51, 7327–7343. [DOI] [PubMed] [Google Scholar]
- Deng H. F. (2010) Fatty acid amide hydrolase as a therapeutic target. Exp. Opin. Drug Discovery 5, 961–970. [DOI] [PubMed] [Google Scholar]
- Otrubova K.; Ezzili C.; Boger D. L. (2011) The discovery and development of inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 21, 4674–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitron-Resendiz S.; Gombart L.; Cravatt B. F.; Henriksen S. (2001) Effect of oleamide on sleep and its relationship to blood pressure, body temperature, and locomotor activity in rats. J. Exp. Neurol. 172, 235–243. [DOI] [PubMed] [Google Scholar]
- Huitron-Resendiz S.; Sanchez-Alavez M.; Will D. N.; Cravatt B. F.; Henriksen S. J. (2004) Characterization of the sleep-wake patterns in mice lacking fatty acid amide hydrolase. Sleep 27, 857–865. [DOI] [PubMed] [Google Scholar]
- Cheer J. F.; Cadogan A. K.; Marsden C. A.; Fone K. C. F.; Kendall D. A. (1999) Modification of 5-HT2 receptor mediated behavior in the rat by oleamide and the role of cannabinoid receptors. Neuropharmacology 38, 533–541. [DOI] [PubMed] [Google Scholar]
- Leggett J. D.; Aspley S.; Beckett S. R. G.; D’Antona A. M.; Kendall D. A. (2004) Oleamide is a selective endogenous agonist of rat and human CB1 cannabinoid receptors. Br. J. Pharmacol. 141, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox B. J.; Ritenour-Rodgers K. J.; Asser A. S.; Baumgart L. E.; Baumgart M. A.; Boger D. L; Patterson J. E.; DeBlassio J. L.; deLong M. A.; Glufke U.; Henz M. E.; King L. III; Merkler K. A.; Robleski J. J.; Vederas J. C.; Merkler D. J. (1999) N-Acylglycine amidation: implications for the biosynthesis of fatty acid primary amides. Biochemistry 38, 3235–3245. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Patterson J. E.; Jin Q. (1998) Structural requirements for 5-HT2A and 5-HT1A receptor potentiation by the biologically active lipid oleamide. Proc. Natl. Acad. Sci. U.S.A. 95, 4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Patterson J. E.; Guan X.; Cravatt B. F.; Lerner R. A.; Gilula N. B. (1999) Chemical requirements for inhibition of gap junction communication by the biologically active lipid oleamide. Proc. Natl. Acad. Sci. U.S.A. 95, 4810–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X.; Cravatt B. F.; Ehring G. R.; Hall J. E.; Boger D. L.; Lerner R. A.; Gilula N. B. (1997) The sleep-inducing lipid oleamide deconvolutes Gap junction communication and calcium wave transmission in glial cells. J. Cell Biol. 139, 1785–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli M. P.; Cravatt B. F. (1999) Fatty acid amide hyrolase degrades bioactive amides and esters through a nonconventional catalytic mechanism. Biochemistry 38, 14125–14130. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Cravatt B. F. (2000) Clarifying the catalytic roles of conserved residues in the amidase signature family. J. Biol. Chem. 275, 19177–19184. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Lovato M. A.; Cravatt B. F. (1999) Chemical and mutagenic investigations of fatty acid amide hydrolase: evidence for a family of serine hydrolases with distinct catalytic properties. Biochemistry 38, 9804–9812. [DOI] [PubMed] [Google Scholar]
- McKinney M. K.; Cravatt B. F. (2003) Evidence for distinct roles in catalysis for residues of the serine-serine-lysine catalytic triad of fatty acid amide hydrolase. J. Biol. Chem. 278, 37393–37399. [DOI] [PubMed] [Google Scholar]
- Bracey M. H.; Hanson M. A.; Masuda K. R.; Stevens R. C.; Cravatt B. F. (2002) Adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 298, 1793–1796. [DOI] [PubMed] [Google Scholar]
- Yanagisawa I.; Yoshikawa H. (1973) A bromine compound isolated from human cerebrospinal fluid. Biochim. Biophys. Acta 329, 283–294. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Patterson J. P.; Boger D. L.; Cravatt B. F. (1998) An endogenous REM sleep inducing compound is a potent competitive inhibitor of fatty acid amide hydrolase (FAAH). Bioorg. Med. Chem. Lett. 8, 613–618. [DOI] [PubMed] [Google Scholar]
- Patterson J. E.; Ollmann I. R.; Cravatt B. F.; Boger D. L.; Wong C.–H.; Lerner R. A. (1996) Inhibition of oleamide hydrolase catalyzed hydrolysis of the endogenous sleep-inducing lipid: cis-9-octadecenamide. J. Am. Chem. Soc. 118, 5938–5945. [Google Scholar]
- Koutek B.; Prestwich G. D.; Howlett A. C.; Chin S. A.; Salehani D.; Akhavan N.; Deutsch D. G. (1994) Inhibitors of arachidonoyl ethanolamide hydrolysis. J. Biol. Chem. 269, 22937–22940. [PubMed] [Google Scholar]
- Boger D. L.; Sato H.; Lerner A. E.; Austin B. J.; Patterson J. E.; Patricelli M. P.; Cravatt B. F. (1999) Trifluoromethyl ketone inhibitors of fatty acid amide hydrolase: a probe of structural and conformational features contributing to inhibition. Bioorg. Med. Chem. Lett. 9, 265–270. [DOI] [PubMed] [Google Scholar]
- Edwards P. D.; Meyer E. F. J.; Vijayalakshmi J.; Tuthill P. A.; Andisik D. A.; Gomes B.; Strimpler A. (1992) Design, synthesis, and kinetic evaluation of a unique class of elastase inhibitors, the peptidyl α-ketooxazoles, the x-ray crystal structure of the covalent complex between porcine pancreatic elastase and Ac-Ala-Pro-Val-2-benzoxazole. J. Am. Chem. Soc. 114, 1854–1863. [Google Scholar]
- Boger D. L.; Sato H.; Lerner A. E.; Hedrick M. P.; Fecik R. A.; Miyauchi H.; Wilkie G. D.; Austin B. J.; Patricelli M. P.; Cravatt B. F. (2000) Exceptionally potent inhibitors of fatty acid amide hydrolase: the enzyme responsible for degradation of endogenous oleamide and anandamide. Proc. Natl. Acad. Sci. U.S.A. 97, 5044–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Miyauchi H.; Hedrick M. P. (2001) α-Keto heterocycle inhibitors of fatty acid amide hydrolase: carbonyl group modification and α-substitution. Bioorg. Med. Chem. Lett. 11, 1517–1520. [DOI] [PubMed] [Google Scholar]
- Leung D.; Du W.; Hardouin C.; Cheng H.; Hwang I.; Cravatt B. F.; Boger D. L. (2005) Discovery of an exceptionally potent and selective class of fatty acid amide hydrolase inhibitors enlisting proteome-wide selectivity screening: concurrent optimization of enzyme inhibitor potency and selectivity. Bioorg. Med. Chem. Lett. 15, 1423–1428. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Patricelli M. P.; Cravatt B. F. (1999) Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. U.S.A. 96, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli M. P.; Giang D. K.; Stamp L. M.; Burbaum J. J. (2001) Direct visualization of serine hydrolase activities in complex proteomes using fluorescent active site-directed probes. Proteomics 1, 1067–1071. [DOI] [PubMed] [Google Scholar]
- Kidd D.; Liu Y.; Cravatt B. F. (2001) Profiling serine hydrolase activity in complex proteomes. Biochemistry 40, 4005–4015. [DOI] [PubMed] [Google Scholar]
- Leung D.; Hardouin C.; Boger D. L.; Cravatt B. F. (2003) Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat. Biotechnol. 21, 687–691. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Miyauchi H.; Du W.; Hardouin C.; Fecik R. A.; Cheng H.; Hwang I.; Hedrick M. P.; Leung D.; Acevedo O.; Guimaráes C. R. W.; Jorgensen W. L.; Cravatt B. F. (2005) Discovery of a potent, selective, and efficacious class of reversible α-ketoheterocycle inhibitors of fatty acid amide hydrolase as analgesics. J. Med. Chem. 48, 1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman A. H.; Leung D.; Shelton C. C.; Saghatelian A.; Hardouin C.; Boger D. L.; Cravatt B. F. (2004) Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J. Pharmacol. Exp. Ther. 311, 441–448. [DOI] [PubMed] [Google Scholar]
- Romero F. A.; Hwang I.; Boger D. L. (2006) Delineation of a fundamental α-ketoheterocycle substituent effect for use in the design of enzyme inhibitors. J. Am. Chem. Soc. 68, 14004–14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L.; Luo L.; Palmer J. A.; Sutton S.; Wilson S. J.; Barbier A. J.; Breitenbucher J. G.; Chaplan S. R.; Webb M. (2006) Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br. J. Pharmacol. 148, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey S. G.; Long J. Z.; O’Neal S. T.; Abdulla R. A.; Poklis J. L.; Boger D. L.; Cravatt B. F.; Lichtman A. H. (2009) Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J. Pharmacol. Exp. Ther. 330, 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg J. E.; Boger D. L.; Cravatt B. F.; Lichtman A. H. (2009) Endocannabinoid modulation of scratching response in an acute allergenic model: new prospective neural therapeutic target for pruritus. J. Pharmacol. Exp. Ther. 329, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker L.; Kinsey S. G.; Abdullah R. A.; Blankman J. L.; Long. J. Z.; Ezzili C.; Boger D. L.; Cravatt B. F.; Lichtman A. H.. The FAAH inhibitor PF-3845 acts in the nervous system to reverse lipopolysaccharide-induced tactile allodynia in mice. Br. J. Pharmacol. 2011, in press. [DOI] [PMC free article] [PubMed]
- Romero F. A.; Du W.; Hwang I.; Rayl T. J.; Kimball F. S.; Leung D.; Hoover H. S.; Apodaca R. L.; Breitenbucher J. G.; Cravatt B. F.; Boger D. L. (2007) Potent and selective α-ketoheterocycle-based inhibitors of the anandamide and oleamide catabolizing enzyme, fatty acid amide hydrolase. J. Med. Chem. 50, 1058–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMartino J. K.; Garfunkle J.; Hochstatter D. G.; Cravatt B. F.; Boger D. L. (2008) Exploration of a fundamental substituent effect of α-ketoheterocycle enzyme inhibitors: potent and selective inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 18, 5842–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardouin C.; Kelso M. J.; Romero F. A.; Rayl T. J.; Leung D.; Hwang I.; Cravatt B. F.; Boger D. L. (2007) Structure–activity relationships of α-ketooxazole inhibitors of fatty acid amide hydrolase. J. Med. Chem. 50, 3359–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball F. S.; Romero F. A.; Ezzili C.; Garfunkle J.; Rayl T. J.; Hochstatter D. G.; Hwang I.; Boger D. L. (2008) Optimization of α-ketooxazole inhibitors of fatty acid amide hydrolase. J. Med. Chem. 51, 937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfunkle J.; Ezzili C.; Rayl T. J.; Hochstatter D. G.; Hwang I.; Boger D. L. (2008) Optimization of the central heterocycle of α-ketoheterocycle inhibitors of fatty acid amide hydrolase. J. Med. Chem. 51, 4393–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaráes C. R. W.; Boger D. L.; Jorgensen W. L. (2005) Elucidation of fatty acid amide hydrolase inhibition by potent α-ketoheterocycle derivatives from Monte Carlo simulations. J. Am. Chem. Soc. 127, 17377–17384. [DOI] [PubMed] [Google Scholar]
- Slaymaker I. M.; Bracey M.; Mileni M.; Garfunkle J.; Cravatt B. F.; Boger D. L.; Stevens R. C. (2008) Correlation of inhibitor effects on enzyme activity and thermal stability for integral membrane protein fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 18, 5847–5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzili C.; Mileni M.; McGlinchey N.; Long J. Z.; Kinsey S. G.; Hochstatter D. G.; Stevens R. C.; Lichtman A. H.; Cravatt B. F.; Bilsky E. J.; Boger D. L. (2011) Reversible competitive α-ketoheterocycle inhibitors of fatty acid amide hydrolase containing additional conformational constraints in the acyl side chain: orally active, long acting analgesics. J. Med. Chem. 54, 2805–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileni M.; Garfunkle J.; DeMartino J. K.; Cravatt B. F.; Boger D. L.; Stevens R. C. (2009) Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by α-ketoheterocycle inhibitors revealed from cocrystal structures. J. Am. Chem. Soc. 131, 10497–10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileni M.; Garfunkle J.; Ezzili C.; Kimball F. S.; Cravatt B. F.; Stevens R. C.; Boger D. L. (2010) X-ray crystallographic analysis of α-ketoheterocycle inhibitors bound to a humanized variant of fatty acid amide hydrolase. J. Med. Chem. 53, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileni M.; Garfunkle J.; Ezzili C.; Cravatt B. F.; Stevens R, C.; Boger D. L. (2011) Fluoride-mediated capture of a noncovalent bound state of a reversible covalent enzyme inhibitor: X-ray crystallographic analysis of an exceptionally potent α-ketoheterocycle inhibitor of fatty acid amide hydrolase. J. Am. Chem. Soc. 133, 4092–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]