

Abstract
All animals need to sense temperature to avoid hostile environments and to regulate their internal homeostasis. A particularly obvious example is that animals need to avoid damagingly hot stimuli. The mechanisms by which temperature is sensed have until recently been mysterious, but in the last couple of years, we have begun to understand how noxious thermal stimuli are detected by sensory neurons. Heat has been found to open a nonselective cation channel in primary sensory neurons, probably by a direct action. In a separate study, an ion channel gated by capsaicin, the active ingredient of chili peppers, was cloned from sensory neurons. This channel (vanilloid receptor subtype 1, VR1) is gated by heat in a manner similar to the native heat-activated channel, and our current best guess is that this channel is the molecular substrate for the detection of painful heat. Both the heat channel and VR1 are modulated in interesting ways. The response of the heat channel is potentiated by phosphorylation by protein kinase C, whereas VR1 is potentiated by externally applied protons. Protein kinase C is known to be activated by a variety of inflammatory mediators, including bradykinin, whereas extracellular acidification is characteristically produced by anoxia and inflammation. Both modulatory pathways are likely, therefore, to have important physiological correlates in terms of the enhanced pain (hyperalgesia) produced by tissue damage and inflammation. Future work should focus on establishing, in molecular terms, how a single ion channel can detect heat and how the detection threshold can be modulated by hyperalgesic stimuli.
Organisms sense temperature for all sorts of reasons. Highly accurate thermosensation is required to set the body temperature of a mammal. Simpler animals sense the external temperature to seek out favorable environments for feeding or for mating. Damaging extremes of temperature must be avoided, of course, and for this purpose, pain-sensitive nerve terminals detect very low and very high temperatures and induce an avoidance response. In all these instances, temperature must be detected—but how? In many cases, the detection mechanism involves a specialized temperature-sensitive nerve terminal, which, on application of a temperature change, generates a depolarization and a resulting train of action potentials in the sensory nerve axon. So it is perhaps obvious to state that temperature must gate an ion channel in the sensory nerve terminal. But how does it work? One could imagine a temperature-sensitive biochemical pathway that modulates an internal transmitter and in turn gates the ion channel. There is some evidence for such a system in at least one temperature-sensitive pathway in the nematode Caenorhabditis elegans (see below). But in the only other instance of which we have any understanding, the action of temperature on the ion channel seems instead to be direct. To the existing voltage-gated, ligand-gated, and mechanosensitive ion channels, we can therefore add a fourth major category of ion channels, namely, heat-sensitive ion channels. This article reviews our understanding to date of this newly characterized class of ion channels.
Heat-Sensitive Ion Channels in Primary Sensory Neurons.
The most direct way to study the detection of hot stimuli is in situ, either by asking subjects at what temperature a sensation of warmth changes to a sensation of pain or alternatively by recording the frequency of action potentials in the axon of a primary pain-sensitive neuron (a nociceptor) while a thermal stimulus is applied to the receptive field. Experiments like these have shown that, as the temperature is raised, a sensation of warmth changes to pain at around 43–45°C and that the intensity of the pain sensation increases steeply thereafter (1). Recordings of action potentials from nociceptive nerve fibers show a similar picture, namely of a threshold for initiation of action potentials at 43–45°C and a steep increase in firing frequency as the temperature is increased further (1, 2).
To take things much further, for instance to study the pathways controlling ionic currents involved in the transduction process, a preparation of isolated nociceptors is needed. Other sensory receptors can be isolated more or less intact, and the study of isolated photoreceptors, auditory receptors, olfactory receptors, etc. has told us a great deal about their mechanisms of operation. Nociceptors are unfortunately a much more difficult proposition. The sensory terminals are extremely fine and are embedded in a cellular matrix whose disruption during dissection releases the very signaling molecules that the nociceptor nerve terminal is designed to detect. The difficulty of isolating intact nociceptive nerve terminals has meant that studies on isolated nociceptors have all been on neuronal cell bodies. In a typical procedure, the neuronal cell body is isolated by enzymatic treatment and is cultured for a few days before use (3, 4). The sensory terminals are, of course, completely removed during the isolation procedure, and we must hope that the properties of those terminals are recreated in the cultured cell body and dendrites. When they are acutely isolated, nociceptive cell bodies often fail to respond to noxious stimuli, but in a process that is poorly understood, nociceptive properties characteristic of the sensory terminal reappear after a few days in culture in the presence of serum and nerve growth factor (3–6). This preparation of cultured nociceptors has been used for almost all experiments investigating the cellular and molecular basis of detection of painful stimuli. The complexity of the procedure for isolating nociceptors nonetheless makes it essential that we check very carefully that our nociceptors’ responses resemble those in vivo.
An example of the response of the membrane potential of a cultured nociceptor to application of a 49°C heat stimulus is shown in Fig. 1 (6, 7). As is seen in nociceptive nerve terminals in vivo (8), the heat stimulus causes a rapid depolarization and initiates a train of action potentials. The fact that this response is present in isolated nociceptors shows that no other cells are necessary to produce the response to heat; there is no signal molecule released from adjacent damaged cells to which the nociceptor responds. Nor is the response due to damage to the nociceptive neuron itself—as might occur if heat were causing a breakdown in the plasma membrane and a consequent depolarization—because the depolarization and action-potential firing ceases immediately when the stimulus is withdrawn and because similar responses can be elicited again and again on repeated application of the heat stimulus.
Figure 1.

Depolarization and a train of action potentials initiated in a nociceptive neuron in culture by application of a brief heat stimulus. Membrane potential recorded by using whole-cell patch clamp (see ref. 6 for details).
The reproducibility of the response in isolated nociceptors resembles that in other sensory receptors. Interestingly, though, the behavior of nociceptors in vivo is different. Repeated application of a strong stimulus leads to a progressive increase in the response in nociceptors in vivo but not in isolated nociceptors nor in other sensory receptors. This process, known as sensitization or hyperalgesia, is characteristic of nociceptors in vivo and has obvious protective value for the organism as a whole, in that the pain caused by a damaging stimulus becomes more urgent if the stimulus is repeated or maintained. The fact that sensitization is not observed in isolated nociceptors suggests that the phenomenon is not intrinsic to the neuron but instead has its origin in extracellular signals released from nearby damaged or inflamed tissue (9). Recent advances in our understanding of this process of sensitization are discussed below.
The membrane current induced by heat in a voltage-clamped nociceptor is shown in Fig. 2A. The current is activated rapidly (but not instantaneously) by heat, with a mean time to half activation of ≈35 ms at 50°C (6, 7). By contrast, neurons insensitive to heat show only a small current change, which occurs as rapidly as the solution change and therefore probably has a simple physical origin such as a temperature dependence of membrane leakage resistance. When the temperature dependence of the heat-sensitive current is examined (Fig. 2B), the current can be seen to be activated above ≈42°C and to increase exponentially as the temperature is raised further, much as is observed in nociceptors in vivo (1, 2).
Figure 2.

Responses of membrane current in isolated nociceptors to heat. (A) Application of a rapid step change in temperature (from room temperature to 49°C; time course shown by the top trace) elicits an inward current with a short delay (≈35 ms) in a nociceptive neuron (lower of the two membrane current traces). In heat-insensitive neurons, a much smaller current change is elicited with no delay (top current trace). Neurons were voltage-clamped by the whole-cell patch-clamp method at −70 mV. (B). Current as a function of temperature in a heat-sensitive neuron (lower trace) and in a heat-insensitive neuron (upper trace). Modified from ref. 6.
Experiments examining the ionic selectivity of the heat-activated current (6, 7) have shown that the heat-activated channel discriminates poorly amongst monovalent alkali cations, in common with many other ion channels such as those gated by glutamate, acetylcholine, or cyclic nucleotides. Calcium ions can by themselves carry current through the channel but, in addition, have the effect of partially blocking a current carried by monovalent ions. The channel must therefore possess a binding site in the pore region with a higher affinity for Ca2+ than for monovalent cations. Contrary to early reports (10), the channel does not seem to be blocked by Cs+ ions. The current-voltage relation shows outward rectification and a reversal potential of around 0 mV under physiological conditions (6, 10, 11).
Single heat-activated channels have a conductance of around 30–40 pS (10, 11). The single channel conductance itself is only weakly temperature-dependent, in common with other ion channels, and the pronounced dependence of current on temperature is caused by a strong temperature dependence of the probability of channel opening. The time constants of channel opening can be deduced from the characteristics of the current noise produced when several channels are present simultaneously in a cell-attached membrane patch (see Fig. 3). Application of heat activates inward current and current noise in heat-sensitive but not in heat-insensitive neurons (Fig. 3A). The power spectrum of heat-induced noise (Fig. 3B) indicates the existence of two Lorentzian components with time constants τ1 = 17 ms and τ2 = 0.49 ms. A simple two-state model reproducing these features is
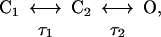 |
where τ1 is the time constant of transition from the closed state C1 to C2 and τ2 is the time constant of transition from the closed state C2 to the open state (O). The similarity of τ1 to the half-time of channel opening after application of a heat jump (Fig. 2A) suggests that it is the transition between C1 and C2 that is the main temperature-dependent event.
Figure 3.

Noise associated with opening of heat-activated ion channels. (A) Examples of cell-attached patch-clamp recordings of the responses of a heat-insensitive neuron (top recording) and a heat-sensitive neuron (bottom recording) to temperature steps. Patch pipette contained only 154 mM NaCl/10 mM Hepes to maximize current through heat-sensitive ion channels and was held at 0 mV. Heat-sensitive currents through channels outside the patch were prevented by bathing the rest of the cell in solution free of permeant ions (154 mM N-methyl glucamine/10 mM Hepes). (B) Power spectrum of cell-attached current activated by a temperature step to 49°C. For each experiment, 10 consecutive traces (16,384 samples per trace at 10 kHz; filtered at 4 kHz) were acquired, first at room temperature then at 49°C, and the power spectra were calculated. Heat-induced power spectrum (points) was calculated as the difference between the two spectra. Power spectrum fitted with the sum of two Lorentzian functions (sum shown as a solid line; component spectra as a dashed line) with half-power frequencies as shown, corresponding to time constants of 17 ms and 0.49 ms (V.V., P.C., and P.A.M., unpublished data).
Electrophysiologists who work on cultured sensory neurons tend to think of their cells as a bimodal population, consisting of nociceptive and nonnociceptive neurons. Whole-animal physiologists who work on nociceptors in situ know differently; nociceptors come in many different varieties, with properties such as heat, mechanical, and chemical sensitivity present to variable extents in different single-unit recordings. The main division, of course, is between slowly conducting, unmyelinated nociceptive nerve fibers, which commonly respond to a wide range of stimuli (polymodal fibers) and more rapidly conducting myelinated nerve fibers, which frequently respond to a smaller subset of noxious stimuli, but amongst which heat-sensitive units are also commonly encountered (12, 13). A corresponding division of nerve-cell bodies is seen, both in dorsal root ganglia and in cultured preparations, into small-diameter dark neurons and large-diameter pale neurons (14). Heat sensitivity is, perhaps reassuringly, seen in both cell types in culture (10, 15), but there is a quantitative difference: the smaller cells have a threshold of around 45°C, whereas larger cells form a different population with a threshold of 51°C (15). Interestingly, only the former population responds to capsaicin, suggesting that there is more than one heat-sensitive channel at work in this diverse population of nociceptors (see further discussion below).
Thermosensation in C. elegans.
The nematode worm C. elegans is capable of seeking out a preferred temperature at which to feed, and mutants unable to detect temperature can therefore be selected by isolating individuals that stray from preferred-temperature areas. These worms have a mutation either in a gene, tax-4, that codes for the α-subunit of an ion channel gated by cyclic nucleotides (16) or in a second gene, tax-2, that codes for a β-subunit (17). The fact that these channels can be gated by cyclic nucleotides suggests (but does not prove) that the mechanism of thermosensation is the modulation of the pathway that controls the level of cyclic nucleotides, rather than the direct action of heat on the ion channel itself. In this respect, detection of nonnoxious temperatures in C. elegans is different from mammalian noxious heat sensation. The latter depends only on expression of a heat-sensitive ion channel, which can be seen to function in isolated membrane patches and therefore is not gated by diffusible messengers controlled by intracellular signaling pathways (11, 18, 19), whereas thermosensation in C. elegans seems to depend on cyclic nucleotides as intracellular messengers, and the thermosensitive element is therefore presumably some stage in the pathway modulating the level of cyclic nucleotides. There are many forms of mammalian thermosensation, as outlined in the introduction, and it is quite possible that signaling pathways are involved in some of these, even though they do not seem to be directly responsible for heat sensation in the nociceptors of higher vertebrates.
Sensitization of Nociceptors.
The process of sensitization (or hyperalgesia) is familiar to us all: a stimulus strong enough to cause tissue damage hurts more with time, and even after the stimulus has been removed, the damaged area is hypersensitive to touch and to temperature. This phenomenon can be attributed partly to changes in pain transmission in the spinal cord or at higher levels, but an important component results from processes occurring at the site of injury. A large number of molecules released by tissue damage are known to act as mediators of hyperalgesia. Examples include neuropeptides, prostaglandins, histamine, platelet-activating factor, and bradykinin (9, 20).
With so many different factors able to cause hyperalgesia, it is perhaps no surprise that more than one cellular mechanism is involved. One recently elucidated mechanism involves activation of protein kinase A. External inflammatory messengers such as prostaglandins, serotonin, and adenosine activate adenylate cyclase and consequently increase the level of cAMP, leading to activation of protein kinase A (21, 22). The principal physiologically important target of protein kinase A seems to be a recently identified voltage-sensitive Na channel (23), which, unlike the more usual neuronal Na channel, is not blocked by tetrodotoxin. The effect of phosphorylation of the tetrodotoxin-resistant Na channel is to lower its threshold, thereby making it more likely that an action potential will be elicited (21, 22). This membrane ionic current is probably not the only one modulated by cAMP, as actions on a K+ current and on a voltage and cyclic nucleotide-gated conductance have also been identified (24, 25). All of these cAMP-dependent mechanisms, however, operate in the same direction, in that they sensitize the nociceptive nerve terminal to any stimulus that is capable of exciting it, because the effect is to reduce the threshold for action potential firing, rather than on the specific receptor current induced by the stimulus.
A second and more specific mechanism uses activation of protein kinase C (PKC) to sensitize the response to heat. Fig. 4A shows that the inflammatory mediator bradykinin potently increases the membrane current activated by a heat pulse (6). Bradykinin is known to activate both phospholipase C and phospholipase A2, thereby releasing a number of potential intracellular signaling molecules. The enhancement of the heat response can be shown to be due to activation of PKC, however, and not to other possible intracellular mediators, because it can be mimicked by phorbol esters, which are specific activators of PKC (Fig. 4B). Fig. 4C shows that activation of PKC increases the current activated by a heat stimulus and shifts the relation between temperature and membrane current to lower temperatures. These observations predict that normally innocuous temperatures, such as body warmth, will therefore become painful after sensitization, an observation that corresponds well both with experiments on intact preparations and with our personal experience that even the warmth of a hand can cause a sensation of pain when applied to an injured area of the body. Other evidence supporting the identity of PKC as an intracellular mediator of sensitization includes the findings that sensitization can be reversed by PKC inhibitors and can be prolonged by phosphatase inhibitors, which prevent dephosphorylation after a protein target has been phosphorylated by PKC (6). The possibility that mediators other than bradykinin may also employ the PKC pathway to induce sensitization has not yet been investigated and certainly deserves to be.
Figure 4.

Phosphorylation by PKC sensitizes the heat response of nociceptors. (A) Response of membrane current to a 49°C heat pulse before and after exposure to bradykinin (Bk). (B) Similar effect to that seen in A is observed after treatment by the specific PKC activator phorbol myristate acetate (PMA). (C) Current vs. temperature relations before and after PMA treatment, showing sensitization of the heat response. [Reproduced with permission from ref. 6 (Copyright 1996, Proceedings of the National Academy of Sciences of the United States of America)].
Desensitization of Nociceptors.
When a long pulse of moderate heat, insufficiently strong to cause cell damage and to release the extracellular mediators responsible for sensitization, is applied to a heat-sensitive nociceptor in vivo, gradual adaptation or desensitization in the firing frequency is observed (26). A similar phenomenon is seen in isolated nociceptors (Fig. 5), showing that desensitization, unlike sensitization, is intrinsic to the nociceptor. Recent experiments in our lab have shown that desensitization is triggered by an influx of calcium ions from the external medium through the heat-sensitive ion channel. In this respect, desensitization of the heat response resembles the desensitization in response to prolonged application of capsaicin (27), which is triggered by activation of the calcium-dependent phosphatase calcineurin by an influx of calcium through the capsaicin-gated channel itself. These observations suggest that both the heat-activated channel and the capsaicin-gated channel are desensitized when dephosphorylated by calcineurin, one of many similarities between the two (see below). The molecular mechanisms of sensitization of the heat-activated channel (phosphorylation by PKC; see above) and desensitization (dephosphorylation by calcineurin) may therefore be simply complementary aspects of the same process, in which the heat sensitivity of the channel is regulated by phosphorylation (see Fig. 5 and discussion below).
Figure 5.

The heat-sensitive current in a nociceptor undergoes desensitization in response to a maintained pulse of heat (P.C., A.M., V.V., and P.A.M., unpublished data).
The Capsaicin Receptor, Vanilloid Receptor Subtype 1 (VR1), and Its Relation to Heat Sensation.
Capsaicin, the active ingredient of chili peppers, has been known for some time to depolarize nociceptive nerve terminals by a direct action on an ion channel (28). Capsaicin is not part of the normal environment of most animals. Therefore, it had always been supposed that the capsaicin receptor was gated physiologically by an endogenous agonist, just as the morphine-receptor family is activated physiologically not by morphine but by endogenous opiates. Capsaicin-responding neurons can be activated by low pH, and, as pH can drop considerably during inflammation, hydrogen ions were a plausible candidate for the physiological agonist activating these nociceptors (28).
The capsaicin receptor VR1 has recently been cloned by an ingenious strategy by using imaging of the increase in internal calcium caused by application of capsaicin to detect expression of capsaicin-receptor clones (18). The expressed receptor is indeed sensitive to low pH, but, perhaps more interestingly, it responds to heat like the native heat receptor, as outlined above. The main points of resemblance are as follows (see refs. 18 and 19). (i) The current passing through both channels is zero at room temperature and increases sharply above about 42°C. (ii) The capsaicin receptor is a cation channel with an ionic selectivity similar to that of the native heat receptor. (iii) The single-channel conductance and the current-voltage relation are similar. (iv) The open time constant of VR1, 0.9 ms, is similar to the fast open time constant of the heat-activated channel (0.5 ms; see above). (v) The actions of capsaicin and heat are synergistic on both VR1 and the native heat receptor (19, 29). (vi) VR1 is expressed exclusively in small neurons of primary sensory ganglia.
There may, however, be one crucial point of difference: the ion current through VR1 is blocked by the capsaicin-channel antagonists capsazepine and ruthenium red, whether the current is elicited by capsaicin application or by heat (19). However, in cultured nociceptors, the current induced by capsaicin is blocked by these agonists (28), but the response to heat does not seem to be (30).
One particularly interesting feature of VR1 is the interaction between its heat sensitivity and its pH sensitivity. At normal pH, VR1 is activated only at temperatures above ≈42°C. Low pH acts as a sensitizing agent, which reduces the threshold for activation by heat to ≈30°C at pH 6.3 (19). The sensitizing effect of pH explains the observation that capsaicin receptors are activated by low pH; at a sufficiently low pH, room temperature is adequate to induce channel openings (19). In inflamed or anoxic tissue, the pH can drop to as low as 6.0, and at this pH, body temperature would be sufficient to activate VR1. The pain of inflammation and anoxia may therefore be explained at least partly by a combined effect of low pH and normally innocuous temperature on VR1.
Is VR1 the only heat-detecting mechanism in nociceptors? Probably not, in view of the observation by Nagy and Rang (15) that the two properties of heat sensitivity and capsaicin sensitivity (and therefore presumably expression of VR1) are not absolutely colocalized in sensory neurons, contrary to an earlier report based on a smaller number of experiments (31). A recent study (32) reports the cloning of a vanilloid receptor-like channel (VRL-1) that is not sensitive to capsaicin but is gated by temperatures above 52°C. Expression of this channel may explain the responses to higher temperatures observed in some capsaicin-insensitive neurons (15).
How Does the Heat Receptor Work?
How ligand-gated or voltage-gated ion channels might work is intuitively fairly obvious, at least in terms of general principles. Ligand-gated channels operate like a lock and key; insertion of the key (the ligand) stabilizes the open state of the channel. Voltage-gated channels possess a charged gating unit within the membrane field, such that changes in the membrane potential move this unit and thereby induce a conformational change that gates the channel open or closed. How small elevations in temperature might shift the heat-sensitive channel from the closed to the open state is less intuitively obvious but must depend on the well known thermodynamic equation
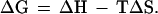 |
The change in the equilibrium between closed and open states of the channel, which depends on the Gibbs free energy change (ΔG), can be markedly temperature-dependent only if there is a large entropy difference (ΔS) between the two states. Elevations in temperature (T) must therefore cause the heat-sensitive ion channel to change from an ordered to a more disordered state, as occurs during melting of ice or dissolving of a salt in water. It does not seem that any accessory protein or signaling pathway is needed to gate the channel, because heat-sensitive ion channels can be seen in cell-free membrane patches from nociceptors (11) and because VR1 functions as a heat receptor when heterologously expressed in HEK 293 cells or in Xenopus oocytes (18, 19). The temperature-sensitive gating unit is likely, therefore, to be intrinsic to the heat-sensitive channel protein.
The process of sensitization, which shifts the relation between temperature and channel opening to lower temperatures (see Fig. 3C), must act by stabilizing the more disordered, higher-temperature state of the channel in such a way that lower temperatures are needed to induce channel opening. How might this interesting and physiologically important process be operating? One possibility is that opposite changes in the charge on either side of the membrane may be important. The work of Tominaga et al. (19) has shown that protonation of an external site of VR1 induces sensitization, and work in our own lab has shown that phosphorylation of an internal site of the heat-sensitive receptor induces an apparently identical sensitized state (6). If VR1 and the heat-sensitive receptors are one and the same, as is suggested by most lines of evidence (see above), then we can put these two observations together in a simple (and speculative) model of the sensitization process (Fig. 6). In this model, addition of positive charge to the external face of the membrane or addition of negative charge to the internal face have equivalent effects, with both manipulations leading to stabilization of a disordered state of the protein and consequently to sensitization of the response to heat.
Figure 6.

Possible model for sensitization of the heat response. Acidification of the external solution causes protonation of an external site on the heat-sensitive ion channel and consequent sensitization (19). Phosphorylation at the internal surface has a similar effect (6). Both effects are reversible. PP, phosphatase.
ABBREVIATIONS
- PKC
protein kinase C
- VR1
vanilloid receptor subtype 1
References
- 1.Treede R D, Meyer R A, Raja S N, Campbell J N. Prog Neurobiol. 1992;38:397–421. doi: 10.1016/0301-0082(92)90027-c. [DOI] [PubMed] [Google Scholar]
- 2.Belmonte C, Gallar J. In: Neurobiology of Nociceptors. Belmonte C, Cervero F, editors. Vol. 6. Oxford: Oxford Univ. Press; 1996. pp. 146–183. [Google Scholar]
- 3.Rang H P, Bevan S, Dray A. In: Textbook of Pain. Melzack R, Wall P, editors. Vol. 3. Edinburgh: Churchill Livingstone; 1994. pp. 57–78. [Google Scholar]
- 4.Baccaglini P I, Hogan P G. Proc Natl Acad Sci USA. 1983;80:594–598. doi: 10.1073/pnas.80.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilabert R, McNaughton P A. J Neurosci Methods. 1997;71:191–198. doi: 10.1016/s0165-0270(96)00144-6. [DOI] [PubMed] [Google Scholar]
- 6.Cesare P, McNaughton P A. Proc Natl Acad Sci USA. 1996;93:15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cesare P, McNaughton P A. Curr Opin Neurobiol. 1997;7:493–499. doi: 10.1016/s0959-4388(97)80028-1. [DOI] [PubMed] [Google Scholar]
- 8.Treede R D, Meyer R A, Raja S N, Campbell J N. J Physiol. 1995;483:747–758. doi: 10.1113/jphysiol.1995.sp020619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kress M, Reeh P W. In: Neurobiology of Nociceptors. Belmonte C, Cervero F, editors. Vol. 11. Oxford: Oxford Univ. Press; 1996. pp. 258–297. [Google Scholar]
- 10.Reichling D B, Levine J D. Proc Natl Acad Sci USA. 1997;94:7006–7011. doi: 10.1073/pnas.94.13.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagy I, Rang H P. J Physiol. 1998;507:29P. [Google Scholar]
- 12.Besson J M, Chaouch A. Physiol Rev. 1987;67:67–155. doi: 10.1152/physrev.1987.67.1.67. [DOI] [PubMed] [Google Scholar]
- 13.Lynn B, Faulstroh K, Pierau F K. Eur J Neurosci. 1995;7:431–437. doi: 10.1111/j.1460-9568.1995.tb00339.x. [DOI] [PubMed] [Google Scholar]
- 14.Lawson S N, Perry M J, Prabhakar E, McCarthy P W. Brain Res Bull. 1993;30:239–243. doi: 10.1016/0361-9230(93)90250-f. [DOI] [PubMed] [Google Scholar]
- 15.Nagy I, Rang H P. Neuroscience. 1999;88:995–997. doi: 10.1016/s0306-4522(98)00535-1. [DOI] [PubMed] [Google Scholar]
- 16.Komatsu H, Mori I, Rhee J, Akaike N, Ohshima Y. Neuron. 1996;17:707–718. doi: 10.1016/s0896-6273(00)80202-0. [DOI] [PubMed] [Google Scholar]
- 17.Coburn C M, Bargmann C I. Neuron. 1996;17:695–706. doi: 10.1016/s0896-6273(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 18.Caterina M J, Schumacher M A, Tominaga M, Rosen T A, Levine J D, Julius D. Nature (London) 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 19.Tominaga M, Caterina M J, Malmberg A B, Rosen T A, Gilbert H, Skinner K, Raumann B E, Basbaum A I, Julius D. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 20.Levine J D, Fields H L, Basbaum A I. J Neurosci. 1993;13:2273–2286. doi: 10.1523/JNEUROSCI.13-06-02273.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gold M S, Reichling D B, Shuster M J, Levine J D. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.England S, Bevan S, Docherty R J. J Physiol. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akopian A N, Sivilotti L, Wood J N. Nature (London) 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 24.Ingram S L, Williams J T. J Physiol. 1996;492:97–106. doi: 10.1113/jphysiol.1996.sp021292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicol G D, Vasko M R, Evans A R. J Neurophysiol. 1997;77:167–176. doi: 10.1152/jn.1997.77.1.167. [DOI] [PubMed] [Google Scholar]
- 26.Treede R D. Ann Med. 1995;27:213–216. doi: 10.3109/07853899509031961. [DOI] [PubMed] [Google Scholar]
- 27.Docherty R J, Yeats J C, Bevan S, Boddeke H W G M. Pflügers Arch Eur J Physiol. 1996;431:828–837. doi: 10.1007/s004240050074. [DOI] [PubMed] [Google Scholar]
- 28.Bevan S, Geppetti P. Trends Neurosci. 1994;17:509–512. doi: 10.1016/0166-2236(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 29.Dittert I, Vlachova V, Knotkova H, Vitaskova Z, Vyklicky L, Kress M, Reeh P W. J Neurosci Methods. 1998;82:195–201. doi: 10.1016/s0165-0270(98)00051-x. [DOI] [PubMed] [Google Scholar]
- 30.Hepworth M B, Pinnock R D. J Physiol. 1998;513:133P. [Google Scholar]
- 31.Kirschstein T, Busselberg D, Treede R D. Neurosci Lett. 1997;231:33–36. doi: 10.1016/s0304-3940(97)00533-8. [DOI] [PubMed] [Google Scholar]
- 32.Caterina M J, Rosen T A, Tominaga M, Brake A J, Julius D. Nature (London) 1999;398:436–441. doi: 10.1038/18906. [DOI] [PubMed] [Google Scholar]