

Abstract
Catalytic quantities of bismuth(III) triflate efficiently initiate the rearrangement of epoxides to aldehydes which subsequently react with (Z)-δ-hydroxyalkenylsilanes to afford 2,6-disubstituted-3,6-dihydro-2H-pyrans. Isolated yields of desired products using Bi(OTf)3 were compared with yields when the reactions were run with TfOH and TMSOTf in the presence and absence of several additives. These studies, as well as NMR spectroscopic analyses, indicate an initial Lewis acid/base interaction between Bi(OTf)3 and substrates providing TfOH in situ.
Keywords: Silyl-Prins, dihydropyran, oxocarbenium ion, vinylsilane, bismuth, Bi(OTf)3, oxonia-Cope, triflic acid, Brønsted acid, Lewis acid
Introduction
A variety of bismuth compounds have been used in organic reactions over the last 10-15 years because of low toxicities, a wide range of reactivities and low cost.1 Catalytic quantities of Bi(III) compounds are effective in promoting allylations and cyanations,2 etherification,3 Diels-Alder,4 and protection/deprotection reactions. Furthermore, asymmetric total syntheses of leucascandrolide, 5 centrolobine,6 mucocin7 and bistramide C8 have included Bi(III)-mediated sequences. As part of a program to increase the uses of Bi(III) in the synthesis of cyclic ethers, we have described two- and three-component reactions toward tetrahydropyrans9 and dihydropyrans10,11 using catalytic quantities of bismuth bromide (BiBr3) at room temperature. Extension of this work to a cascade reaction involving epoxide rearrangement12 followed by a tandem addition/silyl-Prins reaction is reported herein along with comparisons of BiX3 (X = Br, OTf), trimethylsilyltrifluoromethanesulfonate (TMSOTf) and trifluoromethanesulfonic acid (TfOH) as initiators/catalysts.
Although a wide variety of protocols have been used for the synthesis of dihydropyran ring systems, the Prins13 and silyl-Prins14 reactions remain widely used and studied. Dobbs' InCl3-mediated synthesis,15 Rychnovsky's segmented Prins reaction16 and Markó's intramolecular silyl-modified Sakurai reaction (ISMS)17 are all chemically related to the BiBr3-mediated reaction shown in eq 1.11
Despite the drastic increase of Bi(III) salts in synthesis, debate continues over their exact role in catalytic cycles. Several authors have noted that Bi(III) salts generate Brønsted acids in situ18 whereas Shibasaki et al.,19 Kadam and Kim,20 as well as Kobayashi and co-workers21 propose that the bismuth is acting as a Lewis acid in amination, substitution, and asymmetric Mukaiyama aldol reactions, respectively. In addition, both Cook22 and Donnelly23 report that allylic halide substitution reactions occur readily in the presence of catalytic quantities of bismuth compounds and attribute the success of these reactions to the Lewis acid character of Bi(III).24,25,26 The Lewis acidic nature of Bi(III) compounds is usually attributed to the tendency for expanded coordination, availability of unoccupied orbitals (Bi-X σ* and d), and the presence of electron-withdrawing atoms, or groups bonded to the metal center.1f,27 Finally, Lambert and coworkers recently proposed a mechanism for furan synthesis in which in situ production of TMSOTf participated in a multi-catalytic system along with TfOH and bismuth triflate (Bi(OTf)3).28
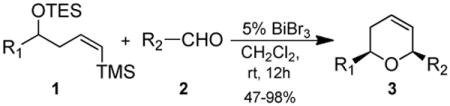 |
(1) |
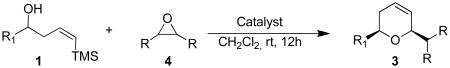
In order to both expand the repertoire of reactions that Bi(III) compounds catalyze as well as investigate the role(s) of this metal ion, we chose to examine the cascade reaction shown in eq 2. Epoxides add versatility to the synthesis since they are common, useful reagents which are easy to handle and prone to neither aldol condensations nor oxidation to the corresponding carboxylic acids during storage. Although a limited number of examples have been implied for this type of reaction with InCl3,15 to the best of our knowledge, no expanded studies or investigations of the catalytic species for this type of transformation have been performed.
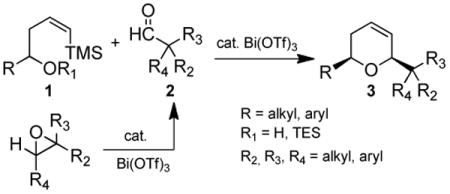 |
(2) |
Results and Discussion
Comparison Studies
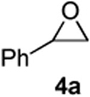
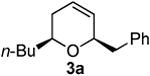
Prior to attempting the complete one pot synthesis, we studied the effect of different catalysts with differing ratios on the tandem cyclization (Table 1). Although the triethysilyloxy- analogs of (Z)-δ-hydroxyalkenylsilanes 1 were attempted first, the Bi(OTf)3 mediated reactions afforded more complex mixtures than those conducted with BiBr3. Therefore, the free alcohol version of the (Z)-δ-hydroxyalkenylsilane 1 was added to phenylacetaldehyde 2a (product which results from epoxide rearrangement of styrene oxide), giving 6-benzyl-2-butyl-3,6-dihydro-2H-pyran 3a as the product (Table 1). Each reaction was carried out with both 1 mol% and 5 mol% catalyst. The following catalysts were chosen for comparison: TfOH, Bi(OTf)3•nH2O prepared according to Dubac's method,29 and both Bi(OTf)3 and BiBr3 obtained from Sigma-Aldrich, Inc. Triflic acid was chosen because of its potential production from the hydrolysis of bismuth triflate (eq. 3).30
Table 1.
Comparison of catalyst loading on tandem reaction of (Z)-δ-hydroxyalkenylsilane 1a and phenylacetaldehyde.
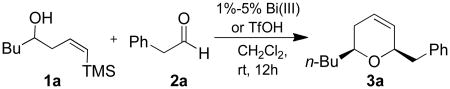 | ||
|---|---|---|
| catalyst | mol % | Yield (%)a |
| TfOH | 1% | 87 |
| Bi(OTf)3•nH2O | 1% | 45 |
| Bi(OTf)3 | 1% | 50 |
| BiBr3 | 1% | 61 |
| TfOH | 5% | 82 |
| Bi(OTf)3•nH2O | 5% | 78 |
| Bi(OTf)3 | 5% | 66 |
| BiBr3 | 5% | 75 |
Isolated yields of pure cis-3a after flash chromatography.
| (3) |
Both the Bi(OTf)3•4H2O synthesized in this work and the commercially available Bi(OTf)3 gave moderate yields of desired dihydropyran product 3a at 1 mol%. When catalyst loading was increased to 5 mol%, bismuth triflate afforded good yields of 3a. The increased yield could be due to either an increased amount of Bi(OTf)3 as a Lewis acid or the formation of an increased amount of triflic acid produced in situ from hydrolysis of bismuth triflate.
After establishing that Bi(OTf)3 can be used for the tandem addition/ISMS sequence, we once again examined both triethylsilyl- protected and unprotected (Z)-δ-hydroxyalkenylsilanes in the cascade involving epoxide rearrangement, intermolecular addition, and an intramolecular silyl-modified Sakurai (ISMS) sequence. We again compared BiBr3, Bi(OTf)3, and TfOH as catalysts/initiators.
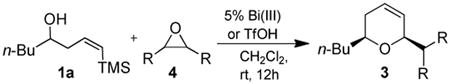
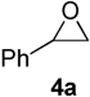
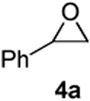
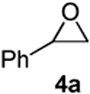
As with the reactions in Table 1, reaction of the unprotected (Z)-δ-hydroxyalkenylsilane 1a with styrene oxide 4a and isobutylene oxide 4b afforded less of the problematic silane byproducts (e.g., disiloxanes, TES-O-TES, TMS-O-TES, TMS-O-TMS as well as unidentified silanes). Although efficient for reactions between δ-silyloxy-,11 or δ-hydroxyalkenylsilanes and aldehydes, BiBr3 failed to induce initial epoxide ring opening to give the aldehyde needed for the addition/ISMS sequence. As confirmed by NMR spectroscopic analysis, starting materials predominantly remained after 12h for the reaction run with 5 mol% BiBr3. This indicates that BiBr3 (or HBr) is not a strong enough acid to facilitate the rearrangement.
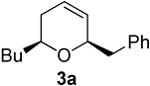
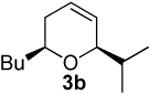
In contrast, bismuth triflate promoted the epoxide rearrangement followed by the addition/ISMS cascade to afford 3,6-dihydro-2H-pyrans 3a-b in 40-59 % isolated yields. As with the reactions between δ-hydroxyalkenylsilanes and aldehydes (Table 1, vide supra), TfOH also initiated the cascade reaction sequence (Table 2), and in the case of styrene oxide, afforded DHP 3a in 78% isolated yield.
Table 2.
Epoxide rearrangement followed by addition and ISMS using the unprotected δ-hydroxyalkenylsilanes.
 | |||
|---|---|---|---|
| epoxide | product | catalyst | yield (%)a |
 |
 |
BiBr3 | NR |
| Bi(OTf)3•nH2O | 41 | ||
| Bi(OTf)3 | 40 | ||
| TfOH | 78 | ||
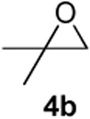 |
 |
BiBr3 | NR |
| Bi(OTf)3•nH2O | 59 | ||
| Bi(OTf)3 | 44 | ||
| TfOH | 52 | ||
Isolated yields of pure cis-isomers after flash chromatography on silica gel.
Since a number of authors argue that TfOH is the real catalyst in Bi(OTf)3-mediated reactions, we synthesized fifteen 2,6-disubstitued-3,6-dihydro-2H-pyrans using both Bi(OTf)3•nH2O and TfOH as catalysts/initiators (Table 3). At least two equivalents of TfOH are possible from hydrolysis of each Bi(III) salt (eq 3), so 5 mol% Bi(OTf)3•nH2O and 10 mol% TfOH were compared directly. In most cases, the yields of isolated, pure products were similar for Bi(OTf)3•nH2O and TfOH. Regardless of its actual role in the catalytic cycle, however, stable, easily handled Bi(OTf)3•nH2O provides a convenient, practical alternative to corrosive and dangerous TfOH.
Table 3.
Synthesis of 2,6-disubstituted 3,6-dihydropyrans.
 | |||||
|---|---|---|---|---|---|
| entry | vinylsilane | epoxide | product | yield(%)a,b | yield(%)a,c |
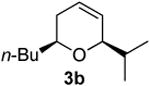
| 1 | 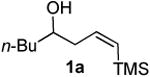 |
 |
 |
56 | 58 |
| 2 | 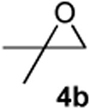 |
 |
66 | 70 | |
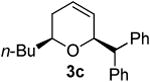
| 3 | 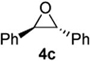 |
 |
31 | 27 | |
| 4 | 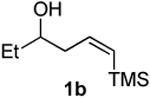 |
 |
 |
53 | 50 |
| 5 | 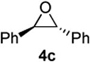 |
 |
28 | 29 | |
| 6 | 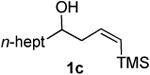 |
 |
 |
59 | 74 |
| 7 | 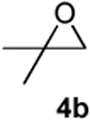 |
 |
57d | 65d | |
| 8 | 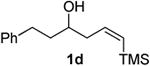 |
 |
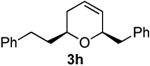 |
71 | 62 |
| 9 | 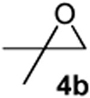 |
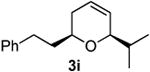 |
42 | 60 | |
| 10 | 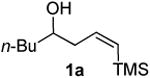 |
 |
 |
47 | 45 |
| 11 | 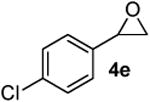 |
 |
49 | 50 | |
| 12 | 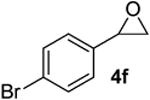 |
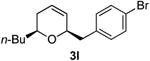 |
41 | 46 | |
| 13 |  |
 |
 |
45 | 60 |
| 14 | 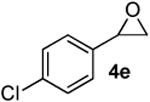 |
 |
54 | 56 | |
| 15 | 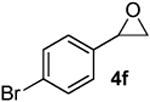 |
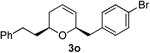 |
59 | 53 | |
Isolated yield of pure cis-isomers only, after flash column chromatography. Reactions were carried out on a ∼0.5 mmol scale in 5.0 mL CH2Cl2 at room temperature using 2.0 equiv of the corresponding epoxide.
5 mol% Bi(OTf)3•H2O.
10 mol% TfOH.
8/2 mixture of product and inseparable 2,6-diheptyl-3,6-dihydro-2H-pyran.
Reaction Scope
We evaluated the utility of the protocol by testing a variety of substrates under the developed reaction conditions (Table 3). Phenyl-, halogenated aryl- and 1,1-dialkyl-alkyl epoxides could be used, but each requires substituents that can stabilize an intermediate cation necessary for hydride rearrangement to occur. Overall yields range from moderate to good, and only cis-diastereomers were isolated after column chromatography with no trans-diastereomers detected by either 1H NMR spectroscopy or GC MS analyses of the crude reaction mixtures.
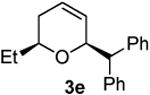
The cis- relative stereochemistry was further confirmed by X-ray crystallographic analysis of isolated product 3e shown in entry 5 of Table 3 (see Supporting Information). The diastereoselectivity is in agreement with the stereochemistry reported by Dobbs and coworkers in their InCl3 mediated study15 and consistent with that reported by Speckamp's group14e and Rychnovsky and co-workers in other Prins-type reactions.13b,16
The overall mechanism (Scheme 1) involves intermolecular attack of an alcohol (or silyloxy) oxygen on an activated aldehyde or ketone, formation of oxocarbenium ion A (2,6-substituents pseudoequatorial), and intramolecular attack of pendant π-electrons to afford an intermediate carbocation which either deprotonates for X = H (Prins) or loses silane for X = SiMe3 (silyl-Prins) to form a dihydropyran product.
Scheme 1.
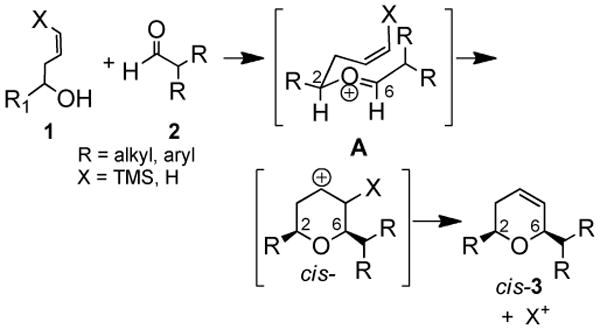
Overall mechanism for silyl-Prins reaction affording cis-dihydropyrans.
The moderate yields as well as the formation of several unidentified byproducts prompted the examination of crude reaction mixtures to determine alternative reaction pathways which were detracting from desired products. The lowest yields invariably resulted when trans-stilbene oxide 4c was used as an aldehyde surrogate. Despite the efficiency of the stilbene oxide rearrangement to 2,2-diphenylacetaldehyde (99%), this epoxide afforded low yields of DHP products 3 regardless of the (Z)-δ-hydroxyalkenylsilane 1 to which it was subjected. (Table 3, entries 3 and 5). The low yields are likely due to steric hindrance within the intermediate (Figure 1) necessary to achieve the requisite silyl-Prins cyclization. We observed the same low yields when bypassing the epoxide rearrangement altogether and conducting the addition/ISMS reaction with freshly-purified 2,2-diphenylacetaldehyde.31
Figure 1.
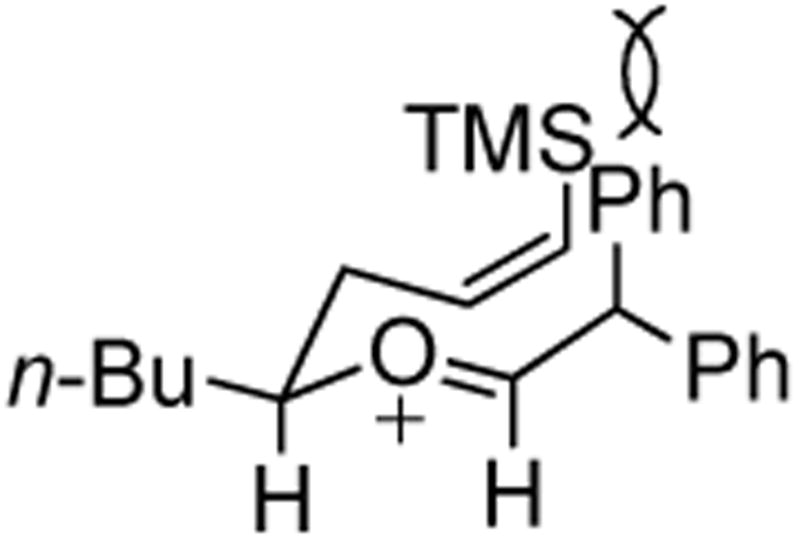
Proposed steric hindrance limiting the transformation of trans-Stilbene oxide to the desired DHP Product.
The reaction shown in entry 7 of Table 3 resulted in a mixture containing 3g and a minor amount of an inseparable second dihydropyran (Scheme 2, G). As reported by Rychnovsky and co-workers, immediately following the formation of oxocarbenium ion A there is competition between the π-electrons of the olefin and other molecules with nucleophilic character (e.g., H2O) for attack of the electrophilic carbon. Reaction of the olefinic π electrons affords the desired cyclization whereas re-addition of water to the intermediate ion would result in the regeneration of the starting materials, C and D. Intramolecular oxonia-Cope rearrangement of the intermediate affords a different oxocarbenium ion B, and results in the formation of new aldehyde E and hydroxyallylsilane F. The resulting mixture of aldehydes (C and E) and hydroxysilanes (D and F) can then undergo competing addition/ISMS sequences to potentially product thee additional dihydropyran byproducts G-I. GC MS analysis of dihydropyran 3g, however, showed no change after exposure to catalyst indicating that once the silyl-Prins step is complete, no further rearrangements or exchanges occur.32
Scheme 2.

Side-chain exchange reactions of oxocarbenium ion intermediates.
Probing the role of Bi(OTf)3
In an effort to explore the nature of Bi(OTf)3 in the overall reaction scheme, experiments were run using a number of additives (Table 4). Initial studies focused on the production of HBr and TfOH in situ.33 Consistent with the hypothesis that the purported Lewis acid acts as a source of HX which in turn functions as the catalyst or initiator, addition of 15 mol% of the hindered, non-nucleophilic base, 2,6-di-tert-butyl-4-methylpyridine (DTBMP) completely suppressed the reaction (entries 2, 3, 7, 14 and 15).30a Given Ollevier and co-workers' report that DTBMP-TfOH efficiently catalyzed the three-component Mannich reaction,34 we added 5 and 10 mol% of this salt to a solution of the epoxide and (Z)-δ-hydroxyalkenylsilane (entries 10-11). This resulted in no observable reaction. To confirm the efficacy of the DTBMP as an acid scavenger, all reactivity was suppressed when the protocol was run with TfOH as the catalyst (entry 15).
Table 4.
Effects of additives on the addition/ISMS reaction sequence.
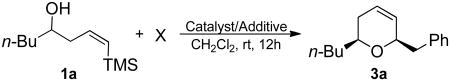 | |||||
|---|---|---|---|---|---|
| entry | X | catalyst | mol% | additive | (%) yielda |
| 1 | 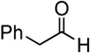 |
BiBr3 | 15 | – | 66 |
| 2 | " | " | DTBMPb | 0 | |
| 3 | TMSOTf | 10 | DTBMPb | 0 | |
| 4 | 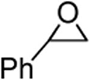 |
Bi(OTf)3•nH2O | 5 | – | 41 |
| 5 | " | " | 3Å sievesc | 48 | |
| 6 | " | " | " | 4Å sievesc | 53 |
| 7 | " | " | " | DTBMPb | 0 |
| 8 | " | Bi(OTf)3•nH2Od | 10 | – | 0 |
| 9 | " | Bi(OTf)3 | " | 4Å sievesc | 47 |
| 10 | " | DTBMP•HOTfe | 5 | – | 0 |
| 11 | " | " | 10 | – | 0 |
| 12 | " | TfOH | " | – | 58 |
| 13 | " | TMSOTf | " | – | 57 |
| 14 | " | TMSOTf | " | DTBMPb | 0 |
| 15 | " | TfOH | " | DTBMPb | 0 |
Isolated yields of pure cis-isomers only, after flash column chromatography. All the reactions were carried out on ∼0.5 mmol scale in 5.0 mL CH2Cl2 at rt using 2.0 equiv of the corresponding epoxide.
15 mol % 2,6-di-tert-butyl-4-methylpyridine
Activated by heating with a propane torch under high vacuum.
Bi(OTf)3•nH2O was stirred in dichloromethane for 30 min and filtered. Only filtrate was used in reaction flask.
2,6-Di-tert-butyl-4-methylpyridinium triflate.
Adventitious water was eliminated by running the reaction with both 3 Å and 4 Å activated molecular sieves. Neither of these had any effect on the reaction, resulting in moderate yields of product (entries 5, 6 and 9). If the Lewis acid had been producing the active catalytic species (HX) through hydrolytic interactions with adventitious H2O, then presumably, both the sieves and acid scavenger would have suppressed the reaction.
As shown in Scheme 3, a Lewis acid/Lewis base interaction between Bi(III) and the hydroxyl group on the (Z)-δ-hydroxyalkenylsilane could explain the inability of molecular sieves to suppress the sequence. To confirm that adventitious water alone does not contribute to the in situ production of TfOH, we stirred a suspension of Bi(OTf)3•nH2O in CH2Cl2 for 30 minutes and filtered the solids through a 0.2 μm PTFE syringe filter. The resulting solution failed to induce rearrangement of the epoxide or the addition/ISMS sequence (Table 4, entry 8). The Lewis acid/Lewis base coordination results in the loss of a ligand (–OTf) from the bismuth compound and produces TfOH in situ. Consistent with the observed experimental results, mol sieves would have no effect on the reaction under this model, while DTBMP would neutralize any HX produced by either simple hydrolysis or the interaction of substrate with Bi(OTf)3.
Scheme 3.
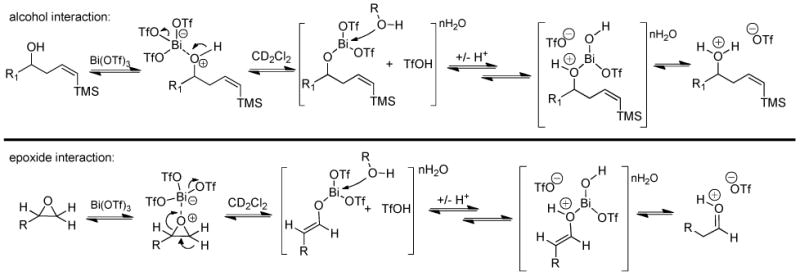
In situ generation of ionic triflates by interaction of substrates and Bi(OTf)3.
Due to Lambert and co-workers' hypothesis28 that Bi(OTf)3 catalyzed reactions afford TMSOTf and that both species are participating in the overall mechanism, we initially presumed that TMSOTf was the active species since at 10 mol% it promotes the reaction to afford product in moderate yield (Table 4, entry 13). Additionally, desilylation of (Z)-1-(trimethylsilyl)oct-1-en-4-ol 1a occurs readily in the presence of TfOH to produce TMSOTf in situ. Only after addition of excess acid do reactions other than protodesilylation occur and cause significant decomposition of the starting material; this most likely occurs through formation of carbocationic intermediates from protonation of the hydroxyl moiety as proposed by Keramane et al. in mechanistic studies of BiCl3-catalyzed chlorination reactions of benzylic alcohols35 and the benzylation of alcohols.36 In both of these cases, however, the authors argue that the Bi(III) atoms are acting as Lewis acids.
At 10 mol% loading levels, TMSOTf, Bi(OTf)3•H2O, and BiBr3 all promote the protodesilylation (GC MS) of the olefin, generating TMSBr or TMSOTf in situ (eq 4). Since all reactivity ceases when the Lewis acid, TMSOTf, is added in the presence of DTBMP, Brønsted acid (TfOH) must be the actual catalyst. Any protodesilylation of the starting material results only in a small amount of TMSOTf as a byproduct.
A number of NMR experiments were also performed to further validate that the reaction is catalyzed by triflic acid. We found that Bi(OTf)3•nH2O is insoluble in CD2Cl2 at room temperature even when H2O is present in small, but detectable amounts in the NMR solvent. No resonances were observed in the 1H, 19F or 13C NMR spectra even with prolonged acquisition times. If any Bi(OTf)3•nH2O is slightly soluble, it is below the limit of detection in NMR spectroscopic experiments. The signature 19F peaks for TfOH (−76.3 ppm), TMSOTf (−77.4 ppm), and Bu4N+ –OTf (−78.7 ppm) were determined using authentic standards. Upon addition of either alkenylsilane or styrene oxide to both 5 and 10 mol% Bi(OTf)3 in CD2Cl2, a 19F peak was observed corresponding to the triflate anion (δ −78.8). This same resonance was observed when either substrate was subjected to 10% TfOH and 10% TMSOTf.
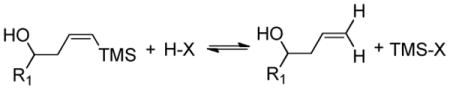 |
(4) |
To further test the argument that the water present in the Bi(OTf)3•nH2O simply reacts in the reaction medium to afford TfOH in situ, a suspension of bismuth triflate was stirred rapidly at room temperature in CD2Cl2 for 90 minutes under an argon atmosphere. The suspension was then filtered through a plug of fine glass fibers to remove the suspended solids, but to allow dissolved species to remain in solution. Unlike the case in which phenylacetaldehyde was added directly to an unfiltered suspension and underwent some aldol reaction, addition of the same aldehyde to the filtered solvent showed no propensity to react. When a similar experiment with filtered Bi(OTf)3•nH2O was performed with the alkenylsilane, no desilylation was observed even after 48h at room temperature. These two experiments once again indicate that intimate contact of the organic substrates and bismuth salt is necessary to activate the suspended Bi(OTf)3 and facilitate dissolution of the solid to afford TfOH which catalyzes either reaction. Therefore, the Lewis acidity of the Bi(OTf)3 does play an important role in the tandem reaction even though the bismuth compound is not the catalytic species.
The observance of the triflate anion peak is likely due to the presence of an ion pair with the triflate anion acting as the counter ion to the protonated substrate (see Scheme 4, vide infra). In order to further assess this possibility, we dissolved cyclohexanol in CD2Cl2, then added 10 mol% Bi(OTf)3 followed by 10 mol% TfOH. Upon addition of bismuth triflate, a 19F signal was observed at −78.9 ppm; addition of TfOH to the same tube increased the intensity of this same resonance. Similar spectroscopic signals were observed for interactions of cyclohexanone with both Bi(OTf)3 and TfOH. As a whole, these studies provide compelling evidence that the overall reaction is TfOH catalyzed, but that the Lewis acidic nature of the Bi(OTf)3 is still important as an initiation step.
Scheme 4.
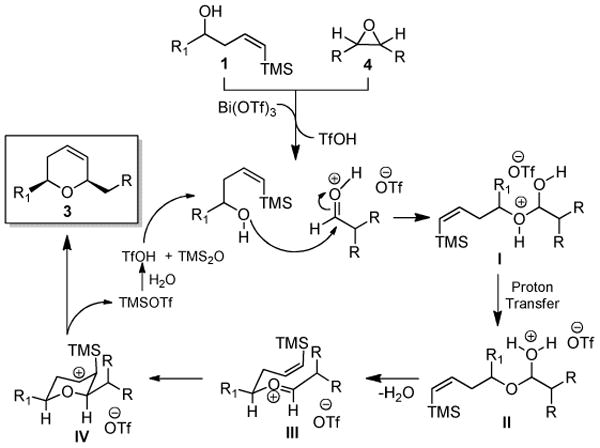
Proposed mechanism for combined rearrangement, addition, and ISMS reaction to dihydropyrans, 3.
Mechanistic Considerations
A key finding of our experiments is that a Lewis acid/base interaction between the substrates and Bi(OTf)3•nH2O occurs prior to formation of any significant quantities of TfOH. Once catalytic quantities of TfOH are present, the reaction proceeds in the same manner as briefly explained in Scheme 1 (vide supra). As more completely shown in Scheme 4, protonation of the aldehyde electrophile is followed by attack of the hydroxyl- moiety of the (Z)-δ-hydroxyalkenylsilane 1 to afford protonated hemiacetal I. Proton-transfer and elimination of water generates (E)-oxocarbenium ion III, which is completely analogous to intermediate A from Scheme 1. The pseudoequatorial disposition of the two side-chains, and pseudoaxial orientation of the silyl- group14e all favor formation of the cis-2,6-disubstituted carbocation IV and elimination of TMSOTf provides product 3. Hydrolysis of the TMSOTf then regenerates TfOH and forms disiloxane (TMSOTMS) as a byproduct. As previously shown, oxocarbenium ion III can undergo [3,3] sigmatropic oxonia-Cope rearrangements to provide some side-chain exchange products which are certainly more prevalent when reactions are run with Bi(OTf)3 instead of BiBr3.32a
Conclusions
We have developed a convenient Bi(OTf)3-initiated cascade reaction involving epoxide rearrangement to an aldehyde electrophile, intermolecular addition of a (Z)-δ-hydroxyalkenylsilane 1, and ISMS reaction to afford cis-2,6-disubstituted-3,6-dihydro-2H-pyrans, 3, in moderate to good yields. NMR spectroscopic evaluation of Bi(OTf)3 in the presence of substrates indicate that Lewis acid/base interactions are necessary to liberate TfOH, the catalytic species, in solution.
Experimental Section
Materials
All reagents and substrates were used as received unless otherwise noted. Dichloromethane was distilled from CaH2. Trifluoromethane-sulfonic acid (TfOH) was purchased from Acros Organics, Inc. Bismuth bromide and bismuth triflate were purchased from Aldrich Chemical Company, Inc. Trimethylsilyltrifluoromethanesulfonate was obtained from GFS Chemicals, Inc. Bismuth triflate tetrahydrate (Bi(OTf)3•4H2O) was synthesized from triflic acid and triphenylbismuth.29 Compounds 1a-1d were synthesized as previously described.9 NMR spectra (13C) were recorded with the aid of an APT experiment in which methylene (2H) and quaternary carbons (0 H) are even (e) and methyl (3H) and methyne (1H) carbons are odd (o). Coupling constants were determined by the method outlined by Hoye and Zhao.37 Single crystal X-ray determinations were carried out using a Bruker SMART Apex II diffractometer using graphite-monochromated Cu Kα radiation. Chromatography was performed using 200-475 MESH silica gel. Unless otherwise noted, all experiments were conducted under argon atmosphere. All compounds were judged to be >95% homogeneous by 1H NMR spectroscopy.
NMR Spectroscopy Studies
1H and 13C NMR chemical shifts are reported as δ using CD2Cl2 (Acros Organics) as a solvent and are referenced to dichloromethane or trimethylsilane. 19F NMR chemical shifts are reported as δ using trichlorofluoromethane (Aldrich) as an internal standard (δ 0.0). NMR samples were prepared by addition of 5-10% catalyst to a solution of substrate (25-50 mg), 0.75ml of deuterated dichloromethane and 10μL of trichlorofluoromethane. In the case of the addition of multiple catalysts, the catalysts (10 mol% each addition) were added sequentially to the same NMR sample, recording both 1H and 19F spectra between additions.
General procedure for the synthesis of dihydropyrans (3a-3o)
Catalyst (TfOH, TMS-OTf, BiOTf3, BiBr3 0.010-0.050 equiv) was weighed into 15 mL round bottom flask and 5 mL CH2Cl2 was added via syringe. The solution was cooled to 0 °C using an ice water bath. δ-Hydroxyalkenylsilane, 1a-1d (1.0 equiv) and epoxide 4a-4f (1.1-2.0 equiv) were added sequentially. The mixture was warmed to rt slowly and stirred for 12 h. The suspension was then filtered through a small SiO2 pipette column with CH2Cl2 as eluent and concentrated in vacuo again. The product was then purified by column chromatography.
Preparation of cis-6-benzyl-2-butyl-3,6-dihydro-2H-pyran (3a)
According to the general procedure, (Z)-1-(trimethylsilyl)oct-1-en-4-ol, 1a (0.100 g, 0.50 mmol, 1.00 equiv) was treated with styrene oxide (0.120 g, 1.00 mmol, 2.0 equiv) to provide 0.067 g (58%) of 3a as a colorless oil, after purification by column chromatography (95:5 petroleum ether:ethyl ether, Rf = 0.57): IR (neat) 3029(m), 2955(s), 2929(s), 2860(m), 1454(m), 1184(m), 1085(s), 1064(s) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.18-7.31 (m, 5H), 5.76-5.81 (m, 1H), 5.61 (dm, J = 10.3 Hz, 1H), 4.31 (m, 1H), 3.48-3.54 (m, 1H), 2.99 (dd, J = 13.5, 7.0 Hz, 1H), 2.68 (dd, J = 13.5, 7.0 Hz, 1H), 1.92-1.98 (m, 2H), 1.22-1.64 (m, 6H), 0.89 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 138.5(e), 129.7(o), 129.4(o), 128.2(o), 126.2(o), 125.2(o), 76.0(o), 74.3(o), 42.3(e), 36.1(e), 31.6(e), 28.0(e), 23.1(e), 14.5(o); Anal. calcd. For C16H22O (230.35): C 83.43, H 9.63; Found: C 83.51, H 9.67.
Preparation of cis-2-butyl-6-isopropyl-3,6-dihydro-2H-pyran (3b)
According to the general procedure, (Z)-1-(trimethylsilyl) oct-1-en-4-ol, 1a (0.100 g, 0.500 mmol, 1.00 equiv) was treated with isobutylene oxide (0.072 g, 1.00 mmol, 2.0 equiv) to provide 0.057 g (63%) of 3b as a colorless oil, after purification by column chromatography (95:5 petroleum ether:ethyl ether, Rf = 0.62): IR (neat) 3031(m), 2957(s), 2929(s), 2868(s), 1465(m), 1366(m), 1185(m), 1080(s) cm−1; 1H NMR (400 MHz, CDCl3) δ 5.80-5.85 (m, 1H), 5.66 (dq, J = 10.3, 1.5 Hz, 1H), 3.84-3.88 (m, 1H), 3.44-3.51 (m, 1H), 1.90-1.94 (m, 2H), 1.71-1.79 (dq, J = 6.6, 5.5 Hz, 1H), 1.25-1.60 (m, 6H), 0.93 (d, J = 5.5 Hz, 3H), 0.92 (d, J = 5.5 Hz, 3H), 0.90(t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 128.6(o), 125.6(o), 79.8(o), 74.0(o), 36.2(e), 33.0(o), 31.8(e), 28.1(e), 23.1(e), 18.3(o), 14.5(o); Anal. calcd. For C12H22O (182.30): C 79.06, H 12.16; Found: C 79.06, H 12.22.
Preparation of cis-6-benzhydryl-2-butyl-3,6-dihydro-2H-pyran (3c)
According to the general procedure, (Z)-1-(trimethyl-silyl)oct-1-en-4-ol, 1a (0.100 g, 0.500 mmol, 1.00 equiv) was treated with trans-stilbene oxide (0.108 g, 0.55 mmol, 1.1 equiv) to provide 0.047 g (31 %) of 3c as a colorless powder, after purification by column chromatography (95:5 petroleum ether:ethyl ether, Rf = 0.43) IR (neat) 3060(m), 3026(m), 2954(s), 2926(s), 2858(s), 1599(m), 1495(s), 1451(s), 1369(m), 1187(m), 1094(s), 745(s), 699(s) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.13-7.37 (m, 10H), 5.74-5.78 (m, 1H), 5.55 (dq, J = 10.3, 1.5 Hz, 1H), 4.78-4.83 (m, 1H), 3.97 (d, J = 8.4 Hz, 1H), 3.52-3.59 (m, 1H), 1.88-1.99 (m, 2H), 1.18-1.54 (m, 6H), 0.84 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 142.5(e), 142.1(e), 129.01(o), 128.97(o), 128.93(o), 128.4(o), 127.9(o), 126.4(o), 126.12(o), 126.06(o), 76.9(o), 74.5(o), 56.6(o), 35.9(e), 31.7(e), 28.1(e), 22.8(e), 14.5(o); Anal. calcd. For C22H26O (306.44): C 86.23, H 8.55; Found: C 85.85, H 8.53
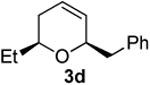
Preparation of cis-6-benzyl-2-ethyl-3,6-dihydro-2H-pyran (3d)
According to the general procedure, (Z)-6-(trimethylsilyl)hex-5-en-3-ol, 1b (0.100 g, 0.580 mmol, 1.0 equiv.) was reacted with styrene oxide (0.139 g, 1.16 mmol, 2.0 equiv) to provide 0.062 g (53%) of 3d as a pale yellow oil, after purification by column chromatography (95:5 petroleum ether:ethyl ether, Rf = 0.52): IR (neat) 3030(m), 2963(s), 1496(m), 1185(m), 1086(s) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.25-7.37 (m, 5H), 5.85 (ddd, J = 10.2, 1.6 Hz, 1H), 5.67 (dd, J = 10.2, 1.6 Hz, 1H), 4.36-4.39 (m, 1H), 3.51 (q, J = 6.6 Hz, 1H), 3.05 (dd, J = 13.7, 6.6 Hz, 1H), 2.75 (dd, J = 13.5, 7.2 Hz, 1H), 2.00-2.03 (m, 2H), 1.51-1.71 (m, 2H), 1.00 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 138.6(e), 129.8(o), 129.6(o), 128.3(o), 126.3(o), 125.3(o), 76.0(o), 75.6(o), 42.3(e), 31.0(e), 29.2(e), 10.1(o); HRMS (CI) calcd for (C14H18O)Na (M + Na)+ = 225.1250, Found = 225.1250.
Preparation of cis-6-benzhydryl-2-ethyl-3,6-dihydro-2H-pyran (3e)
According to the general procedure (Z)-6-(trimethylsilyl)hex-5-en-3-ol, 1b (0.100 g, 0.58 mmol, 1.0 equiv.) was treated with trans-stilbene oxide (0.228g, 1.16 mmol, 2.0 equiv) to provide 0.047 g (29%) of 3e as colorless crystals after purification by column chromatography (95:5 petroleum ether: ethyl ether, Rf = 0.47): IR (neat) 3055(m), 2985(s), 2306(s), 2306(s), 1599(s), 1495(s), 1265(m) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.16-7.37(m, 10H), 5.74-5.79(m, 1H), 5.56 (dd, J = 10.2, 1.6 Hz, 1H), 4.80-4.83 (m, 1H), 3.98 (d, J = 8.2 Hz, 1H), 3.48 (q, J = 7.0 Hz, 1H), 1.92-1.96 (m, 2H), 1.34-1.52 (m, 2H), 0.87 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 142.7(e), 142.2(e), 129.2(o), 129.1(o), 128.6(o), 128.0(o), 126.5(o), 126.2(o), 76.9(o), 75.9(o), 56.6(o), 31.2(e), 29.2(e), 10.3(o); HRMS (CI) calcd for (C20H22O)Na (M + Na)+ = 301.1563, Found = 301.1563.
Preparation of cis-6-benzyl-2-heptyl-3,6-dihydro-2H-pyran (3f)
According to the general procedure (Z)-1-(trimethylsilyl)undec-1-en-4-ol, 1c (0.075 g, 0.31 mmol, 1.0 equiv) was treated with styrene oxide (0.041 g, 0.34 mmol, 1.1 equiv.) to provide 0.050 g (59%) of 3f as a pale yellow liquid after column chromatography (95:5 petroleum ether: ethyl ether, Rf = 0.56): IR (neat) 3029(s), 2858(s), 1604(s), 1495(m), 1185(m),1085(s) cm−1; 1H NMR (400 MHz, CDCl3) δ 7.19-7.31 (m, 5H), 5.79 (dm, J = 10.2 Hz, 1H), 5.60 (dm, J = 10.0 Hz, 1H), 4.29-4.33 (m, 1H), 3.48-3.54 (m, 1H), 2.99 (dd, J = 13.7, 6.6 Hz, 1H), 2.68 (dd, J = 13.7, 7.4 Hz, 1H), 1.93-1.98 (m, 2H), 1.27-1.62 (m, 12H), 0.89 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 138.7(e), 129.8(o), 129.6(o), 128.3(o), 126.3(o), 125.3(o), 76.0(o), 74.3(o), 42.3(e), 36.3(e), 32.0(e), 31.5(e), 29.8(e), 29.5(e), 25.7(e), 22.9(e), 14.4(o); HRMS (CI) calcd for (C19H28O)Na (M + Na)+ = 295.2032, Found = 295.2033.
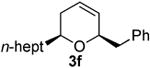
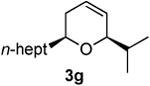
Preparation of cis-2-heptyl-6-isopropyl-3,6-dihydro-2H-pyran (3g)
According to the general procedure (Z)-1-(trimethylsilyl)undec-1-en-4-ol, 1c (0.150 g, 0.62 mmol, 1.0 equiv.) was treated with isobutylene oxide (0.089 g, 1.23 mmol, 2.0 equiv.) to provide 0.072 g (52% yield) of 3g and 0.018 g (11%) of inseparable 2,6-diheptyl-3,6-dihydro-2H-pyran as a pale yellow liquid, after column chromatography (95:5 petroleum ether: ethyl ether, Rf = 0.72): IR (neat) 3035(s), 2972(s), 1467(s), 1186(s), 1076(m) cm−1; 1H NMR (400 MHz, CDCl3) δ 5.80-5.86 (m, 1H), 5.67 (dq, J = 10.16, 1.6 Hz, 1H), 3.83-3.88 (m, 1H), 3.44-3.51 (m, 1H), 1.89-1.94 (m, 2H), 1.75 (dh, J = 5.5, 1.4 Hz, 1H), 1.20-1.60 (m, 12H), 0.86-0.96 (m, 9H); 13C NMR (100 MHz, CDCl3) δ 128.8(o), 125.8(o), 79.8(o), 74.0(o), 36.4(e), 32.9(o), 32.1(e), 31.7(e), 29.9(e), 29.5(e), 25.7(e), 22.9(e), 18.2(o), 14.3(o) HRMS (CI) calcd for (C15H28O)2Na (2M + Na)+ = 471.4173, Found = 471.416
Preparation of cis-6-benzyl-2-phenethyl-3,6-dihydro-2H-pyran (3h)
According to the general procedure, (Z)-1-phenyl-6-(trimethylsilyl)hex-5-en-3-ol 1d (0.100 g, 0.40 mmol, 1.00 equiv) was treated with styrene oxide (0.100g, 0.80 mmol, 2.0 equiv), to provide 0.069g (62%) of 3h as a colorless oil, after purification by column chromatography (95:5 petroleum ether:diethyl ether, Rf = 0.64): IR (neat) 3062(s), 3028(s), 2923(s), 2858(s), 1603(s), 1495(s), 1454(s), 1084(s), 1064(s), 747(s), 699(s); 1H NMR (400 MHz, CDCl3) δ 7.07 – 7.37 (m, 10H), 5.78 – 5.82 (m, 1H), 5.65 (dq, J = 10.4, 1.4 Hz, 1H), 4.27 – 4.29 (1H, m), 3.47 (1H, ddd, J = 14, 9.2, 4.8Hz), 2.98 (1H, dd, J = 13.4, 7.8Hz), 2.64 – 2.81(3H, m), 1.74 – 2.08 (4H, m); 13C NMR (100 MHz, CDCl3) δ 142.4(e), 138.9(e), 129.8(o), 128.7(o), 128.5(o), 128.3(o), 126.4(o), 125.8(o), 125.3(o), 76.0(o), 72.9(o), 42.3(e), 37.7(e), 31.7(e), 31.5(e)
Preparation of cis-2-phenethyl-6-isopropyl-3,6-dihydro-2H-pyran (3i)38
According to the general procedure, (Z)-1-phenyl-6-(trimethylsilyl)hex-5-en-3-ol, 1d (0.100 g, 4.00 mmol, 1.00 equiv) was treated with isobutylene oxide (0.06 g, 0.80 mmol, 2.0 equiv) to provide pure product 0.054g (60%) of 3i as a colorless oil, after purification by column chromatography (95:5 petroleum ether:diethyl ether; Rf = 0.67): IR (neat) 3028.0(s), 2959.6(s), 2649.2(s), 2360.9(s), 1940.7(s), 1603.8(m) 1096.7(s); 1H NMR (400 MHz, CDCl3) δ 7.17 – 7.31 (5H, m), 5.80 – 5.84 (1H, m), 5.67 (1H, dt, J = 10, 1.2 Hz), 3.85 (1H, dd, J = 3.4, 1.4 Hz), 3.48 (1H, dddd, J = 14, 4.4 Hz), 2.69 – 2.86 (2H, m), 1.72 – 2.04 (5H, m), 0.97 (6H, d, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 142.6(e) 129.0(o), 128.8(o), 128.5(o), 125.8(o), 125.6(o), 79.7(o), 72.6(o), 37.8(e), 33.0(o), 31.8(e), 31.7(e), 18.4(o), 18.1(o); HRMS (CI) calcd for (C16H22O)Na (M + Na)+ = 253.1563, Found = 253.1556.
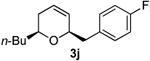
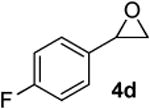
Preparation of cis-2-butyl-6-(4-fluorobenzyl)-3,6-dihydro-2H-pyran (3j)
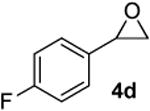
According to the general procedure, (Z)-1-(trimethylsilyl)oct-1-en-4-ol, 1a (0.100 g, 0.50 mmol, 1.00 equiv) was treated with 2-(4-fluorophenyl)oxirane, 4d (0.140g, 1.00 mmol, 2.00 equiv), to provide 0.058g (47%) of 3j as a colorless oil, after purification by column chromatography (95:5 petroleum ether:ethyl ether, Rf: 0.51): IR (neat) 3035(s), 2932(s), 2861(s), 1601(s), 1510(s), 1222(s), 1157(s), 1072(s), 843(s), 713(s); 1H NMR (400 MHz, CDCl3) δ 7.23 (t, J = 6.8 Hz, 2H), 6.99 (t, J = 8.8 Hz, 2H), 5.79-5.84 (m, 1H), 5.61 (d, J = 10.2 Hz, 1H), 4.26-4.32 (m, 1H), 3.48-3.56 (m, 1H), 2.93 (dd, J = 7.0, 6.6 Hz, 1H), 2.70 (dd, J = 7.0, 6.6 Hz, 1H), 1.92-1.99 (m, 2H), 1.28-1.65 (m, 6H), 0.91 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 161.7(e) (C-F; q, J = 244.0 Hz), 134.3(e), 131.2(o), 131.1(o), 129.4(o), 125.6(o), 115.1(o), 114.9(o), 75.8(o), 74.3(o), 41.3(e), 36.0(e), 31.4(e), 27.9(e), 22.9(e), 14.3(o); HRMS (CI) calcd for (C16H21FO)Na (M + Na)+ = 271.1469, Found = 271.1462.
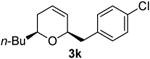
Preparation of cis-2-butyl-6-(4-chlorobenzyl)-3,6-dihydro-2H-pyran (3k)
According to the general procedure, (Z)-1-(trimethylsilyl)oct-1-en-4-ol, 1a (0.100 g, 0.50 mmol, 1.00 equiv) was treated with 2-(4-chlorophenyl)oxirane (0.12 mL, 1.00 mmol, 2.00 equiv), to provide 0.065g (49%) of 3k as a colorless oil, after purification by column chromatography (95:5 petroleum ether:ethyl ether, Rf: 0.56): IR (neat) 3032(s), 2933(s), 2861(s), 1493(s), 1184(s), 1091(s), 833(s); 1H NMR (400 MHz, CDCl3) δ 7.21 (dd, J = 14.0, 7.0 Hz, 4H), 5.76-5.82 (m, 1H), 5.58 (d, J = 10.2 Hz, 1H), 4.27 (s, 1H), 3.45-3.54 (m, 1H), 2.89 (dd, J = 7.0, 6.6 Hz, 1H), 2.68 (dd, J = 7.4, 6.3 Hz, 1H), 1.91-1.96 (m, 2H), 1.25-1.62 (m, 6H), 0.91 (t, J = 6.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 137.2(e), 132.1(e), 131.1(o), 129.4(o), 128.3(o), 125.7(o), 75.6(o), 74.3(o), 41.5(e), 36.0(e), 31.4(e), 27.9(e), 22.9(e), 14.3(o); HRMS (CI) calcd for (C16H21ClO)Na (M + Na)+ = 287.1173, Found = 287.1170.
Preparation of cis-6-(4-bromobenzyl)-2-butyl-3,6-dihydro-2H-pyran (3l)
According to the general procedure, (Z)-1-(trimethylsilyl)oct-1-en-4-ol, 1a (0.100 g, 0.50 mmol, 1.00 equiv) was treated with 2-(4-bromophenyl)oxirane (0.200g, 1.00 mmol, 2.00 equiv), to provide 0.063g (41%) of 3l as a colorless oil, after purification by column chromatography (95:5 petroleum ether:ethyl ether, Rf: 0.46): IR (neat) 3032(s), 2959(s), 2861(s), 1652(s), 1489(s), 1368(s), 1184(s), 1072(s), 1012(s), 712(s); 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.2, Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 5.77-5.84 (m, 1H), 5.58 (d, J = 10.2 Hz, 1H), 4.25-4.31 (m, 1H), 3.46-3.54 (m, 1H), 2.89 (dd, J = 7.4, 6.3 Hz, 1H), 2.68 (dd, J = 7.0, 6.6 Hz, 1H), 1.92-1.97 (m, 2H), 1.25-1.63 (m, 6H), 0.90 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 137.7(e), 131.6(o), 131.3(o), 129.3(o), 125.7(o), 120.2(e), 75.5(o), 74.3(o), 41.6(e), 36.0(e), 31.4(e), 27.9(e), 22.9(e), 14.3(o); HRMS (CI) calcd for (C16H21BrO)Na (M + Na)+ = 331.0668, Found = 331.0664.
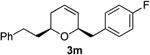
Preparation of cis-6-(4-fluorobenzyl)-2-phenethyl-3,6-dihydro-2H-pyran (3m)
According to the general procedure, (Z)-1-phenyl-6-(trimethylsilyl)hex-5-en-3-ol, 1d (0.124 g, 0.50 mmol, 1.0 equiv) was treated with 2-(4-fluorophenyl)oxirane, 4d (0.138g, 1.00 mmol, 2.0 equiv), to provide 0.088 g (60%) of 3m as a colorless oil, after purification by column chromatography (95:5 petroleum ether:diethyl ether, Rf = 0.53): IR (neat) 3024(s), 2929(s), 1946(s), 1889(s), 1724(s), 1601(s), 1496(s), 1157(s), 838; 1H NMR (400 MHz, CDCl3) δ 6.95 – 7.30 (m, 9H), 5.75 – 5.82 (1H, m), 5.60 (d, J = 10.2 Hz, 1H), 4.18 – 4.25 (m, 1H), 3.39–3.48(m, 1H), 2.88 (dd, J = 13.9, 7.6Hz, 1H), 2.59– 2.77(m, 3H), 1.70 – 2.07 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 161.7(e) (C-F; q, J = 243.3 Hz), 142.3(e), 134.5(e), 131.2(o), 129.6(o), 128.7(o), 128.5(o), 125.9(o), 125.5(o), 115.1(o), 114.9(o), 75.8(o), 72.8(o), 41.3(e), 37.7(e), 31.7(e), 31.4(e); HRMS (CI) calcd for (C20H21FO)Na (M + Na)+ = 319.1469, Found = 319.1465
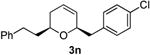
Preparation of cis-6-(4-chlorobenzyl)-2-phenethyl-3,6-dihydro-2H-pyran (3n)
According to the general procedure, (Z)-1-phenyl-6-(trimethylsilyl)hex-5-en-3-ol, 1d (0.124 g, 0.50 mmol, 1.0 equiv) was treated with 2-(4-chlorophenyl)oxirane (0.155 g, 1.00 mmol, 2.0 equiv), to provide 0.087g (56%) of 3n as a pale yellow oil, after purification by column chromatography (95:5 petroleum ether:diethyl ether, Rf = 0.43): IR (neat) 3024(s), 2924(s), 1721(s), 1602(s), 1493(s), 1368(s), 1185(s), 1017(s), 835(s), 746(s); 1H NMR (400 MHz, CDCl3) δ 7.09 – 7.32 (m, 9H), 5.77 – 5.83 (m, 1H), 5.62 (dt, J = 10.2, 1.2 Hz, 1H), 4.23-4.28 (m, 1H), 3.42-3.50 (m, 1H), 2.98 (dd, J = 13.4, 7.8 Hz, 1H), 2.64 – 2.81(m, 3H), 1.74 – 2.08 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 142.3(e), 137.3(e) 132.1(e), 131.2(o), 129.5(o), 128.7(o), 128.5(o), 128.4(o), 125.9(o), 125.6(o), 75.6(o), 72.9(o), 41.5(e), 37.7(e), 31.7(e), 31.4(e); HRMS (CI) calcd for (C20H21ClO)Na (M + Na)+ = 335.1173, Found = 335.1171.
Preparation of cis-6-(4-bromobenzyl)-2-phenethyl-3,6-dihydro-2H-pyran (3o)
According to the general procedure, (Z)-1-phenyl-6-(trimethylsilyl)hex-5-en-3-ol, 1d (0.124 g, 0.50 mmol, 1.0 equiv) was treated with 2-(4-bromophenyl)oxirane (0.199 g, 1.0 mmol, 2.0 equiv), to provide 0.099 g (53%) of 3o as a pale yellow oil, after purification by column chromatography (95:5 petroleum ether:diethyl ether, Rf = 0.48): IR (neat) 3024(s), 1652(s), 1489(s), 1454(s), 1368(s), 1185(s), 746(s); 1H NMR (400 MHz, CDCl3) δ 7.08 – 7.50 (m, 9H), 5.78 – 5.83 (m, 1H), 5.61 (dm, J = 10.16 Hz, 1H), 4.25 (dm, J = 1.6 Hz, 1H), 3.42-3.55 (m, 1H), 2.60 – 2.95(m, 4H), 1.72– 2.10 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 142.3(e), 137.8(e), 131.6(o), 131.3(o), 129.5(o), 128.7(o), 128.5(o), 125.9(o), 125.6(o), 120.2(e), 75.5(o), 72.9(o), 41.6(e), 37.7(e), 31.7(e), 31.4(e); HRMS (CI) calcd for (C20H21BrO)Na (M + Na)+ = 379.0668, Found = 379.0663.
Supplementary Material
Acknowledgments
We thank the AREA Program of the National Institutes of Health (RGM072525-01A2), the Camille and Henry Dreyfus Foundation (Henry Dreyfus Teacher-Scholar Award for R.J.H), the Beckman Scholars' Program (R.F.L), and the Roy R. Charles Center of the College of William & Mary (R.F.L. and S.E.A). Partial support of this work was also provided by a Howard Hughes Medical Institute grant through the Undergraduate Biological Sciences Education Program to the College of William & Mary via student research fellowships (S.E.A). The NSF (CHE-0443345) and the College of William & Mary provided funds for the purchase of the X-ray diffractometer.
Footnotes
Supporting Information Available. 1H and 13C APT NMR spectra for 3a-3o, table of 19F resonances for substrates and triflate species, and X-ray crystallographic data for 3e. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.(a) Bothwell JM, Krabbe SW, Mohan RS. Chem Soc Rev. 2011;40:4649–4704. doi: 10.1039/c0cs00206b. [DOI] [PubMed] [Google Scholar]; (b) Sandhu S, Sandhu JS. Rasãyan J Chem. 2011;4:73–85. [Google Scholar]; (c) Salvador JAR, Pinto RMA, Silvestre SM. Curr Org Synth. 2009;6:426–470. [Google Scholar]; (d) Hua R. Curr Org Synth. 2008;5:1–27. [Google Scholar]; (e) Komatsu N. In: Organobismuth Chemistry. Suzuki H, Matano Y, editors. Elsevier; New York: 2001. pp. 371–440. [Google Scholar]; (f) Gaspard-Iloughmane H, Le Roux C. Eur J Org Chem. 2004:2517–2532. [Google Scholar]; (g) Leonard NW, Wieland LC, Mohan RS. Tetrahedron. 2002;58:8373–8397. [Google Scholar]
- 2.Komatsu N, Uda M, Suzuki H, Takahashi T, Domae T, Wada M. Tetrahedron Lett. 1997;38:7215–7218. [Google Scholar]
- 3.Wada M, Nagayama S, Mizutani K, Hiroi R, Miyoshi N. Chem Lett. 2002:248–249. [Google Scholar]
- 4.Garrigues B, Gonzaga F, Robert H, Dubac J. J Org Chem. 1997;62:4880–4882. [Google Scholar]
- 5.Evans PA, Andrews WJ. Angew Chem, Int Ed. 2008;47:5426–5429. doi: 10.1002/anie.200801357. [DOI] [PubMed] [Google Scholar]; (b) Jung HH, Seiders JR, Floreancig PE. Angew Chem, Int Ed. 2007;46:8464–8467. doi: 10.1002/anie.200702999. [DOI] [PubMed] [Google Scholar]
- 6.Evans PA, Cui J, Gharpure SJ. Org Lett. 2003;5:3883–3885. doi: 10.1021/ol035438t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans PA, Cui J, Gharpure SJ, Polosukhin A, Zhang HR. J Am Chem Soc. 2003;125:14702–14703. doi: 10.1021/ja0384734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wipf P, Hopkins TD. Chem Commun. 2005:3421–3423. doi: 10.1039/b505100b. [DOI] [PubMed] [Google Scholar]
- 9.(a) Evans PA, Cui J, Gharpure SJ, Hinkle RJ. J Am Chem Soc. 2003;125:11456–11457. doi: 10.1021/ja036439j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hinkle RJ, Lian Y, Litvinas ND, Jenkins AT, Burnette DC. Tetrahedron. 2005;61:11679–11685. [Google Scholar]
- 10.Hinkle RJ, Lian Y, Speight LC, Stevenson HE, Sprachman MM, Katkish LA, Mattern MC. Tetrahedron. 2009;65:6834–6839. doi: 10.1016/j.tet.2009.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lian Y, Hinkle RJ. J Org Chem. 2006;71:7071–7074. doi: 10.1021/jo060738k. [DOI] [PubMed] [Google Scholar]
- 12.For the efficient rearrangement of epoxides with catalytic Bi(OTf)3, see: Bhatia KA, Eash KJ, Leonard NM, Oswald MC, Mohan RS. Tetrahedron Lett. 2001;42:8129–8132.
- 13.For a few recent examples, see: Wender PA, Schrier AJ. J Am Chem Soc. 2011;133:9228–9231. doi: 10.1021/ja203034k.Gesinski MR, Rychnovsky SD. J Am Chem Soc. 2011;133:9727–9729. doi: 10.1021/ja204228q.Lin HY, Snider BB. Org Lett. 2011;13:1234–1237. doi: 10.1021/ol200119h.Fenster E, Fehl C, Aubé J. Org Lett. 2011;13:2614–2617. doi: 10.1021/ol200725m.Tenenbaum JM, Morris WJ, Custar DW, Scheidt K. Angew Chem, Int Ed. 2011;50:5892–5895. doi: 10.1002/anie.201102037.Wang J, Crane EA, Scheidt KA. Org Lett. 2011;13:3086–3089. doi: 10.1021/ol200987c.Olier C, Kaafarani M, Gastaldi S, Bertrand MP. Tetrahedron. 2010;66:413–445.Reddy U, Saikia AK. Synlett. 2010:1027–1032.Li H, Loh TP. Org Lett. 2010;12:2679–2681. doi: 10.1021/ol100937r.
- 14.(a) Hémery T, Wibbeling B, Fröhlich R, Hoppe D. Synthesis. 2010:329–342. [Google Scholar]; (b) Pham M, Allatabakhsh A, Minehan TG. J Org Chem. 2008;73:741–744. doi: 10.1021/jo7016857. [DOI] [PubMed] [Google Scholar]; (c) Meilert K, Brimble MA. Org Biomol Chem. 2006;4:2184–2192. doi: 10.1039/b604334h. [DOI] [PubMed] [Google Scholar]; (d) Dobbs AP, Guesné SJJ. Synlett. 2005:2101–2103. [Google Scholar]; (e) Semeyn C, Blaauw RH, Hiemstra H, Speckamp WN. J Org Chem. 1997;62:3426–3427. doi: 10.1021/jo971192s. [DOI] [PubMed] [Google Scholar]
- 15.For Dobbs' work on Prins and silyl/Prins reactions, see: Chio FK, Warne J, Gough D, Penny M, Green S, Coles SJ, Hursthouse MB, Jones P, Hassall L, McGuire TM, Dobbs AP. Tetrahedron. 2011;67:5107–5124.Dobbs AP, Guesné SJJ, Martinović S, Coles SJ, Hursthouse MB. J Org Chem. 2003;68:7880–7883. doi: 10.1021/jo034981k.Dobbs AP, Martinović S. Tetrahedron Lett. 2002;43:7055–7057.
- 16.For a recent example in an approach toward SCH 351448 see: Cheung LL, Marumoto S, Anderson CD, Rychnovsky SD. Org Lett. 2008;10:3101–3104. doi: 10.1021/ol8011474.
- 17.Markó and co-workers developed and have extensively examined the ISMS reaction. For fundamental representative examples see: Leroy B, Markó IE. Tetrahedron Lett. 2001;42:8685–8688.Markó IE, Dobbs AP, Scheirmann V, Chellé F, Bayston DJ. Tetrahedron Lett. 1997;38:2899–2902.Markó IE, Bayston DJ. Tetrahedron. 1994;50:7141–7156.
- 18.Several researchers claim that Bi(III) salts produce H-X in situ see: Bouguerne B, Hoffmann P, Lherbet C. Synth Commun. 2010;40:915–926.Pinto RMA, Salvador JAR, Le Roux C, Carvalho RA, Beja AM, Paixão JA. Tetrahedron. 2009;65:6169–6178.Dumeunier R, Markó IE. Tetrahedron Lett. 2004;45:825–829.
- 19.Spectroscopic evidence for the role of Bi(OTf)3 as a Lewis acid was recently claimed: Qin H, Yamagiwa N, Matsunaga S, Shibasaki M. Angew Chem, Int Ed. 2007;46:409–413. doi: 10.1002/anie.200602909.Qin H, Yamagiwa N, Matsunaga S, Shibasaki M. J Am Chem Soc. 2006;128:1611–1614. doi: 10.1021/ja056112d.
- 20.Kadam ST, Kim SS. J Organomet Chem. 2009;694:2562–2566. [Google Scholar]
- 21.Kobayashi S, Ogino T, Shimizu H, Ishikawa S, Hamada T, Manabe K. Org Lett. 2005;7:4729–4731. doi: 10.1021/ol051965w. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi R, Cook GR. Tetrahedron Lett. 2008;49:3888–3890. doi: 10.1016/j.tetlet.2008.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donnelly S, Thomas EJ, Arnott EA. Chem Commun. 2003:1460–1461. doi: 10.1039/b303151a. [DOI] [PubMed] [Google Scholar]
- 24.Ollevier T, Lavie-Compin G. Tetrahedron Lett. 2004;45:49–52. [Google Scholar]
- 25.Lewis acidity is evident in Bi(III) compounds with bridging ligands: Yin SF, Shimada S. Chem Commun. 2009:1136–1138. doi: 10.1039/b819911f.
- 26.Ollevier T, Mwene-Mbeja TM. Tetrahedron Lett. 2006;47:4051–4055. [Google Scholar]
- 27.Salvador JAR, Pinto RMA, Silvestre SM. Mini-Rev Org Chem. 2009;6:241–274. [Google Scholar]
- 28.Kelly BD, Allen JM, Tundell RE, Lambert TH. Org Lett. 2009;11:1381–1383. doi: 10.1021/ol900198r. [DOI] [PubMed] [Google Scholar]
- 29.Labrouillère M, Le Roux C, Gaspard H, Laporterie A, Dubac J. Tetrahedron Lett. 1999;40:285–286. [Google Scholar]
- 30.(a) Wabnitz TC, Yu JQ, Spencer JB. Chem Eur J. 2004;10:484–493. doi: 10.1002/chem.200305407. [DOI] [PubMed] [Google Scholar]; (b) Bajwa JS, Jiang X, Slade J, Prasad K, Repíc O, Blacklock TJ. Tetrahedron Lett. 2002;43:6709–6713. [Google Scholar]
- 31.In order to explain low yields for reactions involving trans-stilbene oxide, a reviewer suggested that 2,2-diphenylacetaldehyde likely exists substantially in the enol form under the reaction conditions, and is, therefore, unavailable to act as an electrophile.
- 32.For extensive mechanistic discussions see: Jasti R, Rychnovsky SD. J Am Chem Soc. 2006;128:13640–13648. doi: 10.1021/ja064783l.Jasti R, Rychnovsky SD. Org Lett. 2006;8:2175–2178. doi: 10.1021/ol0606738.
- 33.Grimster NP, Wilton DAA, Chan LKM, Godfrey CRA, Green C, Owen DR, Gaunt MJ. Tetrahedron. 2010;66:6429–6436. [Google Scholar]
- 34.(a) Ollevier T, Nadeau E, Guay-Bégin AA. Tetrahedron Lett. 2006;47:8351–8354. [Google Scholar]; (b) Ollevier T, Nadeau E. J Org Chem. 2004;69:9292–9295. doi: 10.1021/jo048617c. [DOI] [PubMed] [Google Scholar]
- 35.Keramane EM, Boyer B, Roque JP. Tetrahedron. 2001;57:1909–1916. [Google Scholar]
- 36.Keramane EM, Boyer B, Roque JP. Tetrahedron. 2001;57:1917–1921. [Google Scholar]
- 37.Hoye TR, Zhao H. J Org Chem. 2002;67:4014–4016. doi: 10.1021/jo001139v. [DOI] [PubMed] [Google Scholar]
- 38.Uenishi J, Ohmi M, Ueda A. Tetrahedron: Asymmetry. 2005;16:1299–1303. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.