

Abstract
Oxidative stress and mitochondrial dysfunction have been implicated in the pathogenesis of neurodegenerative diseases, with the latter preceding the appearance of clinical symptoms. The energy failure resulting from mitochondrial dysfunction further impedes brain function, which demands large amounts of energy. Schisandrin B (Sch B), an active ingredient isolated from Fructus Schisandrae, has been shown to afford generalized tissue protection against oxidative damage in various organs, including the brain, of experimental animals. Recent experimental findings have further demonstrated that Sch B can protect neuronal cells against oxidative challenge, presumably by functioning as a hormetic agent to sustain cellular redox homeostasis and mitoenergetic capacity in neuronal cells. The combined actions of Sch B offer a promising prospect for preventing or possibly delaying the onset of neurodegenerative diseases, as well as enhancing brain health.
1. The Role of Mitochondrial Dysfunction and Mitoenergetic Failure in the Development of Age-Related Neurodegenerative Diseases
Although the etiologies of age-related neurodegenerative diseases are different and multifactorial, mitochondrial dysfunction has been recognized as a common factor in the pathogenesis of these diseases [1, 2]. The common mechanistic features of most age-related neurodegenerative diseases involve the mitochondrial-derived free radical generation and the existence of a hypometabolic state (i.e., a cellular energy deficit) which results from mitochondrial functional impairment [3–6].
The brain is critically dependent on energy supply in order to sustain various neuronal processes such as induction of action potentials and neurotransmission, [7]. In this regard, mitochondria generate approximately 90% of the required energy through oxidative phosphorylation, in which the electron transport process unavoidably results in reactive oxygen species (ROS) generation [8]. Thus, while being the sites of ATP generation, mitochondria are also a significant source of ROS such as hydrogen peroxide (H2O2) and superoxide anion (O2 .−) [9]. Functional impairment of mitochondria, resulting in excessive ROS production and mitoenergetic failure, can result in subtle pathological alterations to neuronal cells. In this regard, aberrations at the level of organelles involved in cellular energetics have been implicated in more than 40 different pathological conditions [10].
Emerging evidence has shown that mitochondrial ROS-induced oxidative stress is involved in the pathogenesis of neurodegenerative diseases [2, 11, 12]. Mitochondrial dysfunction involving electron transport chain (ETC) failure and ROS-mediated cellular damage is common features of Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS) [13–16]. ROS can harm cells by causing random oxidative damage to essential cellular components including DNA, proteins, and lipids. The high susceptibility of the brain to oxidative stress is mainly due to the relative deficiency of antioxidant enzymes, such as superoxide dismutase (SOD), Se-glutathione peroxidase (GPX), glutathione reductase (GR), and catalase (CAT) in this tissue [17, 18]. Furthermore, brain mitochondria are particularly sensitive to oxidative damage and show a slow turnover rate; the accumulation of dysfunctional mitochondria, therefore, can further exacerbate the oxidative stress in brain tissue [19].
In addition to serving as a cellular source of energy to brain tissue, mitochondria also play a critical role in other important cellular processes, including intermediary metabolism, calcium homeostasis, intracellular signaling, and apoptosis through the generation of intracellular oxidants such as H2O2 [9]. Mitochondria-derived ROS can affect overall cellular and mitochondrial function by altering glutathione redox status and/or the posttranslational modification of proteins structure and function via oxidative processes [20, 21]. Redox-sensitive signaling pathways, such as glycogen synthase kinase (GSK) insulin signaling, the C-Jun-NH2-terminal kinase (JNK) proapoptotic, and protein kinase B (Akt) prosurvival pathways, are found to be dysregulated during neurodegeneration associated with enhanced mitochondrial ROS production [22–24]. The release of oxidants (O2 .−, H2O2, NO) from mitochondria into the cytosol further results in chemical (posttranslational) modification of intracellular proteins subsequent to the changes in cellular redox status. Under conditions of oxidative/nitrosative stress, exposure of proteins to ROS or reactive nitrogen species (RNS) can result in oxidation/nitrosylation of protein thiols, nitration of tyrosine residues, and S-glutathionylation involving the formation of mixed disulfides between protein sulfhydryls and glutathione, all of which can lead to protein structural and functional alterations. For instance, the posttranslational modification of key enzymes involved in energy metabolism, such as pyruvate dehydrogenase (PDH), aconitase, and succinyl-CoA transferase (SCOT), often causes a loss of protein function and results in glucose hypometabolism and mitoenergetic failure [3, 23, 25]. In this regard, several clinical studies have shown that before the occurrence of any pathological changes in the brain, impaired glucose metabolism in cerebral tissues is the earliest and consistent abnormality observed in AD and mild cognitive impairment (MCI) [26]. At the molecular level, the oxidative stress-induced impairment in mitochondrial energy-transducing capacity can lead to the opening of mitochondrial permeability transition (MPT) pores [27]. This process, which is accompanied by a collapse of mitochondrial membrane potential and energy production, can result in the reverse operation of ATP synthase, thereby further accelerating ATP depletion resulting in the loss of ion homeostasis, and ultimately, necrotic cell death [28]. MPT pore opening also causes the leakage of cytochrome c from mitochondria into the cytoplasm and triggers a cascade of events such as caspase-9 activation that eventually leads to mitochondrion-driven apoptosis [29]. In summary, enhanced oxidative stress and mitoenergetic failure resulting from mitochondrial dysfunction can all collectively contribute to the pathogenesis of neurodegenerative diseases.
In this paper, we will review the neuroprotective effects of schisandrin B (Sch B) and its potential application as a hormetic agent for favorably influencing the course of neurodegenerative diseases. In addition, we will discuss the biochemical basis of the Sch B-afforded neuroprotection and its possible role in preventing mitoenergetic failure, thereby improving brain health.
2. Dietary Schisandrin B Mitigates Age-Related Impairment in Mitochondrial Antioxidant Status and Functional Capacity in Brain Tissues
Sch B is the most abundant dibenzocyclooctadiene derivative found in Fructus Schisandrae (FS), a traditional Chinese herb commonly used for the treatment of viral and chemical hepatitis. According to Traditional Chinese Medicine (TCM) theory, FS is classified as a “Qi-invigoration” herb under the “Yang” family. Holistically, while “Yang” is viewed as a manifestation of body function supported by various organs, “Qi” is regarded as a vital substance, which is fundamental to life and provides energy for the human body [30]. FS is believed to nourish the “Qi” of the heart, kidney, liver, lung, and spleen, with improved energy utilization and increased longevity. In order to validate this time-honored TCM theory, various scientific investigations have been undertaken in several laboratories, including our own, to investigate the effectiveness of Sch B in ameliorating impairments in mitochondrial antioxidant status and functional capacity. Early findings have confirmed the beneficial effect of Sch B on liver function, particularly in enhancing the detoxification of xenobiotics and the regeneration of damaged liver [31]. Emerging evidence demonstrates the protective effect of Sch B against free-radical-induced tissue injury in various tissues, including the brain [32–36].
Data from aging studies provide additional information on the ability of Sch B to mitigate age-related impairments in brain mitochondrial antioxidant status and functional capacity. Progressive impairment in mitochondrial antioxidant status and a decline in mitochondrial function have been documented in brain tissue of aging rodents [37, 38]. Decreases in the levels of mitochondrial reduced glutathione (GSH) and α-tocopherol (α-TOC), as well as the activities of GPX and manganese SOD (MnSOD) in brain tissue are invariably associated with the increased mitochondria-driven production of ROS as a function of aging in experimental animals [37, 38]. In agreement with TCM theory, experimental data has shown that long-term dietary supplementation with Sch B significantly enhances mitochondrial antioxidant status, stimulates mitochondrial respiratory function, and maintains mitochondrial structural integrity, as well as decreasing ROS production in the brain of experimental animals [37, 38]. Correlation analysis has further indicated that the protection afforded by Sch B against age-dependent decline in mitochondrial antioxidant components, particularly MnSOD, may be relevant to survival enhancement [37]. MnSOD has been recognized as a major antioxidant enzyme protecting the brain against both oxidative and nitrative stress by eliminating O2 .− and preventing the generation of highly reactive peroxynitrite (ONOO−) arising from the reaction between NO with O2 .−. High levels of oxidative and nitrative stress have been implicated in pathogenesis of neurodegenerative conditions such as AD, PD, and Huntington's disease (HD) [39]; animals with MnSOD deficiency have been shown to be more vulnerable to the mitochondrial neurotoxin malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), both of which can induce oxidative stress [40]. Furthermore, the alteration of redox status as a result of MnSOD-mediated ROS elimination has also been proposed to regulate specific stress-responsive genes related to antiapoptotic substances, such as cyclin B1, cyclin A, GADD153, and 14-3-3 zeta [41].
The enhancement of mitochondrial function (i.e., respiratory activity) by Sch B has been shown to stimulate mitochondrial ATP generation in aging mouse brains [37]. Improvement in respiratory function has been proposed to stimulate the activity of sirtuin [42], which can activate antiapoptotic, anti-inflammatory, and antistress responses, as well as modulate the aggregation of proteins associated with neurodegenerative conditions, thereby preventing or delaying the onset of neurological damage [43]. Enhanced regeneration of NAD as a result of Sch B-induced respiratory functional improvement can also be beneficial to neuronal health. Studies have demonstrated that exposure of neuronal cells to NAD can prevent axonal degeneration, a pathophysiological process that often precedes the death of neuronal cell bodies in PD and AD, through the activation of the NAD-dependent deacetylase sirtuin-1 (SIRT1) pathway [44, 45].
3. Neuroprotection Afforded by Schisandrin B
In addition to the aforementioned aging research, studies utilizing various models of brain oxidative challenge have further advanced our understanding of the neuroprotective effect of Sch B against oxidative insult. In the mouse model of cerebral damage induced by the prooxidant tertbutylhydroperoxide (t-BHP), Sch B treatment prevented the increase in cerebral lipid peroxidation and impairment in GSH antioxidant status produced by intracerebroventricular injection of t-BHP [34]. The restoration of GSH levels by Sch B is important for neuronal viability because an imbalanced glutathione redox status can trigger the sulfhydryl groups of cysteine sulfinic acids to react with other sulfhydryl groups, including that of glutathione, with the resultant formation of disulfides or mixed disulfides. The modification of protein sulfhydryl groups can lead to alterations in protein function, with disruption in normal cellular physiology, as has been shown in the inactivation of the SH-group containing enzyme glucose 6-phosphate dehydrogenase (G6DPH) by ROS [46]. Transgenic overexpression of G6PDH in mouse dopaminergic nigrostriatal neurons has been shown to reduce their sensitivity to MPTP, presumably through increasing the activity of this rate-limiting enzyme in the hexose monophosphate shunt, which provides NADPH required for the regeneration of GSH from its oxidized form and sulfhydryl groups of oxidatively modified antioxidant enzymes [47].
In a rat model of cerebral ischemia/reperfusion (I/R) injury induced by clamping of both carotid arteries in anesthetized animals, the neuroprotective property of Sch B has been shown to be associated with the maintenance of antioxidant status as well as the structural integrity of mitochondria [32]. The neuronal injury caused by I/R challenge involves pathophysiological mechanisms such as ROS production and intracellular calcium overload; the resultant perturbation of cellular redox homeostasis and mitochondrial permeability transition pore opening eventually leads to necrotic and/or apoptotic cell death [48]. Long-term treatment with Sch B (10 mg/kg/day, p.o., for 15 days) has been shown to afford protection in the abovementioned model of I/R-induced cerebral injury; as evidenced by a significant increase the percentage of viable tissue after I/R challenge, which was determined by quantifying the area of 2,3,8-triphenyl tetrazolium chloride-stained tissue in brains of control and Sch B-pretreated rats [32]. The protection was associated with enhancement of mitochondrial antioxidant components and preservation of mitochondrial integrity, as evidenced by increased GSH, α-TOC levels, and MnSOD activity as well as a reduced sensitivity of the Ca2+-induced permeability transition and a reduction in cytochrome c release [32]. The preservation of mitochondrial integrity by long-term Sch B treatment is of crucial importance for the mitoenergetic capacity of cerebral mitochondria. This notion is supported by the observation that the ATP-generating capacity of cerebral mitochondria was increased by long-term Sch B treatment in brain tissue of both control and I/R-challenged rats [32].
Recently, Giridharan et al. have demonstrated that Sch B prevents memory deficits in mice induced by scopolamine, a muscarinic antagonist that induces central cholinergic blockade [36]. Since cholinergic neurotransmission in the basal forebrain is believed to play an important role in learning and memory, scopolamine toxicity may provide a useful model of cognitive impairment in rodents and humans [36]. The scopolamine-induced cognitive deterioration resembles the memory disturbances observed in AD, which are associated with an increase in acetylcholinesterase (AChE) activity and decreased acetylcholine levels [49]. In the aforementioned study, Sch B treatment significantly inhibited AChE activity, resulting in increased acetylcholine levels in the brain, accompanied by an amelioration of the learning and memory impairment caused by scopolamine. Although the detailed inhibitory mechanism of Sch B on AChE remains to be investigated, it was postulated that metabolites of Sch B (see below) may be responsible for the AChE inhibition [36]. The neuroprotective effects of Sch B were also found to be closely related to its antioxidant activity, in which the antioxidant depletion associated with scopolamine toxicity was ameliorated by Sch B treatment, as indicated by enhancements in GSH levels, GPX, and SOD activities as well as a reduction in malondialdehyde and nitrite levels in brain tissue [36].
Taken together, the beneficial effects of Sch B on mitochondrial antioxidant status and mitochondrial function may suggest a new therapeutic approach for the prevention and/or treatment neurodegeneration, thereby promoting brain health in aging individuals.
4. Biochemical Mechanism Underlying Schisandrin B Cytoprotection Against Oxidant Injury in Neuronal Cells
Sch B is a dibenzocyclooctadiene derivative with one methylenedioxy group which can be dealkylated by a cytochrome P-450 (CYP-) catalyzed reaction (Figure 1). In vivo, Sch B was metabolized to yield three main phase I metabolites, in which several oxidation routes appeared to be involved: (1) hydroxylation of an alkyl substitute and (2) demethylation of the OCH3 groups on the aromatic rings [50]. The metabolites were detectable in bile and urine of rats. Sch B could also be demethylated by demethylase presented in red blood cells and then further metabolized to produce phenolic hydroxyl group [51]. The metabolism of Sch B ultimately leads to the formation of a quinone and a subsequent low level ROS production via redox cycling [52, 53]. The modest amount of ROS generated during Sch B metabolism has been shown to elicit a glutathione antioxidant response, which results in an enhancement of glutathione antioxidant status that is closely associated with a decreased susceptibility of various tissues, including the brain, to oxidative injury [32, 54, 55].
Figure 1.

Chemical structures of schisandrin B.
The causal relationship between Sch B-induced GSH enhancement and its cytoprotection against oxidative injury in neuronal cells is further supported by a recent study in differentiated PC12 dopaminergic neuronal cells [56]. Paraquat (PQ), a Parkinsonism-inducing agent, was used to induce oxidative injury in differentiated PC12 cells, and the extent of neuronal damage, as indicated by GSH depletion and cell death, was significantly reduced by pretreatment with (−)Sch B, a potent Sch B stereoisomer [56]. The cytoprotective effect of (−)Sch B in PQ-challenged cells was abrogated by inhibitors of γ-glutamylcysteine ligase (GCL) and GR, suggesting the crucial involvement of GSH regeneration and GSH synthesis in the protection against oxidative stress [56]. Further investigations in PC12 cells subjected to an acute t-BHP challenge have revealed that while the initial GSH depletion induced by the peroxide was reduced through the GR-catalyzed regeneration of GSH in (−)Sch B-pretreated cells, the subsequent enhancement of GSH recovery was mainly mediated by the GCL-catalyzed synthesis of GSH [56]. The results suggested that (−)Sch B treatment may increase the resistance of dopaminergic cells to oxidative stress both by reducing the extent of oxidant-induced GSH depletion and enhancing GSH recovery. A comprehensive study using cultured cell lines derived from various tissues has further demonstrated that the enhancement of GR-mediated GSH regeneration is a universal protective mechanism afforded by Sch B in response to an acute oxidant challenge. The study has also shown that acute t-BHP-induced GSH depletion is significantly reduced by Sch B treatment, in which the activity of GR was found to be enhanced [57]. This finding is consistent with the postulation that GR-catalyzed GSH regeneration is crucial for the maintenance of cellular glutathione redox status, and hence cell survival, even in the absence of oxidative stress [58, 59].
Although the molecular mechanism underlying Sch B-induced neuroprotection remains to be elucidated, the low levels of ROS produced during CYP-mediated Sch B metabolism have been shown to activate several different redox-sensitive signal transduction pathways, with the eliciting of protective cellular responses [60, 61]. Our laboratory has shown recently that (−)Sch B caused a dose-dependent and sustained increase in ROS production as well as a time-dependent activation of mitogen-activated protein kinase (MAPK), particularly extracellular signal-regulated kinases (ERK)1/2 [60, 61]. The MAPK activation was followed by an enhanced translocation of NF-E2-related factors 2 (Nrf2) to the nucleus and the eliciting of a glutathione-dependent antioxidant response in cultured hepatocytes and cardiomyocytes [60, 61]. Nrf2 functions as a redox-sensitive transcription factor that is translocated from the cytosol to the nucleus upon phosphorylation by upstream kinase and then binds to the antioxidant response element consensus sequence to induce a glutathione antioxidant response through the expression of antioxidant proteins such as GR, GPX, and glutathione transferase (GST). Since the eliciting of adaptive responses to oxidative stress often requires one or more members of the MAPK cascade, the possible involvement of oxidative stress-sensitive c-Jun N-terminal kinases (JNK) and p38 MAPK (p38) kinases in the neuronal model cannot be excluded. In addition to MAPK-mediated phosphorylation, Nrf2 has also been shown to be phosphorylated by protein kinase C (PKC) or phospoinositol-3 kinase (PI3K) under conditions of oxidative stress, and the activation of two or more of these pathways may be required to achieve cytoprotection in a cell type- and stimulant-dependent manner [62]. Therefore, the information obtained from hepatocytes and cardiomyocytes, when viewed in the light of established biochemical mechanism of the Sch B-induced glutathione-dependent protection observed in neurons, suggests a new approach for future investigation of the involvement of JNK, p38, PKC, and/or PI3K in the Nrf2-mediated signaling pathways in Sch B-induced neuroprotection.
A recent study of Sch B protection against cisplatin (cis-diamminedichloroplatinum II, cDDP)-induced neurotoxicity has revealed the involvement of nuclear factor Kappa B (NF-κB) [35]. cDDP is a chemotherapeutic agent used for the treatment of various solid tumors. However, its clinical use is limited by its adverse effects, particularly ROS-mediated neurotoxicity which can produce severe cognitive dysfunction [63]. As assessed by the passive avoidance performance task test, cDDP-treated mice were found to develop both short-term and long-term memory deficits that could be significantly improved by Sch B treatment. In this study, the ability of Sch B to ameliorate memory deficits was associated with its ability to suppress the activation of NF-κB. The inhibition of NF-κB by Sch B was further demonstrated in this study by the reduction of the downstream activation of p53 and caspase 3. NF-κB has been well established as a common mediator of cDDP-induced cytotoxicity and has been implicated in many other neurodegenerative diseases such as AD, PD, and HD [35]. Therefore, the ability of Sch B to modulate NF-κB, p53, and caspase-3 signaling pathways further strengthens the prospect of its potential use for preventing or ameliorating neurodegenerative disorders.
5. Potential Role of Schisandrin B as a Hormetic Agent in Preventing Mitoenergetic Failure and Improving Brain Health
To date, the use of antiapoptotic agents to prevent neurodegenerative disorders has not been successful, mainly due to the toxicity and the associated risk of carcinogenesis of the compounds in question. On the other hand, the effectiveness of free radical scavenging antioxidants in achieving neuroprotection requires high concentrations that are not readily achievable in brain tissue. Therefore, in recent years, there has been a growing interest, as supported by a large volume of experimental evidence, in the possible use of hormetic agents as a novel therapeutic alternative in preventing various pathological conditions, including those associated with neurodegeneration [64].
By definition, hormesis refers to an adaptive response of cells and organism to a moderate stress and a hormetic response is a biphasic dose-response phenomenon in which a chemical/stimulus (e.g., ionizing radiation, heat stress, or ROS) has a beneficial effect on maintaining cellular homeostasis at low doses/levels but causes a toxic effect at high doses/levels [65, 66]. A compelling body of evidence suggests that the low-level mitochondrial ROS-induced oxidative stress produced by caloric restriction, hypothermia, or hyperbaric oxygen conditions elicits adaptive cellular signaling/responses that can result in an increased stress resistance, which maintains homeostasis and promotes longevity, thus providing the basis of mitochondrial hormesis or “mitohormesis” [67–70]. However, all the aforementioned conditions are generally impractical to achieve in humans. Therefore, the approach to achieving mitohormesis using hormetic agents to enhance endogenous mitochondrial antioxidant status and functional capacity may offer a promising prospect for preventing or possibly delaying the onset of age-related neurodegenerative diseases.
In this regard, Sch B acts as a hormetic agent in cultured cells, with cytoprotective effects predominating at low concentrations and cytotoxicity occurring at high concentrations [71, 72]. Pharmacokinetic study on Sch B showed that a mean value of 96.1 ± 14.1 ng/mL (~0.25 μM) of maximum plasma drug concentration and a half-life of about 2 hours can be achieved by oral administration of 15 mg Sch B to healthy male subjects [73]. Toxicology study also demonstrated that the LD50 values of orally or intraperitoneally administered petroleum ether extract of FS, which contains 40% (w/w) lignans, were 10.5 and 4.4 g/kg, respectively [74]. No death was observed by administration of a single oral dose of Sch B at 2 g/kg in rats [75]. Furthermore, an intragastric dose of 200 mg of Sch B for 30 days did not significantly affect body weight, blood parameters, and histological parameters of major organs in mice [75]. When given at 10 mg/kg daily for 4 weeks, Sch B also did not affect appetite, liver, or kidney functions, as well as liver histological parameters in dogs [75]. Taken together, Sch B treatment is generally considered to be safe; a single oral dose (0.8 g/kg), multiple doses (200 mg/kg × 30) as well as dietary supplementation (0.012%, w/w, starting from 9 months of age until death) did not cause any undetectable adverse effects in rodents [34, 37].
As far as bioavailability is concerned, the delivery of therapeutic agents across the blood-brain barrier (BBB) represents a major challenge to therapeutic agents aimed at treating brain disorders. This leads to the failure of therapeutic agents that might otherwise be effective if they can penetrate the BBB. Although whether or not Sch B or its metabolites can penetrate the BBB requires further investigation; the aforementioned observations from Sch B in preventing different types of oxidative stress-induced neuronal injury have provided substantial evidence to support the postulation that Sch B can cross the BBB and exert its actions in brain tissue.
In addition to safety and bioavailability concerns, the ability of a hormetic agent to enhance mitoenergetic capacity is another indispensable property for achieving mitohormesis in brain and promoting brain health. Mitoenergetic failure has emerged as a focus of research on the pathology of various age-related neurodegenerative diseases, with numerous investigations attempting to identify potential therapeutic agents that might enhance mitochondrial functional capacity. While health supplements, such as lipoic acid, curcumin, and resveratrol, have all been shown to increase antioxidant capacity by functioning as free radical scavengers, their effectiveness in enhancing mitochondrial function and thereby helping to maintain neuronal viability has not been fully investigated. On the other hand, the well-established mitochondrial antioxidant and mitoenergetic sparing properties of Sch B make it a particularly promising candidate for promoting brain health. Results from our recent investigation have further demonstrated that Sch B treatment can attenuate the oxidative stress induced by 3-nitroproponic acid, a potent irreversible inhibitor of mitochondrial succinate dehydrogenase that has been used in experimental models of HD, in differentiated PC12 cells and prevent the oxidative stress-induced energy crisis by suppressing the activation of the JNK signaling pathway and the consequent inhibition of PDH [76], a key bioenergetic enzyme which bridges anaerobic and aerobic brain energy metabolism. This observation supports the role of Sch B in enhancing glucose utilization in the brain. Abnormalities in brain glucose metabolism have been implicated in the early stages of various neurodegenerative disease development [77].
As of today, investigations on the beneficial effects of Sch B on neurodegenerative disorders remain predominantly preclinical. However, substantial evidence of Sch B on reducing oxidative stress-induced neuronal injury has shed light on its potential use as a therapeutic agent for neurodegenerative diseases. Recently, clinical studies have adopted the concept of disease modification, in which the improvement on long-term clinical outcomes, presumably by slowing down disease progression, is evaluated in patients. In this connection, the reduction of free radical-induced oxidative stress represents a novel therapeutic approach for retarding the progression of neurodegenerative diseases, such as PD, in that MAO-B inhibitors were used in the therapy in order to prevent oxidative stress generated by the transformation of dopamine to its metabolites 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenyl-acetic acid [78]. This approach has favorably influenced the progression of PD, as assessed by both motor and nonmotor symptoms [79], suggesting the causal relationship between reduction of oxidative stress and neuroprotection. As such, the ability of Sch B to retard disease progression would be a primary endpoint in clinical trial. Clinical parameters that are more relevant to pathogenesis of neurodegenerative diseases should be assessed in the trials.
Taken together, we posit that Sch B can function as a hormetic agent by eliciting a cellular antioxidant response, with resultant enhancement of cellular redox capacity and mitochondrial functional integrity, thereby maintaining cellular homeostasis against oxidative challenge. The occurrence of deleterious posttransitional modification of key bioenergetic enzymes can also be reduced by the improved cellular redox status. The abilities of Sch B to enhance mitoenergetic capacity and fortify antioxidant defense offer a promising prospect in preventing or delaying the onset of neurodegenerative disorders, possibly by inhibiting cell apoptosis (see Figure 2).
Figure 2.
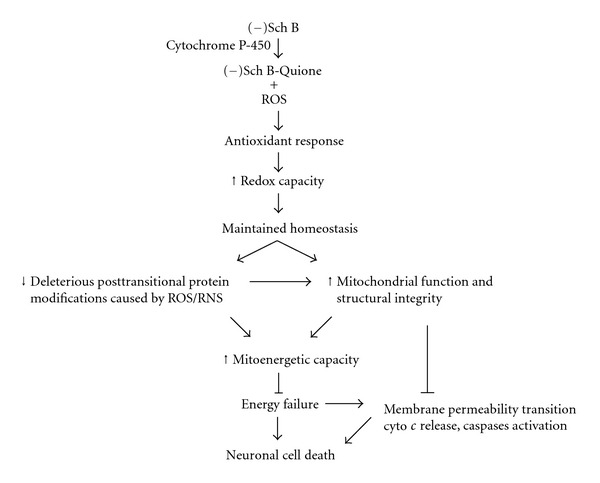
The role of Schisandrin B as a hormetic agent in preventing mitoenergetic failure and neurodegeneration (please refer to the text for details).
References
- 1.Beal MF. Mitochondria, free radicals, and neurodegeneration. Current Opinion in Neurobiology. 1996;6(5):661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 2.Bowling AC, Beal MF. Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sciences. 1995;56(14):1151–1171. doi: 10.1016/0024-3205(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 3.Lam PY, Yin F, Hamilton RT, Boveris A, Cadenas E. Elevated neuronal nitric oxide synthase expression during ageing and mitochondrial energy production. Free Radical Research. 2009;43(5):431–439. doi: 10.1080/10715760902849813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nature Reviews Neuroscience. 2006;7(4):278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson GS, Craft S. The role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. Central Nervous System Drugs. 2003;17(1):27–45. doi: 10.2165/00023210-200317010-00003. [DOI] [PubMed] [Google Scholar]
- 6.Abramov AY, Gegg M, Grunewald A, Wood NW, Klein C, Schapira AH. Bioenergetic consequences of PINK1 mutations in Parkinson disease. PLoS One. 2011;6(10) doi: 10.1371/journal.pone.0025622. Article ID e25622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nogueiras R, Tschop MH, Zigman JM. Central nervous system regulation of energy metabolism: ghrelin versus leptin. Annals of the New York Academy of Sciences. 2008;1126:14–19. doi: 10.1196/annals.1433.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace DC. Mitochondrial DNA in aging and disease. Scientific American. 1997;277(2):40–47. doi: 10.1038/scientificamerican0897-40. [DOI] [PubMed] [Google Scholar]
- 9.Cadenas E. Mitochondrial free radical production and cell signaling. Molecular Aspects of Medicine. 2004;25(1-2):17–26. doi: 10.1016/j.mam.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Storz P. Reactive oxygen species-mediated mitochondria-to-nucleus signaling: a key to aging and radical-caused diseases. Science’s STKE. 2006;2006(332, article re3) doi: 10.1126/stke.3322006re3. [DOI] [PubMed] [Google Scholar]
- 11.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Annals of Neurology. 1995;38(3):357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 12.Beal MF. Energy, oxidative damage, and Alzheimer’s disease: clues to the underlying puzzle. Neurobiology of Aging. 1994;15(supplement 2):S171–S174. doi: 10.1016/0197-4580(94)90198-8. [DOI] [PubMed] [Google Scholar]
- 13.Parker WD, Jr., Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Annals of Neurology. 1989;26(6):719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 14.Parker WD, Jr., Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology. 1990;40(8):1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 15.Parker WD, Jr., Parks J, Filley CM, Kleinschmidt-DeMasters BK. Electron transport chain defects in Alzheimer’s disease brain. Neurology. 1994;44(6):1090–1096. doi: 10.1212/wnl.44.6.1090. [DOI] [PubMed] [Google Scholar]
- 16.Bharath S, Andersen JK. Glutathione depletion in a midbrain-derived immortalized dopaminergic cell line results in limited tyrosine nitration of mitochondrial complex I subunits: implications for Parkinson’s disease. Antioxidants and Redox Signaling. 2005;7(7-8):900–910. doi: 10.1089/ars.2005.7.900. [DOI] [PubMed] [Google Scholar]
- 17.Marklund SL, Westman NG, Lundgren E, Roos G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Research. 1982;42(5):1955–1961. [PubMed] [Google Scholar]
- 18.Hussain S, Slikker W, Jr., Ali SF. Age-related changes in antioxidant enzymes, superoxide dismutase, catalase, glutathione peroxidase and glutathione in different regions of mouse brain. International Journal of Developmental Neuroscience. 1995;13(8):811–817. doi: 10.1016/0736-5748(95)00071-2. [DOI] [PubMed] [Google Scholar]
- 19.Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxidants and Redox Signaling. 2010;12(4):503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. European Journal of Biochemistry. 2000;267(16):4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 21.Inarrea P, Moini H, Rettori D, et al. Redox activation of mitochondrial intermembrane space Cu,Zn-superoxide dismutase. Biochemical Journal. 205;387(part 1):203–209. doi: 10.1042/BJ20041683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor JM, Ali U, Iannello RC, Hertzog P, Crack PJ. Diminished Akt phosphorylation in neurons lacking glutathione peroxidase-1 (Gpx1) leads to increased susceptibility to oxidative stress-induced cell death. Journal of Neurochemistry. 2005;92(2):283–293. doi: 10.1111/j.1471-4159.2004.02863.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Q, Lam PY, Han D, Cadenas E. c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. Journal of Neurochemistry. 2008;104(2):325–335. doi: 10.1111/j.1471-4159.2007.04957.x. [DOI] [PubMed] [Google Scholar]
- 24.Cai F, Wang F, Lin FK, et al. Redox modulation of long-term potentiation in the hippocampus via regulation of the glycogen synthase kinase-3β pathway. Free Radical Biology and Medicine. 2008;45(7):964–970. doi: 10.1016/j.freeradbiomed.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44(36):11986–11996. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 26.Hoyer S. Brain glucose and energy metabolism abnormalities in sporadic Alzheimer disease. Causes and consequences: an update. Experimental Gerontology. 2000;35(9-10):1363–1372. doi: 10.1016/s0531-5565(00)00156-x. [DOI] [PubMed] [Google Scholar]
- 27.Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. The FEBS Letters. 2001;495(1-2):12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- 28.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochemical and Biophysical Research Communications. 2003;304(3):463–470. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 29.Jemmerson R, Dubinsky JM, Brustovetsky N. Cytochrome c release from CNS mitochondria and potential for clinical intervention in apoptosis-mediated CNS diseases. Antioxidants and Redox Signaling. 2005;7(9-10):1158–1172. doi: 10.1089/ars.2005.7.1158. [DOI] [PubMed] [Google Scholar]
- 30.Ko KM, Mak DHF, Chiu PY, Poon MKT. Pharmacological basis of “Yang-invigoration” in Chinese medicine. Trends in Pharmacological Sciences. 2004;25(1):3–6. doi: 10.1016/j.tips.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Ko KM, Mak DH. Schisandrin B and other dibenzocyclooctadiene ligands. In: Lester P, Ong C, Halliwell B, editors. Herbal Medicine and Molecular Books in Health and Disease Management. New York, NY, USA: Marcel Dekker; 2004. pp. 289–314. [Google Scholar]
- 32.Chen N, Chiu PY, Ko KM. Schisandrin B enhances cerebral mitochondrial antioxidant status and structural integrity, and protects against cerebral ischemia/reperfusion injury in rats. Biological and Pharmaceutical Bulletin. 2008;31(7):1387–1391. doi: 10.1248/bpb.31.1387. [DOI] [PubMed] [Google Scholar]
- 33.Lam PY, Chiu PY, Leung HY, Chen N, Leong PK, Ko KM. Schisandrin B co-treatment ameliorates the impairment on mitochondrial antioxidant status in various tissues of long-term ethanol treated rats. Fitoterapia. 2010;81(8):1239–1245. doi: 10.1016/j.fitote.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Ko KM, Lam BYH. Schisandrin B protects against tert-butylhydroperoxide induced cerebral toxicity by enhancing glutathione antioxidant status in mouse brain. Molecular and Cellular Biochemistry. 2002;238(1-2):181–186. doi: 10.1023/a:1019907316129. [DOI] [PubMed] [Google Scholar]
- 35.Giridharan VV, Thandavarayan RA, Bhilwade HN, Ko KM, Watanabe K, Konishi T. Schisandrin B, attenuates cisplatin-induced oxidative stress genotoxicity and neurotoxicity through modulating NF-κB pathway in mice. Free Radical Research. 2012;46(1):50–60. doi: 10.3109/10715762.2011.638291. [DOI] [PubMed] [Google Scholar]
- 36.Giridharan VV, Thandavarayan RA, Sato S, Ko KM, Konishi T. Prevention of scopolamine-induced memory deficits by schisandrin B, an antioxidant lignan from Schisandra chinensis in mice. Free Radical Research. 2011;45(8):950–958. doi: 10.3109/10715762.2011.571682. [DOI] [PubMed] [Google Scholar]
- 37.Ko KM, Chen N, Leung HY, Leong EPK, Poon MKT, Chiu PY. Long-term schisandrin B treatment mitigates age-related impairments in mitochondrial antioxidant status and functional ability in various tissues, and improves the survival of aging C57BL/6J mice. BioFactors. 2008;34(4):331–342. doi: 10.3233/BIO-2009-1086. [DOI] [PubMed] [Google Scholar]
- 38.Chiu PY, Leung HY, Poon MKT, Ko KM. Chronic schisandrin B treatment improves mitochondrial antioxidant status and tissue heat shock protein production in various tissues of young adult and middle-aged rats. Biogerontology. 2006;7(4):199–210. doi: 10.1007/s10522-006-9017-y. [DOI] [PubMed] [Google Scholar]
- 39.Bolanos JP, Almeida A, Stewart V, et al. Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. Journal of Neurochemistry. 1997;68(6):2227–2240. doi: 10.1046/j.1471-4159.1997.68062227.x. [DOI] [PubMed] [Google Scholar]
- 40.Andreassen OA, Ferrante RJ, Dedeoglu A, et al. Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Experimental Neurology. 2001;167(1):189–195. doi: 10.1006/exnr.2000.7525. [DOI] [PubMed] [Google Scholar]
- 41.Guo G, Yan-Sanders Y, Lyn-Cook BD, et al. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Molecular and Cellular Biology. 2003;23(7):2362–2378. doi: 10.1128/MCB.23.7.2362-2378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hipkiss AR. Energy metabolism, altered proteins, sirtuins and ageing: converging mechanisms? Biogerontology. 2008;9(1):49–55. doi: 10.1007/s10522-007-9110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Outeiro TF, Marques O, Kazantsev A. Therapeutic role of sirtuins in neurodegenerative disease. Biochimica et Biophysica Acta. 2008;1782(6):363–369. doi: 10.1016/j.bbadis.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 44.Raff MC, Whitmore AV, Finn JT. Neuroscience: axonal self-destruction and neurodegeneration. Science. 2002;296(5569):868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- 45.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305(5686):1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 46.Reed DJ. Glutathione: toxicological implications. Annual Review of Pharmacology and Toxicology. 1990;30:603–631. doi: 10.1146/annurev.pa.30.040190.003131. [DOI] [PubMed] [Google Scholar]
- 47.Mejias R, Villadiego J, Pintado CO, et al. Neuroprotection by transgenic expression of glucose-6-phosphate dehydrogenase in dopaminergic nigrostriatal neurons of mice. The Journal of Neuroscience. 2006;26(17):4500–4508. doi: 10.1523/JNEUROSCI.0122-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burwell LS, Brookes PS. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxidants and Redox Signaling. 2008;10(3):579–599. doi: 10.1089/ars.2007.1845. [DOI] [PubMed] [Google Scholar]
- 49.Smith CM, Swash M. Possible biochemical basis of memory disorder in Alzheimer disease. Annals of Neurology. 1978;3(6):471–473. doi: 10.1002/ana.410030602. [DOI] [PubMed] [Google Scholar]
- 50.Cui YY, Wang MZ. Aspects of schizandrin metabolism in vitro and in vivo. European Journal of Drug Metabolism and Pharmacokinetics. 1993;18(2):155–160. doi: 10.1007/BF03188790. [DOI] [PubMed] [Google Scholar]
- 51.Zheng RL, Kang JH, Chen FY, Wang PF, Ren JG, Liu QL. Difference in antioxidation for schisandrins and schisantherin between bio- and chemo-systems. Phytotherapy Research. 1997;11(8):600–602. [Google Scholar]
- 52.Lin LY, di Stefano EW, Schmitz DA, et al. Oxidation of methamphetamine and methylenedioxymethamphetamine by CYP2D6. Drug Metabolism and Disposition. 1997;25(9):1059–1064. [PubMed] [Google Scholar]
- 53.Ip SP, Ma CY, Che CT, Ko KM. Methylenedioxy group as determinant of schisandrin in enhancing hepatic mitochondrial glutathione in carbon tetrachloride-intoxicated mice. Biochemical Pharmacology. 1997;54(2):317–319. doi: 10.1016/s0006-2952(97)00164-0. [DOI] [PubMed] [Google Scholar]
- 54.Yim TK, Ko KM. Schisandrin B protects against myocardial ischemia-reperfusion injury by enhancing myocardial glutathione autioxidant status. Molecular and Cellular Biochemistry. 1999;196(1-2):151–156. [PubMed] [Google Scholar]
- 55.Yim TK, Ko KM. Methylenedioxy group and cyclooctadiene ring as structural determinants of schisandrin in protecting against myocardial ischemia-reperfusion injury in rats. Biochemical Pharmacology. 1999;57(1):77–81. doi: 10.1016/s0006-2952(98)00297-4. [DOI] [PubMed] [Google Scholar]
- 56.Lam PY, Ming Ko K. (-)Schisandrin B ameliorates paraquat-induced oxidative stress by suppressing glutathione depletion and enhancing glutathione recovery in differentiated PC12 cells. BioFactors. 2011;37(1):51–57. doi: 10.1002/biof.136. [DOI] [PubMed] [Google Scholar]
- 57.Lam PY, Leong PK, Chen N, Ko KM. Schisandrin B enhances the glutathione redox cycling and protects against oxidant injury in different types of cultured cells. BioFactors. 2011;37(6):439–446. doi: 10.1002/biof.179. [DOI] [PubMed] [Google Scholar]
- 58.Smith AC, Boyd MR. Preferential effects of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) on pulmonary glutathione reductase and glutathione/glutathione disulfide ratios: possible implications for lung toxicity. Journal of Pharmacology and Experimental Therapeutics. 1984;229(3):658–663. [PubMed] [Google Scholar]
- 59.Doroshenko N, Doroshenko P. Ion dependence of cytotoxicity of carmustine against PC12 cells. European Journal of Pharmacology. 2003;476(3):185–191. doi: 10.1016/s0014-2999(03)02191-5. [DOI] [PubMed] [Google Scholar]
- 60.Leong PK, Chiu PY, Chen N, Leung H, Ko KM. Schisandrin B elicits a glutathione antioxidant response and protects against apoptosis via the redox-sensitive ERK/Nrf2 pathway in AML12 hepatocytes. Free Radical Research. 2011;45(4):483–495. doi: 10.3109/10715762.2010.550917. [DOI] [PubMed] [Google Scholar]
- 61.Chiu PY, Chen N, Leong PK, Leung HY, Ko KM. Schisandrin B elicits a glutathione antioxidant response and protects against apoptosis via the redox-sensitive ERK/Nrf2 pathway in H9c2 cells. Molecular and Cellular Biochemistry. 2011;350(1-2):237–250. doi: 10.1007/s11010-010-0703-3. [DOI] [PubMed] [Google Scholar]
- 62.Weng CJ, Chen MJ, Yeh CT, Yen GC. Hepatoprotection of quercetin against oxidative stress by induction of metallothionein expression through activating MAPK and PI3K pathways and enhancing Nrf2 DNA-binding activity. New Biotechnology. 2011;28(6):767–777. doi: 10.1016/j.nbt.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 63.Troy L, McFarland K, Littman-Power S, et al. Cisplatin-based therapy: a neurological and neuropsychological review. Psychooncology. 2000;9(1):29–39. doi: 10.1002/(sici)1099-1611(200001/02)9:1<29::aid-pon428>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 64.Calabrese V, Cornelius C, Dinkova-Kostova AT, et al. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochimica et Biophysica Acta. 2012;1822(5):753–783. doi: 10.1016/j.bbadis.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 65.Kaiser J. Hormesis. Sipping from a poisoned chalice. Science. 2003;302(5644):376–379. doi: 10.1126/science.302.5644.376. [DOI] [PubMed] [Google Scholar]
- 66.Mattson MP. Hormesis defined. Ageing Research Reviews. 2008;7(1):1–7. doi: 10.1016/j.arr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Masoro EJ. Hormesis and the antiaging action of dietary restriction. Experimental Gerontology. 1998;33(1-2):61–66. doi: 10.1016/s0531-5565(97)00071-5. [DOI] [PubMed] [Google Scholar]
- 68.Cypser JR, Johnson TE. Multiple stressors in Caenorhabditis elegans induce stress hormesis and extended longevity. Journals of Gerontology Series A. 2002;57(3):B109–B114. doi: 10.1093/gerona/57.3.b109. [DOI] [PubMed] [Google Scholar]
- 69.Conti B, Sanchez-Alavez M, Winsky-Sommerer R, et al. Transgenic mice with a reduced core body temperature have an increased life span. Science. 2006;314(5800):825–828. doi: 10.1126/science.1132191. [DOI] [PubMed] [Google Scholar]
- 70.Ali SS, Marcondes MCG, Bajova H, Dugan LL, Conti B. Metabolic depression and increased reactive oxygen species production by isolated mitochondria at moderately lower temperatures. The Journal of Biological Chemistry. 2010;285(42):32522–32528. doi: 10.1074/jbc.M110.155432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chiu PY, Leung HY, Poon MKT, Mak DHF, Ko KM. Effects of schisandrin B enantiomers on cellular glutathione and menadione toxicity in AML12 hepatocytes. Pharmacology. 2006;77(2):63–70. doi: 10.1159/000092773. [DOI] [PubMed] [Google Scholar]
- 72.Wu YF, Cao MF, Gao YP, et al. Down-modulation of heat shock protein 70 and up-modulation of Caspase-3 during schisandrin B-induced apoptosis in human hepatoma SMMC-7721 cells. World Journal of Gastroenterology. 2004;10(20):2944–2948. doi: 10.3748/wjg.v10.i20.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ono H, Matsuzaki Y, Wakui Y, et al. Determination of schizandrin in human plasma by gas chromatography-mass spectrometry. Journal of Chromatography B. 1995;674(2):293–297. doi: 10.1016/0378-4347(95)00298-7. [DOI] [PubMed] [Google Scholar]
- 74.Volicer L, Sramka M, Janku I, Capek R, Smetana R, Ditteova V. Some pharmacological effects of Schizandra chinensis . Archives Internationales de Pharmacodynamie et de Therapie. 1966;163:249–262. [Google Scholar]
- 75.Chang HM, But PPH. Pharmacology of Oriental Plants. Singapore. Oxford, UK: Pergamon Press; 1965. [Google Scholar]
- 76.Lam PY, Ko KM. Beneficial effect of (–) Schisandrin B against 3-nitropropionic acid-induced cell death in PC12 cells. doi: 10.1002/biof.1009. BioFactors. In press. [DOI] [PubMed] [Google Scholar]
- 77.Blum-Degen D, Frolich L, Hoyer S, Riederer P. Altered regulation of brain glucose metabolism as a cause of neurodegenerative disorders? Journal of Neural Transmission, Supplement. 1995;(46):139–147. [PubMed] [Google Scholar]
- 78.Muller T. New small molecules for the treatment of Parkinson’s disease. Expert Opinion on Investigational Drugs. 2010;19(9):1077–1086. doi: 10.1517/13543784.2010.504711. [DOI] [PubMed] [Google Scholar]
- 79.Olanow CW, Rascol O, Hauser R, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. New England Journal of Medicine. 2009;361(13):1268–1278. doi: 10.1056/NEJMoa0809335. [DOI] [PubMed] [Google Scholar]