

Abstract
Methylmercury (MeHg) is a ubiquitous environmental contaminant which bioaccumulates in marine biota. Fish constitute an important part of a balanced human diet contributing with health beneficial nutrients but may also contain contaminants such as MeHg. Interactions between the marine n-3 fatty acids eicosapentaenoic acid (20:5n-3, EPA) and docosahexaenoic acid (22:6n-3, DHA) with MeHg-induced toxicity were investigated. Different toxic and metabolic responses were studied in Atlantic salmon kidney (ASK) cell line and the mammalian kidney-derived HEK293 cell line. Both cell lines were preincubated with DHA or EPA prior to MeHg-exposure, and cell toxicity was assessed differently in the cell lines by MeHg-uptake in cells (ASK and HEK293), proliferation (HEK293 and ASK), apoptosis (ASK), oxidation of the red-ox probe roGFP (HEK293), and regulation of selected toxicological and metabolic transcriptional markers (ASK). DHA was observed to decrease the uptake of MeHg in HEK293, but not in ASK cells. DHA also increased, while EPA decreased, MeHg-induced apoptosis in ASK. MeHg exposure induced changes in selected metabolic and known MeHg biomarkers in ASK cells. Both DHA and MeHg, but not EPA, oxidized roGFP in HEK293 cells. In conclusion, marine n-3 fatty acids may ameliorate MeHg toxicity, either by decreasing apoptosis (EPA) or by reducing MeHg uptake (DHA). However, DHA can also augment MeHg toxicity by increasing oxidative stress and apoptosis when combined with MeHg.
1. Introduction
Methylmercury (MeHg) is an environmental contaminant produced from natural or anthropogenic sources of mercury by methylation in widespread sulphate reducing bacteria [1]. MeHg enters the aquatic food chain and accumulates to become a threat for higher-order aquatic mammals and fish, but also to human health through consumption of contaminated fish [2]. MeHg has been shown to be detrimental for human health [3], with many studies emphasizing its neurological toxicity [4, 5]. The molecular pathway by which MeHg exerts its toxicity has been the issue for extensive research. Although MeHg seems to induce specific cytotoxic symptoms, one main route for MeHg molecular toxicity has yet to be elucidated [6, 7]. However, MeHg has a strong affinity for thiol groups, making every cysteine-containing protein a potential target for MeHg-binding and disruption, meaning that there may not exist one specific route of toxicity [8]. In the search for a specific molecular mechanism of MeHg-cytotoxicity, several mechanisms have been suggested for example, oxidative stress [9, 10], excito-toxicological effects [7], microtubule and cell-structural damage [11], genotoxic effects [12], and elevated intracellular Ca2+ leading to apoptosis [11, 13].
The occurrence of MeHg in seafood has led to a debate regarding health promoting nutrients through fish consumption, versus the risk for contaminant exposure [14–16]. Fish serve as an important source of nutrients, vitamins, and minerals and constitute an important part of a balanced diet. Some of the beneficial nutrients in fish are the long chained marine n-3 fatty acids eicosapentaenoic acid (EPA, 20:5n-3) and docosahexaenoic acid (DHA, 22:6n-3), which has shown to be important for optimal cognitive health and neuronal development [17]. But in addition to its nutritional benefits, fish may also accumulate heavy metals and other environmental contaminants in edible parts, posing an exposure risk for higher-order mammals. Many epidemiological studies have investigated the effects of chronic low-dose fetal exposure of MeHg in different geographical locations [6]. Some of these studies report no adverse effects [18, 19], while other studies have reported adverse effects [20]. Myers et al. [21] suggest that dietary effects may be responsible for the discrepancies in MeHg toxicity between different geographical localities. They argue that a study [18, 19], performed at the Seychelles which showed no adverse effects, is based on a mainly fish consuming population, while another, performed at the Pharoe island [20] which shows adverse effects, was based on populations consuming mainly whale meat. Following this argumentation, a fish-based diet may contain certain ameliorating nutrients that will reduce the toxicity of MeHg.
Recently there has been increasing focus on interactions between nutrients and toxicants and how nutrients and the nutrient composition of organisms may affect the toxicity of different environmental contaminants. Reviews have pointed to the lack of research on nutrient-MeHg interactions and suggest that an increased focus on nutrient-MeHg interaction may increase understanding of MeHg toxicological mechanisms [6]. Nutrients can affect MeHg toxicity and retention in fish, as shown by Bjerregaard et al. [22] who demonstrated that dietary selenite decreased MeHg retention in rainbow trout (Oncorhynchus mykiss). Marine n-3 fatty acids, in particular DHA, have also been shown to modulate MeHg-toxicity in rats [23], and to possibly modulate MeHg neurotoxicity, as demonstrated by in vitro studies [24].
The aim of this study was to elucidate possible intervening effects of n-3 marine PUFA (DHA and EPA) compared to the n-6 fatty acid arachidonic acid (ARA, 20:4n-6) on MeHg cytotoxicity in Atlantic salmon kidney (ASK) cells. Human embryonic kidney (HEK293) cells were included in certain aspects of the study, and MeHg-induced toxicity was compared between the two cell types by assessing effects on cell proliferation and death using the xCELLigence system. Interaction effects caused by fatty acids on MeHg toxicity were screened by investigating known mechanistic effects of MeHg, such as uptake of MeHg in both cell lines, apoptosis in ASK cells, and oxidation of roGFP in HEK293 cells. Additionally, we investigated the regulation of transcriptional markers for MeHg toxicity and fatty acids metabolism and how DHA, EPA, and MeHg affected these in ASK cells.
2. Materials and Methods
2.1. General Methodology
2.1.1. Cell Culture
ASK cells (CRL-2643) were purchased from ATCC (London, UK). The cells were grown in Leibowitz L15 media with 20% FBS and 1 × Penicillin/Streptavidin/Amphotericin (all from Sigma, St. Louis, MO, USA) at 20°C without adding CO2. The cells were passaged when a confluence of almost 100% was reached.
HEK293 cells were a gift from Marc Niere (University of Bergen, Norway). The cells were grown in DMEM (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS, 1 × L-glutamine, and 1 × Penicillin/Streptavidin/Amphotericin at 37°C with 5% CO2. The cells were passaged when reaching confluence of almost 100%.
2.1.2. Coupling of Fatty Acids (FAs) to Bovine Serum Albumin (BSA)
Fatty acids were coupled to fatty-acid-free BSA (FAF-BSA) (PAA, Pasching, Austria) as described by [26]. Briefly, the fatty acids were weighed and 0.04 mL chloroform per mg FA was added. The chloroform was evaporated under N2. Furthermore, Potassium hydroxide (KOH) was added in a 1 : 3 relationship and the vial shaken in a whirl mixer for ten minutes. FAF-BSA was added in a 2.5 : 1 ratio to FA. The solution was then shaken 45 minutes, sterile-filtered, and stored anoxic at −80°C until use.
2.1.3. Basic Experimental Design
ASK cells were seeded in concentrations of 24 000 cells per cm2. After 24 hours the cells were preincubated with 300 μM DHA, 300 μM EPA, or 300 μM ARA, all purchased from Sigma, St. Louis, MO, USA. The controls were preincubated with equivalent amounts of BSA as would be found together with fatty acids at concentrations of 300 μM. The cells were preincubated with fatty acids or BSA for 72 hours, and the media were removed before MeHg, diluted in media, or control, just fresh media, was added. Several endpoints were subsequently measured within a time frame of 48 hours.
HEK293 cells were seeded, and similar to ASK they were preincubated with the same fatty acids; however, the incubation time was reduced to 24 hours due to a more rapid cell growth of HEK293 compared to ASK. MeHg, or control, was added and endpoint measurements were performed within a time frame of 48 hours.
2.2. Comparing ASK and HEK293
2.2.1. Uptake of MeHg in FA Preincubated Cells
ASK cells were seeded at a concentration of 24 000 cells per cm2. Cells were treated as described in basic experimental design. 48 hours after MeHg addition, the cells were scraped off using a rubber policeman and washed 3 times in 1 × PBS. The cells were further divided into two aliquots, where one was added to a DMA 80 mercury analyzer. The other half of the cells were lyzed, and total protein was measured using the BCA protein assay (Thermo Scientific Pierce, Rockford, IL, USA) on a Labsystems iEMS Reader (Thermo Fisher Scientific Inc., Waltham, MA, USA).
HEK293 cells were seeded at a concentration of 15 000 cells per cm2. Cells were treated as described in basic experimental design. 24 hours after MeHg addition, cells were harvested and analyzed as described previously for ASK cells.
2.2.2. Cell Toxicity/Impedance Assay
In order to investigate the effect of MeHg on cell viability and proliferation, we implemented the xCELLigence RTCA SP impedance assay from Roche Diagnostics (Mannerheim, Germany) where cell adherence is measured in real time. The instrument was used according to the manufacturer's instructions [27]. The base of the xCELLigence technology is the disposable E-plates, which are similar to normal 96-well microtiter plates. These plates are connected to an RTCA analyzer and computer with RTCA-associated software. The E-plates contain gold-plated sensor electrodes in the bottom of each well, which enables impedance readings from a small current emitted into the system. The distance from one electrode to the next in the xCELLigence system is possibly obstructed by cell media and cells adhering to the electrodes. Cells will act as insulators at the electrodes and will alter impedance measurement based on cell density and cell adherence. Hence, this impedance measurement will correspond to cell growth, cell toxicity, and adherence qualities (morphology) of the cells. A broader introduction of the xCELLigence system and software has previously been presented in [27, 28].
ASK cells were seeded at a concentration of 24 000 cells per cm2 in a 96-well E-plate. Fresh media containing BSA were added 24 hours after seeding, at a concentration of 150 μM (equivalent to the amount incorporated in FA). The cells were incubated another 72 hours, before they were washed twice in fresh media and MeHg was added in concentrations from 1 to 4 μM MeHg dispersed in fresh media. Impedance measurement was recorded every 30 minutes throughout the analysis and up to 48 hours after addition of MeHg.
HEK293 cells were seeded at a concentration of 15 000 cells per cm2 in a 96-well E-plate. Fresh media containing BSA were added, similarly as in ASK cells. The cells were incubated for another 24 hours, before they were washed twice in media, and MeHg dispersed in media was added in concentrations of 1–7 μM. Impedance measurement was recorded every 30 minutes, with the exception of the first hour after any modification to the cells, where it was measured every 15 minutes. Impedance measurements were performed throughout the analysis and up to 48 hours after addition of MeHg.
After addition of MeHg to both ASK and HEK293, the xCELLigence plot showed a pronounced increase in cell adherence. Since this also occurred in the control, it was deemed likely that this specific effect was due to the washing of the cells with the resulting loss of cell adherence and increase in floating cells. When these cells adhered during the next couple of hours, a pseudoincrease in growth curve would be expected. Moreover, the renewal of the media with fresh nutrient was also likely to impose a sudden increase in growth of the cells. In order to focus on the more long-term effects of MeHg, it was chosen to normalize cells when the growth curve stabilized. In order to standardize this normalization, growth curve of control cells in both ASK and HEK cells was smoothed using the Prism 5.04 (Graphpad software Inc., San Diego, CA, USA), to get more continuous curves. After smoothing, the first derivate of the curve was calculated in order to investigate the slope of the curve. At the time point where the first-derivative first approached zero (where cell growth stabilized), the curve was normalized in all treatments for the respective cell type.
2.3. Analyses of ASK Cells
2.3.1. Apoptosis Assay
ASK cells were seeded at a concentration of 24 000 cells per cm2 in 24-well culture plates and further treated as described in basic experimental design. After 48-hour exposure to MeHg, the cells were fixed with 4% Para formaldehyde (Chemi Teknik, Oslo, Norway) and stained with 4 μg mL−1 bisbenzimide H 33342 (Sigma, St. Louis, MO, USA). The percentage apoptosis was counted based on cell morphology in a Nikon fluorescence microscope. The fluorescent imaging was performed at the Molecular Imaging Center (FUGE, Norwegian Research Council), University of Bergen.
2.3.2. Real Time RT-PCR
ASK cells were preincubated with the different FA and exposed to MeHg in 6-well plates at a cell density of 24 000 cells/cm2, as previously described. All treatments were performed in triplicates. After exposure to MeHg for 48 hours, the cells were harvested using a rubber policeman. Cells were lysed, and total RNA was extracted using RNeasy columns (Qiagen, Oslo, Norway). The quantity and quality of the RNA were assessed with the NanoDrop ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). The concentration of RNA in one well of a 6-well plate containing 24 000 cells per cm2 was approximately 50 ng μL−1.
The expression of 11 target genes and two reference genes in ASK cells after FA and MeHg exposure (Table 1), was analyzed using a two-step real-time RT-PCR protocol [29]. The RT reactions were run in triplicates in a 96-well plate, with 250 ng RNA in each sample. For PCR efficiency calculations a 6-fold serial dilution (1000-31 ng) in triplicates was made from a pooled RNA sample. Template controls (ntc) and RT enzyme control (nac) were included in each 96-well plate. The RT reaction was performed according to manufacturer's instructions using TaqMan Reverse Transcription Reagents (Applied biosystems, Foster City, CA, USA) on a GeneAmp PCR 9700 machine (Applied Biosystems, Foster City, CA, USA). Reverse transcription was performed at 48°C for 60 min by using oligo dT primers (2.5 μM) for all genes in 30 μL total volume. The final concentration of the other chemicals in each RT reaction was MgCl2 (5.5 mM), dNTP (500 mM of each), 10X TaqMan RT buffer (1X), RNase inhibitor (0.4 U/μL), and Multiscribe reverse transcriptase (1.67 U/μL) (Applied Biosystems). Diluted cDNA (1 : 2) (2.0 μL cDNA from each RT reaction) was transferred to a new 96-well reaction plate and the qPCR run in 10 μL reactions on the LightCycler 480 Real-Time PCR System (Roche Applied Sciences, Basel, Switzerland). Real-time PCR was performed by using SYBR Green Master Mix (LightCycler 480 SYBR Green master mix kit, Roche Applied Sciences,), which contains FastStart DNA polymerase and gene-specific primers (500 nM). PCR was achieved with a 5 min activation and denaturizing step at 95°C, followed by 45 cycles of a 15 s denaturing step at 95°C, a 60 s annealing step and a 30 s synthesis step, at 72°C. Target gene mean-normalized expression (MNE) was determined using a normalization factor calculated by the geNorm software [30] based on two selected reference genes, ef1aB and arp. geNorm determines the individual stability of a gene within a pool of genes and calculates the stability according to the similarity of their expression profile by pairwise comparison, using the geometric mean as a normalizing factor. The gene with the highest M, that is, the least stabile gene, is then excluded in a stepwise fashion until the most stabile genes are determined. Here, a normalizing factor based on two examined reference genes was used to calculate the MNE.
Table 1.
Primer data.
| Gene | Accession number | Fwd primer (5′–3′) | Rev. primer (5′–3′) | Amplicon size | PCR efficiency |
|---|---|---|---|---|---|
| arp (ref. gen) | AY255630 | GAAAATCATCCAATTGCTGGATG | CTTCCCACGCAAGGACAGA | 101 | 1,813 |
| ef1ab (ref. gene) | BG933853 | TGCCCCTCCAGGATGTCTAC | CACGGCCCACAGGTACTG | 59 | 2,013 |
| fatp | CA373015/AF023258 | TGGGAGCTTGTGGGTTCAA | ACTTTCATGAGGCGGATTGG | 64 | 2,243 |
| actb | BG933897 | CCAAAGCCAACAGGGAGAAG | AGGGACAACACTGCCTGGAT | 92 | 2,007 |
| gst | BQ036247 | ATTTTGGGACGGGCTGACA | CCTGGTGCTCTGCTCCAGTT | 81 | 2,07 |
| tuba | BT049768 | GAGCCAGCCAATCAGATGGT | TGCGCTTGGTCTTGATTGTG | 110 | 2,009 |
| gpx | BQ036408 | TTCTCCACCACACTGGGATCA | GGAAATGGCATCAAGTGGAATT | 101 | 2,011 |
| hsp-70 | BG933934 | CCCCTGTCCCTGGCTATTG | CACCAGGCTGGTTGTCTGAGT | 121 | 1,895 |
| cox2 | AJ238307 | CGTCCTGAGGCAGGAGCAT | TGAGGCGTGTGGTCTGGAA | 62 | 2,18 |
| cpt1 | AM230810 | CTTTGGGAAGGGCCTGATC | CATGGACGCCTCGTACGTTA | 121 | 1,917 |
| bcl-x | NM_001141086 | GCCTGGACGCAGTGAAAGAG | GGACGGCGTGATGTGTAGCT | 107 | 2,127 |
Full gene names: acidic ribosomal protein (arp), elongation factor 1 alpha b (ef1ab), fatty acid transport protein (fatp), β-actin (actb), glutathione-s-transferase (gst), tubulin α (tuba), glutathione peroxidase (gpx), heat shock protein 70 (hsp-70), cyclooxygenase 2 (cox2), carnitine palmitoyltransferase 1 (cpt1), and B-cell lymphoma x (bcl-x).
2.4. Analysis of HEK293 Cells
2.4.1. Red-ox Sensitive GFP (roGFP)
In the fluorescent vector pEGFP-N, already containing a C48S mutation, serine and glutamine at positions 147 and 204 have been replaced by two cysteines to create the roGFP2 C48S/S147C/Q204C [25]. The thiol group in these cysteines reacts to changes in the red-ox environment of the cell, altering the conformation of the roGFP-protein, and subsequently its conformation and fluorescent properties. The roGFP has two emission peaks (Figure 1(a)), which vary in intensity according to red-ox status in the cell. By measuring the emission of the roGFP at these two different excitation wavelengths, a ratiometric value representing the red-ox status in the cell can be obtained [25].
Figure 1.
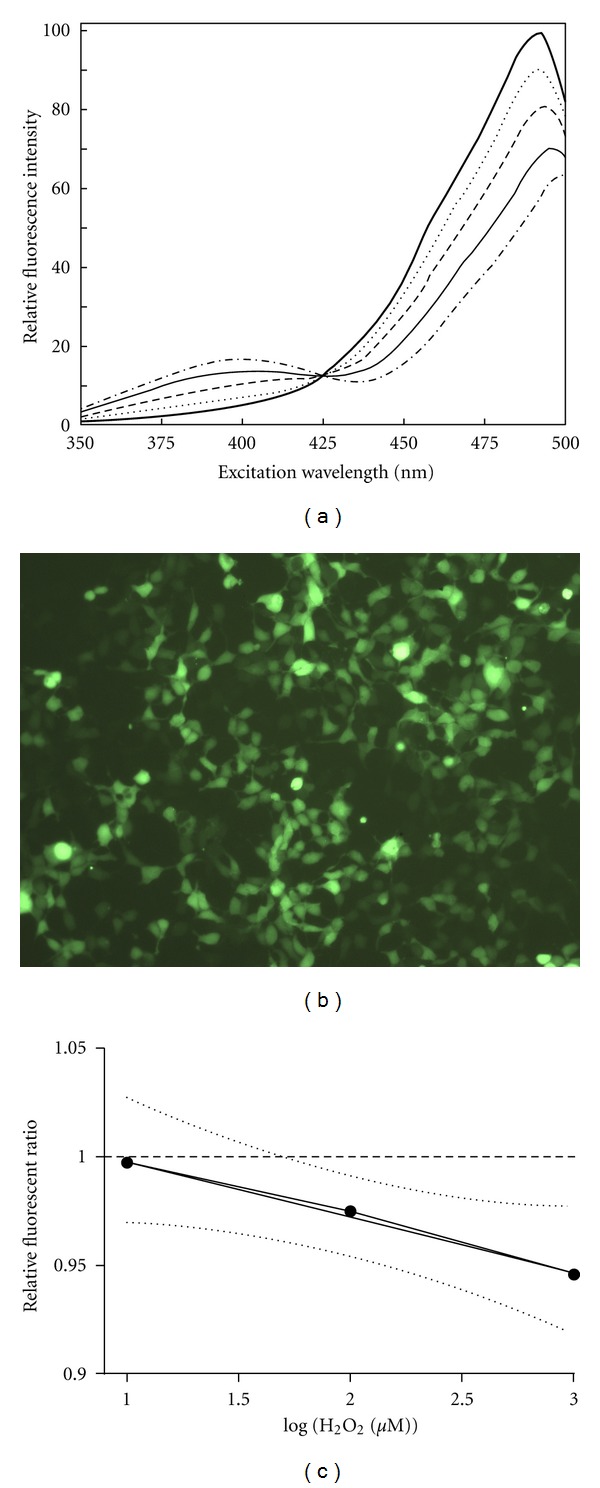
Different aspects of roGFP as marker for oxidative changes in cells. (a) Emission intensity of roGFP measured at serial excitation points. The flat line represents reductive conditions while the semidotted line represents oxidative conditions. The lines in between are different levels of red-ox conditions (image modified from Hanson et al. [25]) (b) Image of HEK293 cells stably transfected with roGFP. (c) Linear regression of log-transformed concentration of H2O2 in roGFP cells. Slope is −0.02617 ± 0.004004, Y intercept when X = 0 is 1.027 ± 0.008649, and R2 is 0.8104.
The plasmid pEGFP-N1/roGFP was purchased from University of Oregon. A stable cell line expressing roGFP (Figure 1(b)) was made by transfecting HEK293 using Fugene HD transfection reagent (Roche diagnostics, Mannheim, Germany) according to manufacturer instructions, and then the cells were selected using 400 μg mL−1 Geneticin (G418) (Invitrogen, By land). After selection, the cells were grown in media containing 200 μg mL−1 Geneticin.
Cells containing roGFP were excited at 400 and 488 nm, and emission at the two different excitation points was measured at 510 nm using a Optima FLUOstar plate reader (BMG labtech, Offenburg, Germany). The emission-ratio between the two excitation points was calculated. A ratiometric value from the emission at the two excitation points was obtained by
| (1) |
When roGFP positive cells grow, the total signal will increase, and since the two peaks measured have different emission maxima, a general increase in signal leads to a small change in the ratio. In order to adjust for this every treated sample was normalized to control at each time point:
| (2) |
In order to reduce systematic errors in the assay, the relative red-ox ratios were also normalized to their relative red-ox ratio at the start of the experiment (t0):
| (3) |
To compare red-ox ratio of fatty acids and MeHg to a known oxidative inducer, a titration curve using H2O2 was run and a standard curve was calculated using linear curve fitting (Figure 1(c)) of the log-transformed H2O2 concentration. Oxidative effects of fatty acids and MeHg were then calculated into corresponding concentrations of H2O2, to visualize oxidative effect.
2.5. Statistical Analyses
All statistic analysis was performed using Statistica 9 (StatSoft Inc., Tulsa, USA) and Graphpad Prism 5.04 (Graphpad software Inc., San Diego, CA, USA). xCELLingence data were processed using regression analysis. Uptake data, apoptosis count, and red-ox measurement were processed using one-way ANOVA followed by post hoc Dunnett's test. Real-time RT-PCR data were processed using the nonparametric Kruskal-Willis with post hoc paired comparisons. Differences were regarded as significant when P < 0.05 with both statistical analysis.
3. Results
3.1. Comparisons between ASK and HEK293
3.1.1. DHA Decrease Uptake of MeHg in HEK293 Cells
Uptake of MeHg was investigated in cells treated with the different fatty acids. Cells were preincubated with the marine fatty acids: DHA or EPA, the n-6 polyunsaturated fatty acid ARA, or BSA as control, before addition of MeHg. ASK cells preincubated with DHA, EPA, or ARA showed no difference in uptake of MeHg compared to MeHg-control (Figure 2(a)). In HEK293 cells, however, DHA significantly decreased the uptake of MeHg (Figure 2(b)).
Figure 2.
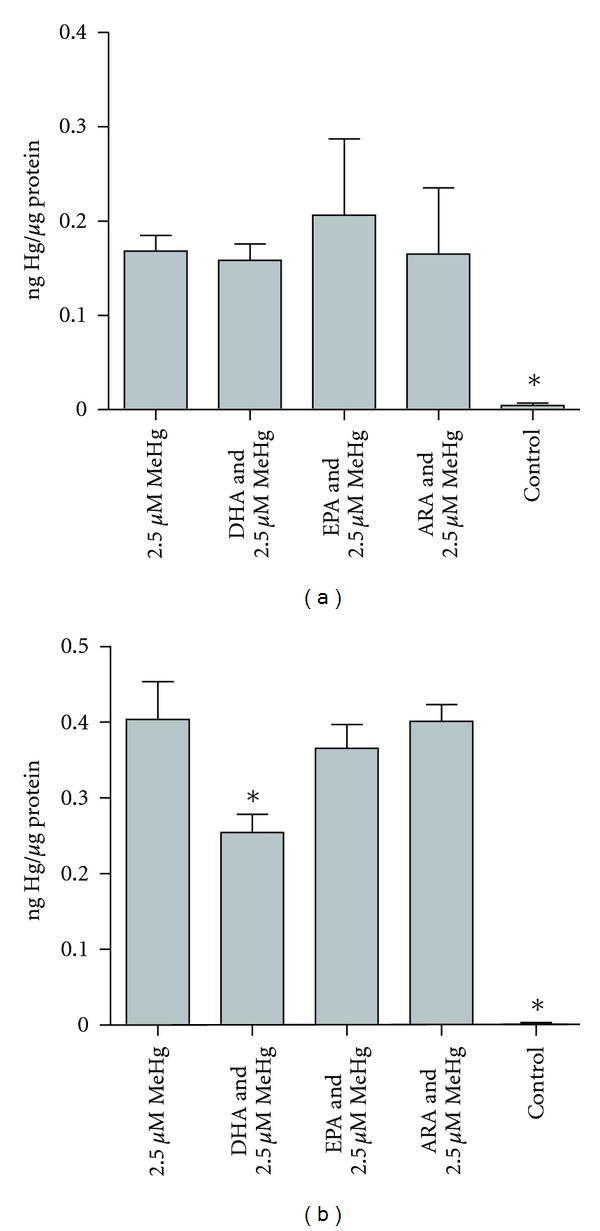
Uptake of MeHg after FA preincubation in ASK cells (a) and HEK cells (b). Results are presented as mean ± SD. Statistical differences are revealed by one-way ANOVA and post hoc Dunnett's test where n = 4, except for Hg- where n = 3. Significant differences compared to 2.5 μM MeHg (P ≤ 0.05) are indicated by asterix.
3.1.2. MeHg Affects Cell Viability Similarly in ASK and HEK293 Cells
Cell toxicity of MeHg in both ASK and HEK293 cells was investigated using xCELLigence impedance measurement. Both cell lines showed a clear dose-response curve after titration versus MeHg (Figure 3). In HEK293, impedance measurements were stabilized at a concentration of 5 μM MeHg, where no further decrease was measured. Therefore, concentrations above 5 μM were considered as too high to be statistically relevant, and only data from 0–5 μM MeHg were used in regression analysis of dose response. Impedance measurement of ASK cells also seemed to reach a plateau as the MeHg concentration increased similar to HEK293, but more noticeable was the early onset of toxicity from control to 1 μM MeHg (Figure 3). Area under curve (AUC) was calculated in ASK cells from time point 102 h–144 h after seeding, as a measurement of cell toxicity, and used in regression analysis. This dose-response curve of ASK AUC followed a nonlinear regression curve with r2 = 0.9824 (Figure 3(c)). AUC was also calculated from HEK cells from time point 53–96 h. The dose-response curve of HEK AUC followed a linear curve fitting with r2 = 0.9491 (Figure 3(d)).
Figure 3.
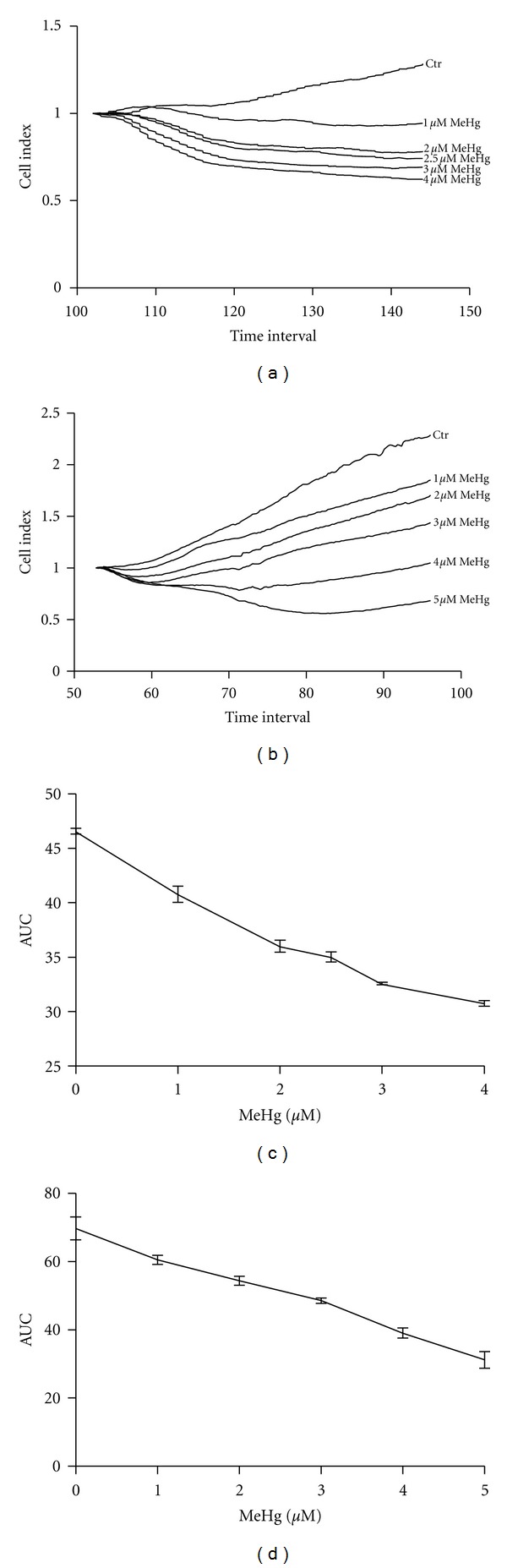
Titration of MeHg in ASK and HEK293 cell culture. (a) Growth pattern of ASK in xCELLigence after normalization. (b) Growth pattern of HEK293 in xCELLigence after normalization. (c) Area under curve from (timespan = 48 h after MeHg exposure), representing loss of adherent cells or decreased growth in ASK cells. The dose response showed a nonlinear curve (r2 = 0.9824). (d) Area under curve (timespan = 48 h after MeHg exposure), representing loss of adherent cells or decreased growth in HEK 293 cells. The dose response showed a linear curve (r2 = 0.9491).
3.2. ASK Cells
3.2.1. Fatty Acids Affect MeHg-Induced Apoptosis
Apoptosis was measured using morphological analysis of ASK cells after fatty acid preincubation and MeHg exposure. A dose-response curve of MeHg was investigated by preincubating cells with BSA, instead of fatty acids, followed by a titration to different MeHg concentrations. Apoptosis count was plotted, and curve fitting was performed (Figure 4(a)). The dose response followed a nonlinear regression curve (r2 = 0.9564). From the dose response curve we chose a concentration of 2.5 μM MeHg to test the effect of fatty acid preincubation on MeHg-induced apoptosis, since this particular concentration of MeHg induced a level of apoptosis which can be remedied by molecular intervention. Cells preincubated with different fatty acids were compared to control cells preincubated with BSA and exposed to MeHg using one-way ANOVA and post hoc Dunnett's test. Preincubating DHA significantly (P = 0.000014) increased while EPA significantly (P = 0.043948) decreased apoptosis induced by 2.5 μM MeHg. ARA showed no effects on apoptosis (Figure 4(b)).
Figure 4.
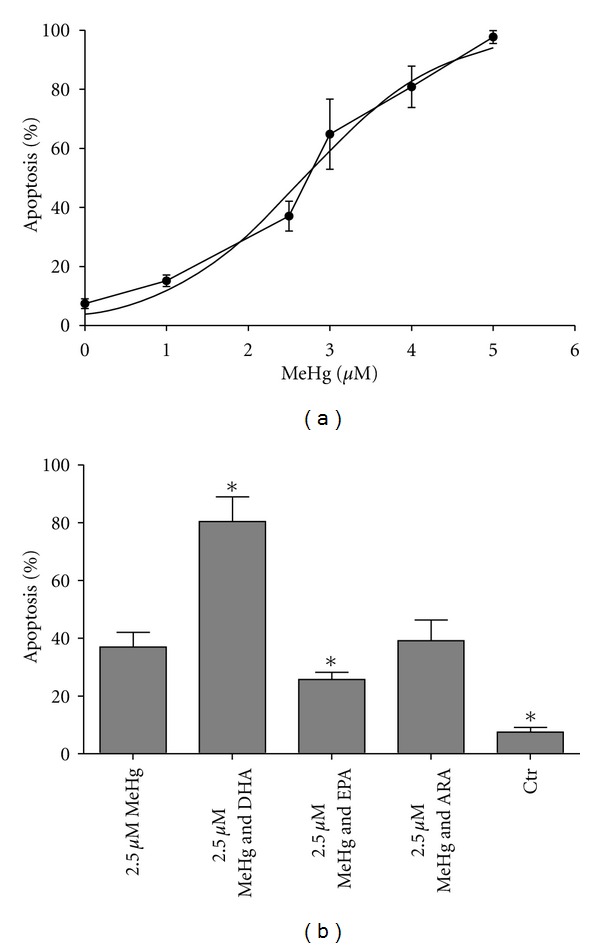
Apoptosis count of ASK cells after treatment with FA and MeHg. (a) MeHg dose-response with curve following a logarithmic nonlinear curve disposition (r2 = 0.9564); from this curve a concentration which gave 40% apoptosis (approx. 2.5 μM MeHg) was used in assay including fatty acids. (b) Apoptosis count after preincubation of cells with fatty acids. Results are presented as mean ± SD. Statistical differences are revealed by one-way ANOVA and post hoc Dunnett's test where n = 4. Significant differences compared to 2.5 μM MeHg (P ≤ 0.05) are indicated by asterix.
3.2.2. MeHg Severely Affected, While Fatty Acids Only Induced, Minor Changes in Gene Expression
Total RNA from ASK cells, preincubated with the different fatty acids and exposed to MeHg, was extracted and the gene expression of selected markers was investigated. When comparing all groups exposed to MeHg versus control (MeHg main effect), the B-cell lymphoma 2 (bcl2) like protein (bclX), heat shock protein 70 (hsp70), and cyclooxygenase 2 (cox2) were all upregulated by MeHg (Figures 5 and 6). Fatty acid transport protein (fatp), carnitine-palmitoyl transferase (cpt1), and tubulin α (tuba) were downregulated by MeHg (Figures 5 and 6). When comparing the different fatty acids (fatty acid main effect) versus control, no effects were observed. When comparing all groups, hsp70 was significantly upregulated in ARA- and MeHg-treated cells compared to control. fatp was significantly downregulated in cells preincubated with DHA compared to the control, while tuba was significantly downregulated in cells preincubated with EPA and exposed to MeHg compared to the control. No significant effects of the treatments were observed in glutathione peroxidase 2 (gpx2) or glutathione-S-transferase (gst) (Figures 5 and 6).
Figure 5.
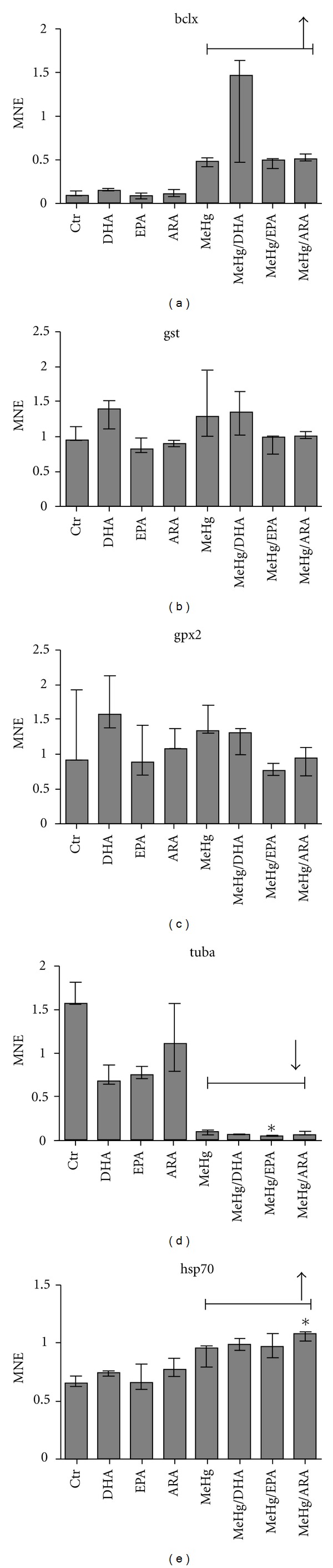
Mean-normalized expression (MNE) of (a) bclx, (b) gst, (c) gpx2, (d) tuba, and (e) hsp70 in ASK cells. Data are represented by median, with error bars representing interquartile range. Data are treated through factorial design, where n = 3 for all groups (columns shown), and significant differences (P ≤ 0.05) are indicated by asterix. MeHg and FA effects were calculated across groups where n = 12 and n = 6, respectively. Significant effects (P ≤ 0.05) of MeHg are shown using bracket lines. No fatty acid effect compared to control was observed. Statistical analysis was performed using nonparametric Kruskal Willis with post hoc paired comparisons.
Figure 6.
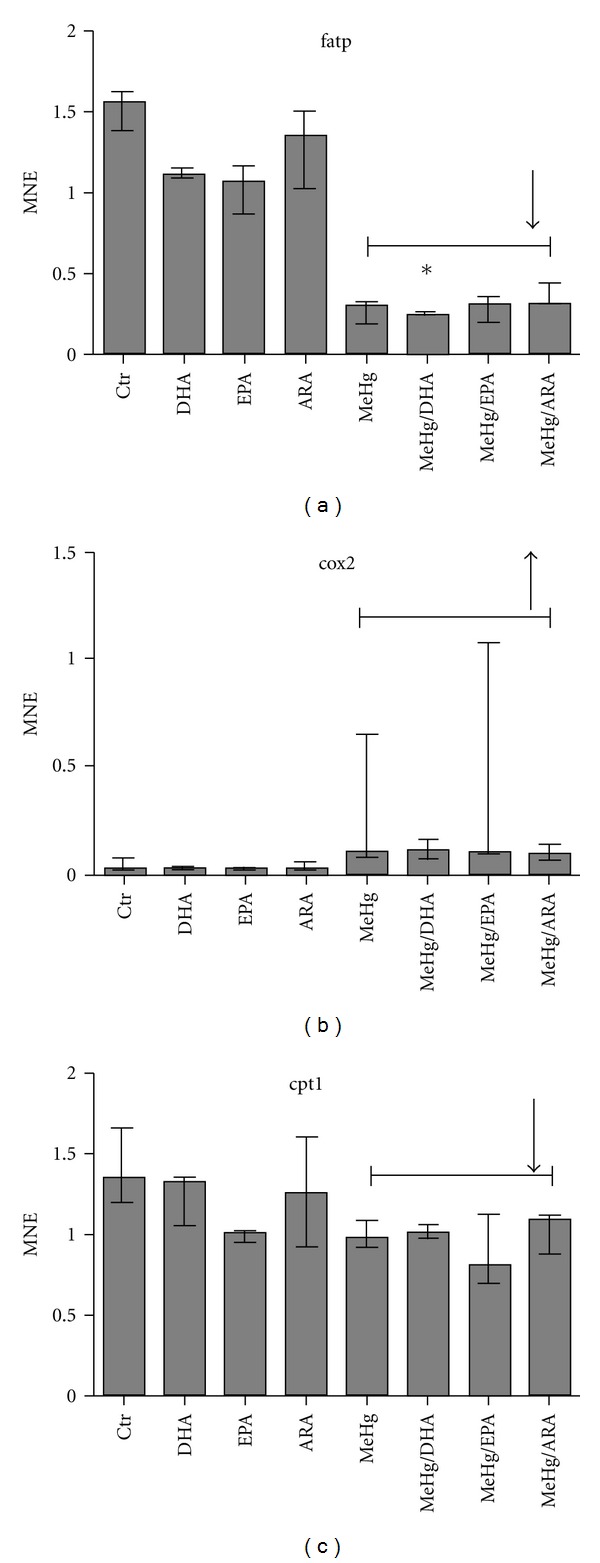
Mean-normalized expression (MNE) of (a) fatp, (b) cox2, and (c) cpt1 in ASK cells. Data are represented by median, with error bars representing interquartile range. Data are treated through factorial design, where n = 3 for all groups (columns shown), and significant differences (P ≤ 0.05) are indicated by asterix. MeHg and FA effects were calculated across groups where n = 12 and n = 6, respectively. Significant effects (P ≤ 0.05) of MeHg are shown using bracket lines. Fatty acid effects compared to control are shown by the letters FA. Statistical analysis was performed using nonparametric Kruskal Willis with post hoc paired comparisons.
3.3. HEK293 Cells
3.3.1. DHA and MeHg Alter Red-Ox Status in HEK293 Cells
HEK293 cells were stably transfected with roGFP plasmid and showed a clear fluorescence after G418-selection (Figure 1(b)). These cells were used to investigate effects of fatty acids and MeHg on oxidative status in cells. Oxidative stress caused by fatty acids and MeHg treatment was compared to equivalent signals generated by the known oxidative inducer H2O2 (Table 2).
Table 2.
Comparison of relative red-ox ratio to the known oxidative inducer H2O2 in HEK293.
| DHA | EPA | ARA | MeHg | DHA and MeHg | EPA and MeHg | ARA and MeHg | |
|---|---|---|---|---|---|---|---|
| Average of relative red-ox ratio | 0.971 | 0.993 | 1.002 | 0.952 | 0.954 | 0.951 | 0.939 |
| Equiv. [H2O2] | 142 | ≈0 | ≈0 | 710 | 606 | 811 | >1000 |
Values below 12 are not significantly different from control, and oxidative stress is assumed almost zero. Calculations are made based on exponential curve fitting and its formula.
“>1000” represents a value that exceeds the standard made from H2O.
Incubating HEK293 cells with EPA and ARA did not induce any change in oxidative status However, both DHA and MeHg significantly induced an oxidative state in the cells after 5 minutes of incubation (Figure 7(a)). DHA steadily kept cells in a state of oxidative stress during its preincubation, but after addition of fresh media a normal oxidative status was recovered (Figure 7(b)). MeHg also rapidly induced oxidation in the cells, but cells recovered their original oxidative status within approximately 1 hour (Figure 7(c)). Preincubating cells with fatty acids did not affect the change in the oxidative status caused by later MeHg exposure (Figure 7(a)).
Figure 7.
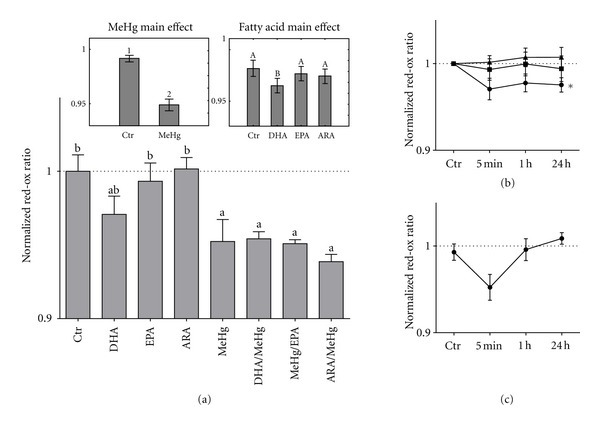
Fatty acid and MeHg effect on red/ox balance in HEK293 cells. (a) Relative fluorescent value of the cells 5 minutes after exposure to the different compounds. Data are treated by two-way ANOVA and main effects analyzed by post hoc Fischer's test. Significant differences (P < 0.05) due to MeHg exposure (MeHg main effect) are represented using lettering. Significant differences (P < 0.05) due to fatty acid treatment (fatty acid main effect) are represented using large letters. Interaction effects were analyzed using post hoc Dunnett's test where significant differences (P < 0.05) compared to control are visualized by small letter a, while significant differences (P < 0.05) compared to MeHg are visualized using small letter b. Dotted line represents normalized control values. (b) Relative fluorescent ratio of fatty acids compared to control over time. Circles represent DHA, squares represent EPA, and triangles represent ARA. The dotted line represents normalized control values. Data are treated with one-way ANOVA followed by a post hoc Dunnett's test; lines significantly different (P < 0.05) from control are marked with ∗. (c) Relative fluorescent ratio of MeHg compared to control over time. Black dots represent MeHg, while the dotted line represents normalized control values. Error margins are signified using standard deviation.
4. Discussion
In this study, the mediating effects of the marine n-3 fatty acids DHA and EPA on MeHg toxicity were studied. EPA ameliorated the apoptotic response of ASK to MeHg, while DHA reduced the uptake of MeHg into HEK293 cells. These results indicate that the metabolism of marine n-3 fatty acids may ameliorate MeHg toxicity at the molecular level and that the nutrient content of a diet may affect the potential negative effect of MeHg-contaminated fish. However, DHA also augmented the MeHg-induced apoptosis in ASK cells, indicating potential potentiating effects of DHA on MeHg toxicity.
4.1. MeHg Affects FA Metabolism in ASK Cells
The metabolism of nutrients into applicable energy for the cell is a vital part in maintaining cell survival. In different exploratory studies performed on fish exposed to MeHg, changes in metabolic genes or proteins have been reported [8, 31]. In our study we found a significant decrease in cpt1 and fatp gene expression as a result of MeHg exposure. cpt1 is responsible for a rate limiting step in mitochondrial β-oxidation of fatty acids: the transport of fatty acids into mitochondria for catabolism [32]. The regulation of this protein is mainly based on its gene transcription [33]. Fatp is a membrane bound protein involved in uptake of fatty acids into cells [34]. The regulation of fatp is thought to be governed through peroxisome proliferator receptors (PPARs) [35], hence being dependent on induction of PPAR through ligand-binding. fatp is also, in part, responsible for the import of fatty acids into mitochondria by interacting with cpt1 [36]. When transcription of both cpt1 and fatp is decreased in our cell system, this may signify a decrease in the availability of fatty acids as substrate for energy metabolism in the mitochondria and a subsequent reduced metabolic throughput. This could be a direct effect of MeHg on the proteins themselves or their regulatory mechanisms, or alternatively it could be that the transcriptional expression of these genes is downregulated as a secondary response to metabolic shutdown after MeHg-induced apoptosis is initiated. Disruption of mitochondrial metabolism by MeHg has previously been reported. Studies have shown that MeHg can disrupt the mitochondrial energy metabolism more directly, possibly through inhibiting phosphorylation of ADP to ATP [37], or depolarizing the mitochondria [10]. The detrimental effect of MeHg on mitochondrial metabolism in the cell may also increase leakage of reactive oxygen species from mitochondria and thereby increasing ROS [38].
An important metabolic pathway for 20 carbon polyunsaturated fatty acids (EPA and ARA) is the eicosanoid pathway, where activation of cytosolic phospholipase A2 (cPLA2) catalyzes the breakdown of membrane phospholipids and releases ARA for subsequent metabolite formation. cPLA2 activity has been shown to be increased by MeHg [39]. An important enzyme in this pathway is cyclooxygenase 2 (cox2), which we showed to be upregulated in response to MeHg exposure. This may be a secondary response to MeHg-induced oxidative stress since cox2 expression has been shown to increase in response to oxidative stress [40, 41]. Cox2 is responsible for the conversion of ARA into prostanoids, such as the prostaglandins. An increase in production of prostaglandins may increase the inflammatory response in the cell, meaning that MeHg may induce proinflammatory metabolites of ARA. Metabolites derived from ARA such as the prostaglandin E2 have also shown to increase intracellular Ca2+ in osteoblast-like cells [42]. Increase of intracellular Ca2+ is also a known toxic effect of MeHg [43], meaning that increased cox 2 leading to increased production of prostaglandins may be part of the underlying cause of MeHg-induced Ca2+ influx, with consequent increase in apoptosis.
4.2. EPA Reduces MeHg-Induced Apoptosis
The enzymes in the eicosanoid pathway have the ability to utilize both ARA and EPA as substrate for production of different metabolites [44]. However, ARA- and EPA-induced eicosanoids exhibit different molecular effects in the cell [45]. In our study, EPA ameliorated the apoptotic effect of MeHg in the ASK cells, while ARA did not. If excess EPA out competes ARA in the eicosanoid pathway, the resulting metabolites show more anti-inflammatory effects than the respective ARA metabolites [45]. EPA might even reduce the inflammatory effects of ARA-metabolites created in the same system [46]. The abandoning of ARAs proinflammatory and possible Ca2+ releasing effects could be part of the explanation why EPA reduces MeHg-induced apoptosis in ASK cells. ARA supplementation did not augment the toxicity of MeHg, which one might expect if the proinflammatory ARA-derived eicosanoids are damaging to the cell. However, the FBS added to the cell media in the MeHg-control probably already contains relatively high ARA-to-EPA ratio, making the production of ARA-derived metabolites saturated, and not dependent on cox2 or the availability of substrate.
Another side of the story is that by replacing ARA with EPA in the eicosanoid system, more EPA-derived eicosanoids will be produced, which could reduce inflammation and affect MeHg toxicity directly, or through apoptosis signaling pathways. The effect of EPA-derived eicosanoids on MeHg toxicity generally, and apoptosis specifically, is not yet known, and further investigations are necessary to elucidate this.
Oxidative effects of fatty acids with and without MeHg were investigated using roGFP-HEK293 cells, and as opposed to DHA, EPA and ARA did not affect the oxidation of roGFP in the cells. These results are consistent with the lack of antioxidant protection against MeHg of fish oil, shown in previous studies [47]. There are, however, studies showing that EPA can cause oxidative stress in muscle and liver cells of Atlantic salmon [48, 49]. However, since we did not see any indications of this, there could be other molecular mechanisms explaining the antioxidant effects noted elsewhere in the literature [50].
EPA, unlike DHA, did not affect the uptake of MeHg into HEK293 cells, suggesting that there must be intracellular molecular mechanisms responsible for EPA's ameliorating effects on MeHg-induced apoptosis.
4.3. DHA Modulates MeHg Uptake and Toxicity
DHA preincubation significantly decreased the uptake of MeHg in HEK293 cells, which is consistent with other studies using human cerebellar astrocytes and cerebellar neurons [51]. No significant difference in uptake of MeHg due to DHA was observed in the ASK cells. However, ASK cells were grown at lower temperature and displayed much lower metabolic rate than HEK293 cells. Given that uptake of MeHg is an active metabolic process linked to carrier mediated processes [52], the effect of DHA on MeHg uptake may be disguised in our experimental settings. Unfortunately, this leaves our results in ASK cells indicative instead of conclusive, and further research of uptake mechanisms affected by DHA, in different cell types, is needed.
The amended uptake of MeHg observed in the HEK293 cells may be due to altered membrane properties after DHA preincubation. PUFAs are important constituents in cell membranes, where they affect several functions such as membrane organization, elasticity, microdomain formation, and permeability of the lipid bilayer. However, due to the difference in chain length and the number of double bonds, DHA is thought to differ in flexibility and conformational freedom compared to EPA and hence will cause different structure disorganization [53]. By changing physical properties of cell membranes, DHA may affect uptake of extracellular compounds, such as MeHg, into cells.
Although DHA decreased the uptake of MeHg in the HEK293 cells, we observed an increase in apoptosis of ASK cells after DHA preincubation and MeHg exposure. This increase was profound, and real-time RT-PCR analysis of the apoptotic regulator bclx in ASK cells suggested a similar effect, though not significant compared to cells only exposed to MeHg. The apoptotic effect seems to be triggered by the addition of MeHg, since DHA treatment alone did not induce increase in apoptosis (results not shown). A possible explanation for the severe apoptotic effect of DHA together with MeHg may be due to changes in the red-ox status in the cells. Although some literature reports DHA as an inhibitor of oxidative stress in certain cells [54], DHA has also been reported to induce oxidative stress and production of ROS [48, 55]. MeHg is known to be a pro-oxidant [56], so the observation can be explained by a potential effect of the compounds. Another possible explanation could be that DHA has inhibited the cells innate antioxidant defense system over time [24]. When we investigated oxidation of roGFP in the HEK293 cells, both MeHg and DHA were observed to oxidize roGFP. DHA kept roGFP in a continuous oxidative state, whilst MeHg affected the oxidation of roGFP moretemporarily. EPA and ARA did not oxidize roGFP in the cells, indicating that the pro-oxidative effect of these fatty acids, compared to DHA, has been obliterated or kept under control in the cells. Conversely, no significant effects of DHA were noted on the transcriptional regulation of the two antioxidant enzymes gpx2 and gst in ASK cells. However, this may be due to normalization of these levels after replenishing control cells with fresh media after the fatty acid preincubation.
5. Conclusion
In this study we have shown that the marine n-3 fatty acid DHA can decrease MeHg uptake in HEK293 cells as well as increase MeHg-induced apoptosis in ASK cells. Furthermore, the proapoptotic effect of DHA may depend on its ability to induce changes in the red-ox environment in the cell. To our knowledge, this is the first time that ameliorating effects of the marine fatty acid EPA on MeHg-induced apoptosis are reported. Taken together, the effects shown by DHA and EPA on MeHg-induced toxicity, in this study, may help explain the differing toxicity observed in response to type of dietary MeHg source [21].
Acknowledgments
This work was funded by the National Research Council (NFR) of Norway, through the project: Seafood and Mental Health: Uptake and Effects of Marine Nutrients and Contaminants Alone or in Combination on Neurological Function (Project no. 186908). The authors would like to thank Ms Aina Cathrine Øvergård (IMR) for feedback on the paper.
References
- 1.Jensen S, Jernelöv A. Biological methylation of mercury in aquatic organisms. Nature. 1969;223(5207):753–754. doi: 10.1038/223753a0. [DOI] [PubMed] [Google Scholar]
- 2.Morel FMM, Kraepiel AML, Amyot M. The chemical cycle and bioaccumulation of mercury. Annual Review of Ecology and Systematics. 1998;29:543–566. [Google Scholar]
- 3.Diez S. Human health effects of methylmercury exposure. Reviews of Environmental Contamination and Toxicology. 1998;198:111–132. doi: 10.1007/978-0-387-09647-6_3. [DOI] [PubMed] [Google Scholar]
- 4.Castoldi AF, Coccini T, Ceccatelli S, Manzo L. Neurotoxicity and molecular effects of methylmercury. Brain Research Bulletin. 2001;55(2):197–203. doi: 10.1016/s0361-9230(01)00458-0. [DOI] [PubMed] [Google Scholar]
- 5.Castoldi AF, Coccini T, Manzo L. Neurotoxic and molecular effects of methylmercury in humans. Reviews on Environmental Health. 2003;18(1):19–31. doi: 10.1515/reveh.2003.18.1.19. [DOI] [PubMed] [Google Scholar]
- 6.Chapman L, Chan HM. The influence of nutrition on methyl mercury intoxication. Environmental Health Perspectives. 2000;108(supplement 1):29–56. doi: 10.1289/ehp.00108s129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aschner M, Syversen T, Souza DO, Rocha JBT, Farina M. Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Brazilian Journal of Medical and Biological Research. 2007;40(3):285–291. doi: 10.1590/s0100-879x2007000300001. [DOI] [PubMed] [Google Scholar]
- 8.Berg K, Puntervoll P, Valdersnes S, Goksøyr A. Responses in the brain proteome of Atlantic cod (Gadus morhua) exposed to methylmercury. Aquatic Toxicology. 2010;100(1):51–65. doi: 10.1016/j.aquatox.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 9.Berntssen MHG, Aatland A, Handy RD. Chronic dietary mercury exposure causes oxidative stress, brain lesions, and altered behaviour in Atlantic salmon (Salmo salar) parr. Aquatic Toxicology. 2003;65(1):55–72. doi: 10.1016/s0166-445x(03)00104-8. [DOI] [PubMed] [Google Scholar]
- 10.Garg TK, Chang JY. Methylmercury causes oxidative stress and cytotoxicity in microglia: attenuation by 15-deoxy-delta 12, 14-prostaglandin J2. Journal of Neuroimmunology. 2006;171(1-2):17–28. doi: 10.1016/j.jneuroim.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Johansson C, Castoldi AF, Onishchenko N, Manzo L, Vahter M, Ceccatelli S. Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotoxicity Research. 2007;11(3-4):241–260. doi: 10.1007/BF03033570. [DOI] [PubMed] [Google Scholar]
- 12.Crespo-López ME, Lima de Sá A, Herculano AM, Rodríguez Burbano R, Martins do Nascimento JL. Methylmercury genotoxicity: a novel effect in human cell lines of the central nervous system. Environment International. 2007;33(2):141–146. doi: 10.1016/j.envint.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Miura K, Imura N. Mechanism of methylmercury cytotoxicity. Critical Reviews in Toxicology. 1987;18(3):161–188. doi: 10.3109/10408448709089860. [DOI] [PubMed] [Google Scholar]
- 14.Bouzan C, Cohen JT, Connor WE, et al. A quantitative analysis of fish consumption and stroke risk. American Journal of Preventive Medicine. 2005;29(4):347–352. doi: 10.1016/j.amepre.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Cohen JT, Bellinger DC, Connor WE, et al. A quantitative risk-benefit analysis of changes in population fish consumption. American Journal of Preventive Medicine. 2005;29(4):325–334. doi: 10.1016/j.amepre.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Ginsberg GL, Toal BF. Quantitative approach for incorporating methylmercury risks and omega-3 fatty acid benefits in developing species-specific fish consumption advice. Environmental Health Perspectives. 2009;117(2):267–275. doi: 10.1289/ehp.11368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hibbeln JR, Ferguson TA, Blasbalg TL. Omega-3 fatty acid deficiencies in neurodevelopment, aggression and autonomic dysregulation: opportunities for intervention. International Review of Psychiatry. 2006;18(2):107–118. doi: 10.1080/09540260600582967. [DOI] [PubMed] [Google Scholar]
- 18.Myers GJ, Marsh DO, Cox C, et al. A pilot neurodevelopmental study of Seychellois children following in utero exposure to methylmercury from a maternal fish diet. NeuroToxicology. 1995;16(4):629–638. [PubMed] [Google Scholar]
- 19.Myers GJ, Marsh DO, Davidson PW, et al. Main neurodevelopmental study of Seychellois children following in utero exposure to methylmercury from a maternal fish diet: outcome at six months. NeuroToxicology. 1995;16(4):653–664. [PubMed] [Google Scholar]
- 20.Grandjean P, Weihe P, White RF, et al. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicology and Teratology. 1997;19(6):417–428. doi: 10.1016/s0892-0362(97)00097-4. [DOI] [PubMed] [Google Scholar]
- 21.Myers GJ, Davidson PW. Prenatal methylmercury exposure and children: neurologic, developmental, and behavioral research. Environmental Health Perspectives. 1998;106(supplement 3):841–847. doi: 10.1289/ehp.98106841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bjerregaard P, Andersen BW, Rankin JC. Retention of methyl mercury and inorganic mercury in rainbow trout Oncorhynchus mykiss (W): effect of dietary selenium. Aquatic Toxicology. 1999;45(2-3):171–180. [Google Scholar]
- 23.Jin X, Lok E, Bondy G, et al. Modulating effects of dietary fats on methylmercury toxicity and distribution in rats. Toxicology. 2007;230(1):22–44. doi: 10.1016/j.tox.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 24.Kaur P, Schulz K, Aschner M, Syversen T. Role of docosahexaenoic acid in modulating methylmercury-induced neurotoxicity. Toxicological Sciences. 2007;100(2):423–432. doi: 10.1093/toxsci/kfm224. [DOI] [PubMed] [Google Scholar]
- 25.Hanson GT, Aggeler R, Oglesbee D, et al. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. The Journal of Biological Chemistry. 2004;279(13):13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 26.Ghioni C, Tocher DR, Sargent JR. The effect of culture on morphology, lipid and fatty acid composition, and polyunsaturated fatty acid metabolism of rainbow trout (Oncorhynchus mykiss) skin cells. Fish Physiology and Biochemistry. 1997;16(6):499–513. [Google Scholar]
- 27.Roche-Diagnostics-GmbH. RTCA SP Instrument Operator's Manual. ACEA Biosciences; 2009. [Google Scholar]
- 28.Urcan E, Haertel U, Styllou M, Hickel R, Scherthan H, Reichl FX. Real-time xCELLigence impedance analysis of the cytotoxicity of dental composite components on human gingival fibroblasts. Dental Materials. 2010;26(1):51–58. doi: 10.1016/j.dental.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Olsvik PA, Kroglund F, Finstad B, Kristensen T. Effects of the fungicide azoxystrobin on Atlantic salmon (Salmo salar L.) smolt. Ecotoxicology and Environmental Safety. 2010;73(8):1852–1861. doi: 10.1016/j.ecoenv.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 30.Vandesompele J, Preter KDe, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology. 2002;3(7) doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klaper R, Carter BJ, Richter CA, Drevnick PE, Sandheinrich MB, Tillitt DE. Use of a 15 k gene microarray to determine gene expression changes in response to acute and chronic methylmercury exposure in the fathead minnow Pimephales promelas Rafinesque. Journal of Fish Biology. 2008;72(9):2207–2280. [Google Scholar]
- 32.Jogl G, Hsiao YS, Tong L. Structure and function of carnitine acyltransferases. Annals of the New York Academy of Sciences. 2004;1033:17–29. doi: 10.1196/annals.1320.002. [DOI] [PubMed] [Google Scholar]
- 33.Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Molecular Aspects of Medicine. 2004;25(5-6):495–520. doi: 10.1016/j.mam.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 34.Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79(3):427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 35.Frohnert BI, Bernlohr DA. Regulation of fatty acid transporters in mammalian cells. Progress in Lipid Research. 2000;39(1):83–107. doi: 10.1016/s0163-7827(99)00018-1. [DOI] [PubMed] [Google Scholar]
- 36.Sebastián D, Guitart M, García-Martínez C, et al. Novel role of FATP1 in mitochondrial fatty acid oxidation in skeletal muscle cells. Journal of Lipid Research. 2009;50(9):1789–1799. doi: 10.1194/jlr.M800535-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bourdineaud JP, Cambier S, Bénard G, et al. At environmental doses, dietary methylmercury inhibits mitochondrial energy metabolism in skeletal muscles of the zebra fish (Danio rerio) International Journal of Biochemistry and Cell Biology. 2009;41(4):791–799. doi: 10.1016/j.biocel.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 38.Yee S, Choi BH. Oxidative stress in neurotoxic effects of methylmercury poisoning. NeuroToxicology. 1996;17(1):17–26. [PubMed] [Google Scholar]
- 39.Shanker G, Hampson RE, Aschner M. Methylmercury stimulates arachidonic acid release and cytosolic phospholipase A2 expression in primary neuronal cultures. NeuroToxicology. 2004;25(3):399–406. doi: 10.1016/j.neuro.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 40.Rockwell P, Martinez J, Papa L, Gomes E. Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cellular Signalling. 2004;16(3):343–353. doi: 10.1016/j.cellsig.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 41.Sun Y, Chen J, Rigas B. Chemopreventive agents induce oxidative stress in cancer cells leading to COX-2 overexpression and COX-2-independent cell death. Carcinogenesis. 2009;30(1):93–100. doi: 10.1093/carcin/bgn242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tokuda H, Miwa M, Oiso Y, Kozawa O. Autoregulation of prostaglandin E2-induced Ca2+ influx in osteoblast-like cells: inhibition by self-induced activation of protein kinase C. Cellular Signalling. 1992;4(3):261–266. doi: 10.1016/0898-6568(92)90065-g. [DOI] [PubMed] [Google Scholar]
- 43.Sue Marty M, Atchison WD. Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicology and Applied Pharmacology. 1997;147(2):319–330. doi: 10.1006/taap.1997.8262. [DOI] [PubMed] [Google Scholar]
- 44.Spector AA. Essentiality of fatty acids. Lipids. 1999;34(6, supplement 3):S1–S3. doi: 10.1007/BF02562220. [DOI] [PubMed] [Google Scholar]
- 45.Calder PC. n-3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. American Journal of Clinical Nutrition. 2006;83(6, supplement):1505S–1519S. doi: 10.1093/ajcn/83.6.1505S. [DOI] [PubMed] [Google Scholar]
- 46.Bagga D, Wang L, Farias-Eisner R, Glaspy JA, Reddy ST. Differential effects of prostaglandin derived from ω-6 and ω-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(4):1751–1756. doi: 10.1073/pnas.0334211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grotto D, Vicentini J, Friedmann Angeli JP, et al. Evaluation of protective effects of fish oil against oxidative damage in rats exposed to methylmercury. Ecotoxicology and Environmental Safety. 2011;74(3):487–493. doi: 10.1016/j.ecoenv.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 48.Kjaer MA, Todorcević M, Torstensen BE, Vegusdal A, Ruyter B. Dietary n-3 HUFA affects mitochondrial fatty acid beta-oxidation capacity and susceptibility to oxidative stress in Atlantic salmon. Lipids. 2008;43(9):813–827. doi: 10.1007/s11745-008-3208-z. [DOI] [PubMed] [Google Scholar]
- 49.Østbye TK, Kjær MA, Rørå AMB, Torstensen B, Ruyter B. High n-3 HUFA levels in the diet of Atlantic salmon affect muscle and mitochondrial membrane lipids and their susceptibility to oxidative stress. Aquaculture Nutrition. 2011;17(2):177–190. [Google Scholar]
- 50.Nakamura N, Kumasaka R, Osawa H, et al. Effects of eicosapentaenoic acids on oxidative stress and plasma fatty acid composition in patients with lupus nephritis. In Vivo. 2005;19(5):879–882. [PubMed] [Google Scholar]
- 51.Kaur P, Heggland I, Aschner M, Syversen T. Docosahexaenoic acid may act as a neuroprotector for methylmercury-induced neurotoxicity in primary neural cell cultures. NeuroToxicology. 2008;29(6):978–987. doi: 10.1016/j.neuro.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 52.Wang W, Clarkson TW, Ballatori N. γ-Glutamyl transpeptidase and L-Cysteine regulate methylmercury uptake by HepG2 cells, a human hepatoma cell line. Toxicology and Applied Pharmacology. 2000;168(1):72–78. doi: 10.1006/taap.2000.9018. [DOI] [PubMed] [Google Scholar]
- 53.Gorjão R, Azevedo-Martins AK, Rodrigues HG, et al. Comparative effects of DHA and EPA on cell function. Pharmacology and Therapeutics. 2009;122(1):56–64. doi: 10.1016/j.pharmthera.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Shimazawa M, Nakajima Y, Mashima Y, Hara H. Docosahexaenoic acid (DHA) has neuroprotective effects against oxidative stress in retinal ganglion cells. Brain Research. 2009;1251:269–275. doi: 10.1016/j.brainres.2008.11.031. [DOI] [PubMed] [Google Scholar]
- 55.Kang KS, Wang P, Yamabe N, Fukui M, Jay T, Zhu BT. Docosahexaenoic acid induces apoptosis in MCF-7 cells In Vitro and In Vivo via reactive oxygen species formation and caspase 8 activation. PLoS One. 2010;5(4) doi: 10.1371/journal.pone.0010296. Article ID e10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lebel CP, Ali SF, McKee M, Bondy SC. Organometal-induced increases in oxygen reactive species: the potential of 2′,7′-dichlorofluorescin diacetate as an index of neurotoxic damage. Toxicology and Applied Pharmacology. 1990;104(1):17–24. doi: 10.1016/0041-008x(90)90278-3. [DOI] [PubMed] [Google Scholar]