

Abstract
The glucagon-like peptide-1 (GLP-1) receptor is one of the best validated therapeutic targets for the treatment of type 2 diabetes mellitus (T2DM). Over several years, the accumulation of basic, translational, and clinical research helped define the physiologic roles of GLP-1 and its receptor in regulating glucose homeostasis and energy metabolism. These efforts provided much of the foundation for pharmaceutical development of the GLP-1 receptor peptide agonists, exenatide and liraglutide, as novel medicines for patients suffering from T2DM. Now, much attention is focused on better understanding the molecular mechanisms involved in ligand induced signaling of the GLP-1 receptor. For example, advancements in biophysical and structural biology techniques are being applied in attempts to more precisely determine ligand binding and receptor occupancy characteristics at the atomic level. These efforts should better inform three-dimensional modeling of the GLP-1 receptor that will help inspire more rational approaches to identify and optimize small molecule agonists or allosteric modulators targeting the GLP-1 receptor. This article reviews GLP-1 receptor physiology with an emphasis on GLP-1 induced signaling mechanisms in order to highlight new molecular strategies that help determine desired pharmacologic characteristics for guiding development of future nonpeptide GLP-1 receptor activators.
1. Introduction
The glucoregulatory role of the gut is demonstrated by studies showing insulin secretion is profoundly more robust following glucose ingestion compared to the insulinotropic response achieved by parenteral administration of intravenously infused glucose [1–5]. This physiologic phenomenon, coined the “incretin effect,” is primarily mediated by two enteric factors known as the incretins: glucagon-like peptide-1 (7-37)/(7-36)-amide (GLP-1) and glucose dependent insulinotropic polypeptide (1-42) (GIP) [6–13]. In addition to glucose, the presence of other macronutrients in a mixed meal, such as lipids and amino acids, in the intestinal lumen stimulate a similar incretin response [14, 15]. When moving through the intestine, nutrients interact directly with sensory receptors and integral membrane channel and transporter proteins localized on the microvillus-rich apical membrane surface of open-type endocrine cells. These cells are embedded in the mucosal lining throughout various regions of the intestinal tract and release the incretins upon nutrient stimulation. In L-cells, located throughout the intestine but predominantly found in the ileum of the distal small intestine and colon [16, 17], GLP-1 is produced by posttranslational cleavage of the 160-amino acid proglucaon precursor protein [18, 19], a process requiring prohormone convertase-1/3 [20–22]. GIP is the single peptide derived from proteolytic processing of a 153-amino acid precursor protein [23] expressed in endocrine K-cells located mainly in the duodenum and proximal jejunum of the upper small intestine [24].
1.1. Physiologic Action of GLP-1
Upon release into the circulation, GLP-1 and GIP facilitate glucose disposal by directly acting on pancreatic islets to enhance postprandial insulin secretion [6–13]. This process is mediated by two class B1 (secretin-like family) seven transmembrane spanning, heterotrimeric G-protein coupled receptors (GPCRs) that signal in response to binding and occupancy by GLP-1 [25, 26] and GIP [27], respectively. Both of these GPCRs predominantly couple to the Gα s subunit which activates adenylyl cyclases to increase intracellular cyclic 3′5′AMP (cAMP). Genetic deletion of both receptors in mice leads to glucose intolerance and defects in glucose stimulated insulin secretion [28]. In addition to ligand stimulated cAMP generation, β-arrestin interaction [29, 30] and signaling pathways that mobilize intracellular calcium are important effectors of incretin action [31, 32].
In humans, the incretin effect is often reduced in patients suffering from type 2 diabetes mellitus (T2DM) [33]. To combat this, initial strategies to develop incretin based “replacement” therapies largely focused on GLP-1 receptor analogs because studies suggested diabetic patients are resistant to GIP treatment [34, 35], while GLP-1 infusion elicits a strong insulin secretory response and can normalize hyperglycemia [36–39]. In contrast to GIP, GLP-1 also induces several additional antidiabetic effects, including inhibition of glucagon secretion [6, 8] and gastric emptying [40–42] (which both help improve postprandial glucose control) and a decrease in appetite and food intake [43–47]. These latter effects are mediated by the GLP-1 receptor expressed in extrapancreatic tissues, most notably those of the gastrointestinal tract and central nervous system.
While infusion regimens demonstrate remarkable antidiabetic pharmacology, elimination metabolism and pharmacokinetic characteristics of native GLP-1 present major hurdles to developing it as an effective pharmaceutical agent. One significant challenge in pursing GLP-1 based molecules is that GLP-1 is rapidly inactivated by dipeptidyl peptidase 4 (DPP4), a plasma membrane bound enzyme that is positioned with its active site orientated towards the extracellular space. This ubiquitously expressed “ectopeptidase” cleaves the N-terminal dipeptide, His7-Ala8, to inactivate GLP-1 [48, 49]. Removal of these residues dramatically reduces the binding affinity of the peptide for the GLP-1 receptor, thus abolishing its ability to effectively activate receptor signaling [50]. DPP4 is highly expressed on the surface of endothelial cells lining blood vessels; consequently, GLP-1 is immediately vulnerable to inactivation following release into the circulation [51]. Upon cleavage, the inactive GLP-1 metabolite is eliminated by the kidney [52]. As a result of rapid postsecretory proteolysis and renal elimination, the biological half-life of GLP-1 is estimated to be between 1 to 2 minutes [53, 54]. These characteristics limit the pharmaceutical potential of native GLP-1.
1.2. GLP-1 Receptor Peptide Agonists
Several efforts pursued novel GLP-1 analogs with improved metabolic properties. A common approach was to introduce N-terminally substituted modifications to reduce DPP4 sensitivity [54–56]. To date, attempts solely focused on amino acid substitutions of native GLP-1 to identify longer acting molecules likely have been hampered by other issues such as renal clearance and secondary degradation by other endopeptidases. However, two alternate approaches proved successful in advancing more stable, degradation resistant GLP-1 receptor agonists. Both exenatide and liraglutide are approved for marketing by several government regulatory agencies for the treatment of T2DM.
Exenatide is a 39-amino acid peptide GLP-1 receptor agonist that is fully efficacious in cellular assays and competitive with native GLP-1 in receptor binding studies [26, 57–59]. It is the synthetic version of exendin-4 which was among several bioactive peptides containing an N-terminal histidine identified in crude venom preparations extracted from perimandibular salivary glands of Helodermatidae lizards [60, 61]. Exendin-4 was isolated from the poisonous venom of the Gila monster, Heloderma suspectum [60], a lizard indigenous to the southwest United States in the Gila River area of New Mexico and Arizona [62]. In addition to mimicking the physiologic glucoregulatory actions of native GLP-1, exendin-4 is a poor DPP4 substrate [63] and is cleared from the body primarily by glomerular filtration in the kidney [64, 65]. Consequently, exenatide has a longer duration of action compared to GLP-1 [66–68] and an estimated biological half-life of approximately 4 hours [64, 69]. In April of 2005, under the brand name Byetta, exenatide became the first enteroendocrine based therapeutic approved by the United States Food and Drug Administration (FDA) for the treatment of T2DM.
The second GLP-1 receptor agonist approved to treat T2DM is liraglutide (NN2211). For this molecule, a “fatty acid derivatization” strategy was used to prolong the in vivo action of GLP-1. This approach attaches a fatty acid moiety to GLP-1 in order to facilitate GLP-1 binding to serum albumin. Liraglutide is acylated on Lys26 with a covalently attached palmitoyl (C16:0) chain [70]. As this modification enables binding to albumin, GLP-1 is then sterically protected from DPP4 degradation [70]. The plasma half-life of liraglutide is estimated to be between 11 and 15 hours [71, 72]. Under the brand name Victoza, liraglutide received marketing approval by the FDA in January of 2010 for the treatment of T2DM. Byetta and Victoza are both commonly prescribed medicines.
2. GLP-1 Receptor Signal Transduction and Second Messenger Pathways
As the best characterized in vivo action of GLP-1 is an acute insulinotropic effect mediated by the GLP-1 receptor in pancreatic β-cells, the signal transduction coupling mechanisms of this receptor primarily have been analyzed using ex vivo preparations of pancreatic islets, transformed pancreatic β-cell lines, and recombinant GLP-1 receptor expressing systems, Accordingly, critical evaluation of GLP-1 receptor signal transduction in extrapancreatic tissues can be made by inference only. Use of the various peptide GLP-1 receptor agonists to define the in vitro pharmacologic properties of the receptor should define key assay systems to enable optimizing small molecule GLP-1 receptor activators.
2.1. GLP-1 Receptor Activation
Both GLP-1 and exendin-4 are α-helical peptides that interact with the GLP-1 receptor by binding multiple extracellular contact points to induce receptor signaling [73–75]. Similar to the “two-step” mechanism proposed for other class B1 GPCRs [76] (Figure 1), the GLP-1 receptor utilizes an N-terminal extracellular domain as an “affinity trap” to recognize and bind peptide ligands [77, 78]. The N-terminal domain of the GLP-1 receptor is conserved among class B1 GPCRs forming an α-β-βα protein fold that has structural homology to the sushi/CCP/SCR protein folds [79, 80] (Figure 2). This structure, referred to as an “ectodomain” (ECD), is a trilayer fold composed of an N-terminal α-helix, a middle section of two antiparallel β strands, and a final lobe composed of two additional antiparallel β sheets and a short α-helical region (βα) (Figure 2). The overall structure of the ECD is stabilized by three pairs of disulfide bonds formed between six conserved cysteine residues that lock the three layers of the ECD together [81] (Figure 2). X-ray crystal structures of exendin-4 and GLP-1 bound to the ECD confirm the “affinity trap” hypothesis showing the C-terminal α-helical region of GLP-1 or exendin-4 is positioned within a binding cleft of the N-terminal ECD [74, 75] (Figure 3). Both GLP-1 and exendin-4 are amphipathic in nature, and this defines their structurally conserved interaction mechanism with the ECD. The hydrophobic faces of GLP-1 and exendin-4 make the majority of interactions with the ECD and likely are the key contributors to ECD/ligand affinity with only a minor contribution of binding energy provided by the hydrophilic regions of GLP-1 receptor agonist peptides [74, 75] (Figure 3).
Figure 1.
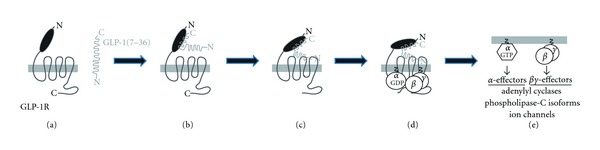
The activation mechanism of the GLP-1 receptor. Biochemical and structural studies have led to a model of class B1 GPCR activation by peptide hormones referred to as the “two-step” mechanism [82]. (a) In the unliganded state, the GLP-1 receptor (GLP-1R) is in a predominately inactive conformation. The natural ligand GLP-1(7-36)-NH2 is freely diffusible in solution and likely has substantial intrinsic α-helical structure. (b) An initial binding event between the globular ectodomain at the N-terminal of the GLP-1R and the C-terminal of the GLP-1(7-36)-NH2 peptide occurs. This “low affinity” interaction acts as a tether or “affinity trap” to localize GLP-1 at the GLP-1R. (c) The weak affinity of the N-terminus of GLP-1(7-36)-NH2 is then able to productively engage with transmembrane domain and loop residues of the receptor to induce a high affinity interaction and likely a conformational change in the GLP-1R. (d) Coincident with agonist binding, the G-protein bound conformation of the GLP-1R is stabilized. This represents the classic high affinity agonist bound state. (e) The high affinity agonist bound state is transient in an intact system as the GLP-1R stimulates guanine nucleotide exchange on the α-subunit of the G-protein heterotrimer, leading to G-protein dissociation and independent or synergistic activation of effector proteins by liberated Gα·GTP and Gβγ.
Figure 2.
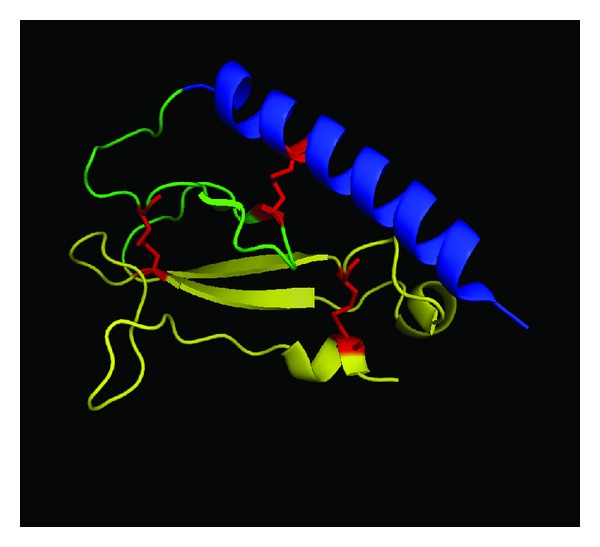
Structure of the GLP-1 receptor ectodomain. The overall structure of the GLP-1 receptor ectodomain is depicted (PDB ID: 3IOL) [74]. The tripartite α-β-βα structure is annotated using color; from N-terminal to C-terminal α (blue), β (green), βα (yellow). The three conserved disulfide bonds that stabilize the tertiary structure of the ectodomain are colored in red.
Figure 3.
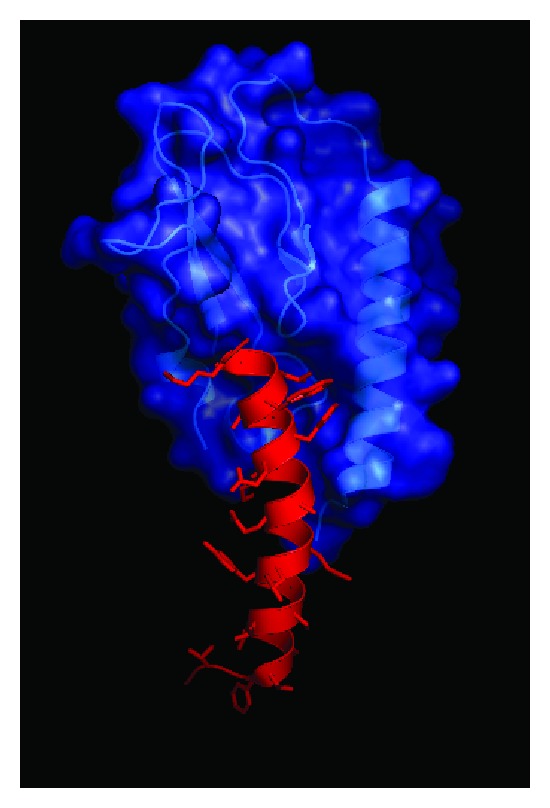
Structural determinants of ligand binding to the GLP-1 receptor ectodomain. (a) Structure of the GLP-1 receptor ectodomain bound to GLP-1. Blue ribbon and space filling model is GLP-1 receptor and the red ribbon and stick model is GLP-1. Data are derived from PDB ID: 3OL.
The second step of the class B1 GPCR activation model predicts the ECD docks the bound peptide in a position that promotes direct contact of N-terminal residues of the ligand with the central activation pocket of the receptor, a region consisting of three interconnecting extracellular loops often referred to as the helical bundle or “J” (juxtamembrane) domain. GLP-1 (or exendin-4) binding to this core region induces a conformational rearrangement of the membrane-spanning α-helices, eliciting a shift of the intracellular receptor loops to stimulate intracellular signal transduction (Figure 1).
Structural information regarding GLP-1 receptor peptide ligands is available primarily from NMR studies. The data are consistent with other class B1 GPCR ligands [83] in that GLP-1(7-36) and exendin-4 peptides are likely α-helical in structure with disordered N-termini, although the artificial environment in which these studies are conducted must be used to qualify any interpretation of the experimental data [84, 85]. Structural studies of class B1 GPCR ECDs have been very informative regarding the molecular mechanisms of peptide ligand selectivity and have generated new hypotheses regarding class B1 GPCR activation mechanisms. However, the field awaits definitive structural data to explain how peptide agonists activate their cognate class B1 GPCRs.
2.2. G-Protein Coupling
The GLP-1 receptor primarily couples to the Gα s heterotrimeric G-protein. Upon ligand binding, the resulting conformational change activates intrinsic guanine nucleotide exchange factor activity of the receptor to catalyze release of bound GDP from the Gα s. The Gα s then rapidly binds GTP which leads to dissociation of Gα s and Gβγ, consequently activating downstream effector pathways. Activated Gα s allosterically stimulates membrane-associated adenylyl cyclases to catalyze conversion of ATP to cAMP, which acts as an intracellular second messenger mediating GLP-1 signaling.
Elevation of cAMP in the pancreatic β-cell is a critical event in the process of glucose dependent insulin secretion and is likely the key mechanism by which GLP-1 and exendin-4 act on β-cells to potentiate insulin secretion [25, 26, 86]. However, early reports highlighted the ability of the GLP-1 receptor to couple to alternative signaling pathways, including phospholipase C (PLC) and the mobilization of intracellular Ca2+ [87, 88], consistent with the known effects of GLP-1 to stimulate Ca2+ mobilization in β-cells [89]. Further, multiple reports indicate GLP-1 receptor couples to Ca2+ mobilization when heterologously expressed [90, 91]. In these systems, it is generally assumed Ca2+ mobilization is a Gα q mediated process. In support of this, studies utilizing the azidoanilide-GTP cross-linking method show the GLP-1 receptor can cause activation of the Gα q- and Gα i-families of G-proteins in GLP-1 receptor expressing CHO cells [88]. Conversely, recent experiments using membrane GTPγS binding assays demonstrate GLP-1 receptor activation does not induce measurable activation of Gα q or Gα i despite the presence of substantial PLC independent Ca2+ mobilization in GLP-1 receptor expressing HEK cells [91]. In vivo, pancreatic β-cell specific dual inactivation of Gα q and Gα 11 does not affect GLP-1 potentiation of glucose stimulated insulin secretion [92], whereas insulinotropic action through known Gα q/11 coupled GPCRs, GPR40 and the M3 muscarinic receptor, is ablated. While this study elegantly demonstrates that Gα q/11 signal transduction is not required for GLP-1 receptor mediated insulin secretion (using perifused islets), it is problematic that a single dose of 100 μM GLP-1 (a concentration greater than 5 orders of magnitude above the K d and peak circulating levels of active GLP-1) was used in the studies [49, 92].
While a role for Gα q/11 signal transduction in β-cell GLP-1 receptor action is generally excluded, a PLC and Ca2+ mobilization pathway may be operant. Experiments using mouse β-cells indicate the elevation of cAMP by GLP-1 receptor signaling results in activated EPAC2 that stimulates PLC [31] and Ca2+ channel recruitment [93] to facilitate calcium induced calcium release, a process integral for robust insulin secretion. These data provide a potential mechanism whereby sole activation of the Gα s pathway induces cAMP- and PLC/Ca2+ dependent responses in β-cells. In light of the contrasting data, it is apparent that the phenotype of GLP-1 receptor signaling may differ according to the cellular context in which the receptor is expressed, a phenomenon now widely recognized but not well understood within the GPCR field [94]. Accordingly, the definitive in vivo G-protein coupling profile of the GLP-1 receptor is unclear, although Gα s induced cAMP accumulation is certainly integral to the biological response of GLP-1 receptor activation. It is of interest that other class B1 GPCRs demonstrate physiologically relevant coupling to multiple G-proteins; the parathyroid hormone receptor is the best characterized example [95]. The use of genetically modified mice, RNA interference, or novel pharmacological tools such as the Gα q/11 inhibitor YM-254890 [96] may serve to answer whether the GLP-1 receptor functionally couples to G-proteins other than Gα s in the endogenous context, particularly in extrapancreatic cell types.
2.3. β-Arrestin Coupling
Despite numerous studies characterizing β-arrestin interactions with GPCRs, only a limited number of reports investigate interactions between incretin receptors and arrestin proteins. Bioluminescence resonance energy transfer studies demonstrate both β-arrestin-1 and -2 interact with the GLP-1 receptor in an agonist dependent manner [97]. Classically, GPCR recruitment of GPCR kinases (GRKs) and β-arrestins is characterized as inducing desensitization of G-protein mediated signal transduction [98]; β-arrestin binding blocks G-protein mediated signaling and facilitates receptor internalization. However, emerging data suggest receptor activated β-arrestins can stimulate signaling pathways independently of G-protein activation [99]. Thus, β-arrestin signaling has physiologic consequences distinct from G-protein coupled signaling [99]. It is, therefore, of great interest to understand the functional outcome of β-arrestin regulation of the GLP-1 receptor.
In INS-1E insulinoma β-cells, siRNA knockdown of β-arrestin-1 reduces GLP-1 induced insulin secretion [30]. These experiments implicate β-arrestin-1 in GLP-1 receptor activity, but the mechanism responsible for the diminution of GLP-1 action is not explicitly characterized. An alternate explanation for lower GLP-1 stimulated insulin secretion could be reduced tonic inhibition from anti-insulinotropic Gα i-coupled GPCRs resulting from β-arrestin-1 removal. Further, in this study, insulin secretion induced by glucose alone is severely attenuated by β-arrestin-1 knockdown making it difficult to establish direct causality between β-arrestin-1 knockdown and GLP-1 receptor dependent signaling.
In studies using MIN6 insulinoma β-cells, GLP-1 receptor stimulation is shown to induce a biphasic activation of ERK [100]. This effect is comprised of an initial cAMP-dependent transient activation of ERK and a prolonged β-arrestin-1 dependent activation of ERK [100]. β-arrestin-1 dependent ERK activity promotes Bad phosphorylation and consequently mediates prosurvival effects of GLP-1 receptor agonists on high glucose induced apoptosis. Because many biochemical mechanisms in MIN6 cells are operant in mouse islets, this report elegantly delineates separable pathways for GLP-1 receptor induced insulin secretion (Gα s-cAMP axis) versus antiapoptotic signaling (β-arrestin-1 → p90RSK → Bad axis). It should be noted, though, that this study also contains experimental caveats; for example, the authors are unable to cause glucotoxic apoptosis in primary islet cultures and thus fail to validate the efficacy of GLP-1 anti-apoptotic signaling in their islet system.
In vivo analysis of insulin secretion in β-arrestin-1 knockout mice indicates glucose stimulated insulin secretion is reduced by over 80% [30, 100]. Accordingly, it is problematic to ascribe any physiologic alterations to GLP-1 receptor agonists in β-arrestin-1 knockout mice as being directly due to the GLP-1 receptor, given that β-arrestins are likely crucial signal regulatory proteins for hundreds of GPCRs. This is exemplified by the recent observation that β-arrestin-2 knockout mice are insulin resistant [101]. A key point to contextualize these studies is that although numerous reports in rodents show positive effects of GLP-1 or exendin-4 on pancreatic β-cell replication, β-cell mass, and function in preclinical models [102, 103], less data are available regarding GLP-1 agonist modulation of β-cell apoptosis or neogenesis in humans (discussed in [104]). One report does, however, demonstrate GLP-1 mediated attenuation of apoptosis and enhanced insulin responsiveness in an ex vivo human islet preparation [105]. Similarly, recent evidence demonstrates that GLP-1 receptor agonism induces β-cell replication in human islet grafts [106].
3. Structural Evaluation of the GLP-1 Receptor
Understanding the molecular mechanisms whereby peptide ligands induce GLP-1 receptor signaling should enhance new efforts to optimize small molecule activators of the GLP-1 receptor. Various approaches are being explored that may ultimately inform more rationale design strategies for small molecule GLP-1 receptor agonists. A comprehensive review of currently disclosed low molecular weight GLP-1 receptor activators is presented in this issue of Experimental Diabetes Research; see Willard et al. for review. Importantly, new strategies to exploit potential small molecule binding to the GLP-1 receptor are advancing as a result of progress in GPCR molecular and structural biology. Extensive site-directed mutagenesis studies have been carried out on the GLP-1 receptor as have structure activity studies on peptide GLP-1 ligands. It is beyond the scope of this review to cover these studies, however, we recognize that such efforts have been a valuable starting point in efforts to understand GLP-1 receptor biochemistry [107, 108].
3.1. Intramolecular Endogenous Peptide Agonists
Dong and colleagues proposed a novel activation mechanism for class B1 GPCRs [109] whereby peptide ligand interaction with the receptor induces a conformational change that exposes an intramolecular “endogenous agonist” epitope within the ECD of a GPCR. The “endogenous agonist” motif is proposed to act at the transmembrane domains to facilitate receptor activation. This hypothesis is largely derived from mutagenesis, peptide cross-linking, and molecular modeling studies using the secretin receptor [109]. In support of the concept, synthetic peptides derived from the ECD of the secretin receptor are shown to be low potency, high efficacy agonists of the secretin receptor. The minimized pharmacophore for the secretin receptor is a tri-peptide Trp70-Asp71-Asn72 [109]. Translation of this finding to the GLP-1 receptor identified an analogous peptide, Asn63-Arg64-Thr65-Phe66-Asp67 (NRTFD), as a low potency but fully efficacious GLP-1 receptor agonist [110]. Although these short peptides have low affinity and poor receptor selectivity, making them unlikely starting points for lead optimization and drug development, the molecules identify a potential binding site for compound action on the GLP-1 receptor. Structural elucidation of these features should be enlightening. The GLP-1 receptor “endogenous agonist” peptide maps to the β1 strand of the GLP-1 receptor ECD crystal structure [74]. However, analyses of these structural data suggest Asn63 is solvent exposed, but the majority of the peptide is not; these results make it difficult at this point to clearly understand the molecular mechanism proposed for the “endogenous agonist” peptides without further information around the structure or dynamics of the GLP-1 receptor. A subsequent report used an elegant cross-linking approach coupled with radiochromatography (see Section 3.3) to identify the site of action of the NRFTD peptide as being extracellular loop 3 in close proximity to transmembrane domain 6 [111]. The mechanism of action of the NRFTD peptide may be analogous to that of the “pepducins” [112]. Pepducins are short peptides derived from the intracellular loops of GPCRs that act allosterically to modulate receptor signaling [113].
3.2. Crystal Structure of the GLP-1 Receptor ECD
Receptor binding and functional studies show both GLP-1 (K d = 0.3 nM) and exendin-4 (K d = 0.1 nM) bind with high affinity and are full agonists at the GLP-1 receptor [26, 57]. In addition, competition binding studies suggest these peptides use the same ligand binding site within the receptor [114]. However, in experiments exploring the initial ligand-receptor binding event, data show exendin-4 binds the isolated soluble form of the GLP-1 receptor ECD with much greater affinity (13 nM) compared to GLP-1 (800 nM) [114, 115]. It was hypothesized that this phenomenon occurs because exendin-4 contains nine additional amino acids at its C-terminus that enable further binding contacts with the ECD [77, 115]. Importantly, the crystal structures of the ECD in complex with either bound GLP-1 or exendin-4(9-39) show these peptides share a very similar mode of binding, and there is no interaction between the last seven residues of the nine amino acid C-terminal extension of exendin-4 with the ECD [74, 75] (Figure 3). Alternatively, biophysical studies suggest the higher propensity of exendin-4 to form helical conformations in solution compared to GLP-1 results in its enhanced binding affinity [73, 114]. However, this area is still in need of further exploration because other reports using a membrane-tethered form of the ECD demonstrate GLP-1 (IC50 = 160 nM) and exendin-4 (IC50 = 20 nM) bind the receptor with closer affinity [116]. Recent studies highlight this incongruity as exendin-4 (K d = 0.9 μM) and GLP-1 (K d = 1 μM) have equivalent affinities for the ECD, as measured by surface plasmon resonance [117]. While studies using purified forms of the ECD are informative, these latter results may highlight the relatively artificial nature of approaches using the soluble form of the ECD. Therefore, experimental methodologies allowing structural characterization of the full length, intact GLP-1 receptor are needed.
3.3. GLP-1 Receptor Photoaffinity Labeling
In the absence of a high resolution crystal structure of the full length GLP-1 receptor, photoaffinity labeling has been used as an alternate approach to identify potentially important ligand-receptor interactions. An inherent advantage of this technique is that whole cells expressing the native receptor or membrane preparations enriched with a receptor of interest are used so the receptor is folded and presented in its proper structural orientation [118–120]. These studies typically use a radio-labeled version of the natural ligand engineered to contain a photoreactive moiety such as p-benzoyl-l-phenylalanine (Bpa). Ultraviolet photolysis of a probe in complex with its receptor covalently labels residues of the receptor that are in close spatial approximations with important structural regions of the ligand. The labeled amino acids within the targeted receptor can then be identified using manual cycles of Edman degradation sequencing of isolated receptor fragments [121]. For the GLP-1 receptor, this technique has established spatial approximations between several ligand-receptor residues that likely occur in the “agonist occupied” receptor conformation. In studies intended to further assess the initial binding event, C-terminal residues, Ala24 and Gly35, of GLP-1 are shown to dock in close proximity near Glu133 and Glu125 of the ECD, respectively [122]. These data are in line with the 2.1 Å resolution crystal structure of the ECD-GLP-1 complex showing GLP-1 binding occurs via a continuous C-terminal α-helix formed by the sequence spanning Thr13 and Val33 with residues between Ala24 and Val33 directly interacting with the ECD [74]. Overall, these data are consistent with the initial binding event proposed by the “two-step” model. Somewhat surprisingly, results from studies testing GLP-1 probes with Bpa incorporated at positions 12 and 16 show docking of this region also near residues contained within the ECD. These data predict Phe12 and Val16 are positioned near Tyr145 and Leu141, respectively, of the ECD, sites located in the distal region of the ECD immediately upstream of the first transmembrane segment of the receptor [123, 124].
Importantly, photoaffinity cross-linking studies have also established potential contacts between the extracellular loops of the receptor with residues of GLP-1. This work is helping provide better insights into the orientation of the ligand bound N-terminal region of GLP-1 with the receptor core. For studies aimed at identifying structural elements involved with the “second step” of GLP-1 binding and receptor activation, an N-terminus labeled photo-labile GLP-1 probe was generated. Because changes to the N-terminal His7 are not well tolerated [125], a probe with Bpa N-terminally attached to His7 (at a new position 6) was used to better understand spatial approximations between the most N-terminal residues of GLP-1 and the receptor. These data show the N-terminus positions near Tyr205 in the first extracellular loop of the receptor [123]. Similarly, using a mid-region position 20 probe, Trp297 within the second extracellular loop of the receptor positions within close proximity to Leu20 of GLP-1 [124]. Taken together, X-ray structural data of the ECD in complex with the C-terminus of GLP-1 and the cumulative results from photoaffinity labeling studies provide experimentally derived information with which to generate a more accurate molecular model of the ligand binding pocket for the GLP-1 receptor [124].
3.4. Emerging Biochemical Technologies
Although progress is continuing, integrated strategies pairing classic receptor pharmacology with newer biophysical and structural biology techniques are needed to progressively refine a model for GLP-1 receptor activation. New techniques aimed at understanding ligand dependent conformational changes in the GLP-1 receptor should aide small molecule discovery pursuits. One emerging approach is the application of solution phase peptide amide hydrogen/deuterium exchange (HDX) coupled with mass spectrometry (MS) for the study of GPCRs. In contrast to photoaffinity labeling, HDX-MS does not require generating probes that require incorporation of a bulky moiety, such as the hydrophobic Bpa, and must retain the binding and activation properties of the natural ligand. Alternatively, HDX-MS is based on the principle that for proteins in solution, amide bond hydrogen atoms are exchangeable, and differences in the rate of exchange are indicative of local accessibility and thus can reflect the conformational status of a protein [126, 127]. Deuterium incorporation into the peptide backbone increases protein mass, and upon protease cleavage, the location and degree of hydrogen/deuterium exchange can be mapped via MS analysis. Although technically difficult to apply to membrane proteins, methods have advanced that better enable purification procedures for isolating GPCRs using detergents that help maintain native structure, protein solubility, and functional activity. From these advances, HDX-MS has now been used to study ligand induced conformational changes of the β2 adrenergic receptor [128, 129]. These biochemical studies demonstrate that distinct receptor conformations are elicited by ligands with different intrinsic efficacies. Further, an elegant parallel MS approach has utilized covalent derivatization of cysteine and lysine residues with stable isotope labeled reactive functionalities to assess dynamic conformational changes elicited by β2 adrenergic receptor ligands [130]. These studies highlight the diversity of receptor conformations induced by ligands with apparently similar functional capacities. Together, this work provides further experimental evidence for the existence of multiple ligand specific conformations of GPCRs.
3.5. Recent Advances in GPCR Crystallography
The first GPCR crystal structure determined was that of Rhodopsin in 2000 [131]. While informative about the general principles of GPCR structure, and having utility as a homology model template for closely related class A GPCRs, this information has not significantly impacted drug discovery activities for class B1 GPCRs. However, recent advances in GPCR biochemistry and macromolecular crystallography have accelerated the pace of structure determination for this important target class [132]. Since 2007, multiple new class A GPCR structures have been determined, including adrenergic, adenosine, chemokine, dopamine, and histamine receptors [132]. In many cases, structures of multiple ligands with cognate GPCRs are solved. Moreover, the recent determination of a G-protein bound complex of an activated GPCR represents a landmark achievement of GPCR crystallography [133]. Several new technological advances have been developed to facilitate these pursuits, including novel detergents [134], creative receptor-fusion proteins [135], camelid nanobodies [136], and lipidic cubic phase crystallization [137]. Consequently, we anticipate that significant effort may now turn toward determining the crystal structures of class B1 GPCRs.
4. Conclusions
It is clear the GLP-1/GLP-1 receptor axis is a key physiologic regulator of glucose metabolism, and diabetic patients treated with degradation resistant GLP-1 receptor peptide agonists, exenatide and liraglutide, experience improved glucose homeostasis. Therefore, efforts to identify orally active small molecule GLP-1 receptor agonists are justifiably being pursued. While several scaffolds are now reported (see Willard et al. in this issue of Experimental Diabetes Research), high-throughput screening campaigns and other discovery approaches have largely failed to identify quality chemical starting points that have been successfully optimized into therapeutic agents. The lack of apparent success is likely due to the inherent difficulty of lower molecular weight, nonpeptide molecules to mimic the complex nature of peptide ligand binding to the GLP-1 receptor ECD and transmembrane regions needed to induce intrinsic structural changes in the receptor to elicit signal transduction.
Fortunately, the availability of peptide ligands for the GLP-1 receptor enables very detailed assessment of GLP-1 receptor signaling pathways, and newer biophysical techniques are helping interrogate the integral mechanisms involved in receptor activation. Further, advances in structural biology methodologies for GPCRs are now rapidly occurring, and application of these techniques to class B1 GPCRs will be groundbreaking. Assimilation of structural information for the full length, intact GLP-1 receptor and improved assay systems with which to monitor the GLP-1 receptor in different conformational states will likely be critical to advancing nascent efforts to identify GLP-1 receptor small molecule ligands. Once novel compounds emerge, it will be important to optimize molecules using testing schemes that incorporate signal transduction mechanisms of GLP-1 physiology, especially GLP-1 receptor stimulation of cAMP production to enhance glucose dependent insulin secretion.
Author Contributions
F. S. Willard and K. W. Sloop contributed equally to this paper.
Conflict of Interest
Both authors are employees of Eli Lilly and Company and may own company stock or possess stock options.
References
- 1.Nauck MA, Homberger E, Siegel EG. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. The Journal of Clinical Endocrinology & Metabolism. 1986;63(2):492–498. doi: 10.1210/jcem-63-2-492. [DOI] [PubMed] [Google Scholar]
- 2.Elrick H, Stimmler L, Hlad CJ, ARAI Y. Plasma insulin response to oral and intravenous glucose administration. The Journal of Clinical Endocrinology & Metabolism. 1964;24:1076–1082. doi: 10.1210/jcem-24-10-1076. [DOI] [PubMed] [Google Scholar]
- 3.Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic sujbjects. Journal of Clinical Investigation. 1967;46(12):1954–1962. doi: 10.1172/JCI105685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mcintyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. The Lancet. 1964;2(7349):20–21. doi: 10.1016/s0140-6736(64)90011-x. [DOI] [PubMed] [Google Scholar]
- 5.Unger RH, Eisentraut AM. Entero-insular axis. Archives of Internal Medicine. 1969;123(3):261–266. [PubMed] [Google Scholar]
- 6.Orskov C, Holst JJ, Nielsen OV. Effect of truncated glucagon-like peptide-1 [Proglucagon-(78–107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology. 1988;123(4):2009–2013. doi: 10.1210/endo-123-4-2009. [DOI] [PubMed] [Google Scholar]
- 7.Holst JJ, Orskov C, Nielsen OV, Schwartz TW. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Letters. 1987;211(2):169–174. doi: 10.1016/0014-5793(87)81430-8. [DOI] [PubMed] [Google Scholar]
- 8.Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7–36: a physiological incretin in man. The Lancet. 1987;2(8571):1300–1304. doi: 10.1016/s0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- 9.Elliott RM, Morgan LM, Tredger JA, Deacon S, Wright J, Marks V. Glucagon-like peptide-1(7–36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post-prandial and 24−h secretion patterns. Journal of Endocrinology. 1993;138(1):159–166. doi: 10.1677/joe.0.1380159. [DOI] [PubMed] [Google Scholar]
- 10.Lauritsen KB, Moody AJ, Christensen KC, Lindkaer Jensen S. Gastric inhibitory polypeptide (GIP) and insulin release after small-bowel resection in man. Scandinavian Journal of Gastroenterology. 1980;15(7):833–840. doi: 10.3109/00365528009181538. [DOI] [PubMed] [Google Scholar]
- 11.Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7–37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. Journal of Clinical Investigation. 1987;79(2):616–619. doi: 10.1172/JCI112855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown JC. Gastric inhibitory polypeptide. Monographs on Endocrinology. 1982;24:1–88. [PubMed] [Google Scholar]
- 13.Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. The Journal of Clinical Endocrinology & Metabolism. 1973;37(5):826–828. doi: 10.1210/jcem-37-5-826. [DOI] [PubMed] [Google Scholar]
- 14.Falko JM, Crockett SE, Cataland S, Mazzaferri EL. Gastric inhibitory polypeptide (GIP) stimulated by fat ingestion in man. The Journal of Clinical Endocrinology & Metabolism. 1975;41(2):260–265. doi: 10.1210/jcem-41-2-260. [DOI] [PubMed] [Google Scholar]
- 15.Thomas FB, Mazzaferri EL, Crockett SE. Stimulation of secretion of gastric inhibitory polypeptide and insulin by intraduodenal amino acid perfusion. Gastroenterology. 1976;70(4):523–527. [PubMed] [Google Scholar]
- 16.Kauth T, Metz J. Immunohistochemical localization of glucagon-like peptide 1. Use of poly- and monoclonal antibodies. Histochemistry. 1987;86(5):509–515. doi: 10.1007/BF00500625. [DOI] [PubMed] [Google Scholar]
- 17.Eissele R, Goke R, Willemer S, et al. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. European Journal of Clinical Investigation. 1992;22(4):283–291. doi: 10.1111/j.1365-2362.1992.tb01464.x. [DOI] [PubMed] [Google Scholar]
- 18.Bell GI, Sanchez Pescador R, Laybourn PJ, Najarian RC. Exon duplication and divergence in the human preproglucagon gene. Nature. 1983;304(5924):368–371. doi: 10.1038/304368a0. [DOI] [PubMed] [Google Scholar]
- 19.Holst JJ. Gut glucagon, enteroglucagon, gut glucagonlike immunoreactivity, glicentin—current status. Gastroenterology. 1983;84(6):1602–1613. [PubMed] [Google Scholar]
- 20.Rouille Y, Kantengwa S, Irminger JC, Halban PA. Role of the prohormone convertase PC3 in the processing of proglucagon to glucagon-like peptide 1. The Journal of Biological Chemistry. 1997;272(52):32810–32816. doi: 10.1074/jbc.272.52.32810. [DOI] [PubMed] [Google Scholar]
- 21.Zhu X, Zhou A, Dey A, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(16):10293–10298. doi: 10.1073/pnas.162352599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothenberg ME, Eilertson CD, Klein K, et al. Processing of mouse proglucagon by recombinant prohormone convertase 1 and immunopurified prohormone convertase 2 in vitro . The Journal of Biological Chemistry. 1995;270(17):10136–10146. doi: 10.1074/jbc.270.17.10136. [DOI] [PubMed] [Google Scholar]
- 23.Takeda J, Seino Y, Tanaka K, et al. Sequence of an intestinal cDNA encoding human gastric inhibitory polypeptide precursor. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(20):7005–7008. doi: 10.1073/pnas.84.20.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchan AM, Polak JM, Capella C. Electronimmunocytochemical evidence for the K cell localization of gastric inhibitory polypeptide (GIP) in man. Histochemistry. 1978;56(1):37–44. doi: 10.1007/BF00492251. [DOI] [PubMed] [Google Scholar]
- 25.Thorens B. Expression cloning of the pancreatic beta cell receptor for the gluco- incretin hormone glucagon-like peptide 1. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(18):8641–8645. doi: 10.1073/pnas.89.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorens B, Porret A, Buhler L, Deng SP, Morel P, Widmann C. Cloning and functional expression of the human islet GLP-1 receptor: demonstration that exendin-4 is an agonist and exendin-(9–39) an antagonist of the receptor. Diabetes. 1993;42(11):1678–1682. doi: 10.2337/diab.42.11.1678. [DOI] [PubMed] [Google Scholar]
- 27.Usdin TB, Mezey E, Button DC, Brownstein MJ, Bonner TI. Gastric inhibitory polypeptide receptor, a member of the secretin- vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology. 1993;133(6):2861–2870. doi: 10.1210/endo.133.6.8243312. [DOI] [PubMed] [Google Scholar]
- 28.Preitner F, Ibberson M, Franklin I, et al. Gluco-incretins control insulin secretion at multiple levels as revealed in mice lacking GLP-1 and GIP receptors. Journal of Clinical Investigation. 2004;113(4):635–645. doi: 10.1172/JCI20518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jorgensen R, Martini L, Schwartz TW, Elling CE. Characterization of glucagon-like peptide-1 receptor beta-arrestin 2 interaction: a high-affinity receptor phenotype. Molecular Endocrinology. 2005;19(3):812–823. doi: 10.1210/me.2004-0312. [DOI] [PubMed] [Google Scholar]
- 30.Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. Beta-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(18):6614–6619. doi: 10.1073/pnas.0710402105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dzhura I, Chepurny OG, Kelley GG, et al. Epac2-dependent mobilization of intracellular Ca2+ by glucagon-like peptide-1 receptor agonist exendin-4 is disrupted in beta-cells of phospholipase C-beta knockout mice. Journal of Physiology. 2010;588, part 24:4871–4889. doi: 10.1113/jphysiol.2010.198424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Islam MS. Calcium signaling in the islets. Advances in Experimental Medicine and Biology. 2010;654:235–259. doi: 10.1007/978-90-481-3271-3_11. [DOI] [PubMed] [Google Scholar]
- 33.Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in Type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29(1):46–52. doi: 10.1007/BF02427280. [DOI] [PubMed] [Google Scholar]
- 34.Jones IR, Owens DR, Moody AJ, Luzio SD, Morris T, Hayes TM. The effects of glucose-dependent insulinotropic polypeptide infused at physiological concentrations in normal subjects and Type 2 (non-insulin-dependent) diabetic patients on glucose tolerance and B-cell secretion. Diabetologia. 1987;30(9):707–712. doi: 10.1007/BF00296993. [DOI] [PubMed] [Google Scholar]
- 35.Krarup T, Saurbrey N, Moody AJ. Effect of porcine gastric inhibitory polypeptide on beta-cell function in type I and type II diabetes mellitus. Metabolism. 1987;36(7):677–682. doi: 10.1016/0026-0495(87)90153-3. [DOI] [PubMed] [Google Scholar]
- 36.Heimesaat MA, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. Journal of Clinical Investigation. 1993;91(1):301–307. doi: 10.1172/JCI116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF. Insulinotropic action of glucagonlike peptide-I-(7–37) in diabetic and nondiabetic subjects. Diabetes Care. 1992;15(2):270–276. doi: 10.2337/diacare.15.2.270. [DOI] [PubMed] [Google Scholar]
- 38.Gutniak M, Orskov C, Holst JJ, Ahren B, Efendic S. Antidiabetogenic effect of glucagon-like peptide-1 (7–36)amide in normal subjects and patients with diabetes mellitus. The New England Journal of Medicine. 1992;326(20):1316–1322. doi: 10.1056/NEJM199205143262003. [DOI] [PubMed] [Google Scholar]
- 39.Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in Type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1993;36(8):741–744. doi: 10.1007/BF00401145. [DOI] [PubMed] [Google Scholar]
- 40.Nauck MA, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. American Journal of Physiology. 1997;273(5, part 1):E981–E988. doi: 10.1152/ajpendo.1997.273.5.E981. [DOI] [PubMed] [Google Scholar]
- 41.Willms B, Werner J, Holst JJ, Ørskov C, Creutzfeldt W, Nauck MA. Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: effects of exogenous glucagon-like peptide-1 (GLP-1)-(7–36) amide in type 2 (noninsulin-dependent) diabetic patients. The Journal of Clinical Endocrinology & Metabolism. 1996;81(1):327–332. doi: 10.1210/jcem.81.1.8550773. [DOI] [PubMed] [Google Scholar]
- 42.Wishart JM, Horowitz M, Morris HA, Jones KL, Nauck MA. Relation between gastric emptying of glucose and plasma concentrations of glucagon-like peptide-1. Peptides. 1998;19(6):1049–1053. doi: 10.1016/s0196-9781(98)00052-7. [DOI] [PubMed] [Google Scholar]
- 43.Meeran K, O’Shea D, Edwards CMB, et al. Repeated intracerebroventricular administration of glucagon-like peptide-1-(7–36) amide or exendin-(9–39) alters body weight in the rat. Endocrinology. 1999;140(1):244–250. doi: 10.1210/endo.140.1.6421. [DOI] [PubMed] [Google Scholar]
- 44.Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. Journal of Clinical Investigation. 1998;101(3):515–520. doi: 10.1172/JCI990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gutzwiller JP, Drewe J, Göke B, et al. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. American Journal of Physiology. 1999;276(5, part 2):R1541–R1544. doi: 10.1152/ajpregu.1999.276.5.R1541. [DOI] [PubMed] [Google Scholar]
- 46.Gutzwiller JP, Göke B, Drewe J, et al. Glucagon-like peptide-1: a potent regulator of food intake in humans. Gut. 1999;44(1):81–86. doi: 10.1136/gut.44.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turton MD, O’Shea D, Gunn I, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. 1996;379(6560):69–72. doi: 10.1038/379069a0. [DOI] [PubMed] [Google Scholar]
- 48.Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. European Journal of Biochemistry. 1993;214(3):829–835. doi: 10.1111/j.1432-1033.1993.tb17986.x. [DOI] [PubMed] [Google Scholar]
- 49.Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo . The Journal of Clinical Endocrinology & Metabolism. 1995;80(3):952–957. doi: 10.1210/jcem.80.3.7883856. [DOI] [PubMed] [Google Scholar]
- 50.Knudsen LB, Pridal L. Glucagon-like peptide-1-(9–36) amide is a major metabolite of glucagon-like peptide-1-(7–36) amide after in vivo administration to dogs, and it acts as an antagonist on the pancreatic receptor. European Journal of Pharmacology. 1996;318(2-3):429–435. doi: 10.1016/s0014-2999(96)00795-9. [DOI] [PubMed] [Google Scholar]
- 51.Hansen L, Deacon CF, Ørskov C, Holst JJ. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology. 1999;140(11):5356–5363. doi: 10.1210/endo.140.11.7143. [DOI] [PubMed] [Google Scholar]
- 52.Deacon CF, Pridal L, Klarskov L, Olesen M, Holst JJ. Glucagon-like peptide 1 undergoes differential tissue-specific metabolism in the anesthetized pig. American Journal of Physiology. 1996;271(3, part 1):E458–E464. doi: 10.1152/ajpendo.1996.271.3.E458. [DOI] [PubMed] [Google Scholar]
- 53.Vilsboll T, Agersø H, Krarup T, Holst JJ. Similar elimination rates of glucagon-like peptide-1 in obese type 2 diabetic patients and healthy subjects. The Journal of Clinical Endocrinology & Metabolism. 2003;88(1):220–224. doi: 10.1210/jc.2002-021053. [DOI] [PubMed] [Google Scholar]
- 54.Siegel EG, Gallwitz B, Scharf G, et al. Biological activity of GLP-1-analogues with N-terminal modifications. Regulatory Peptides. 1999;79(2-3):93–102. doi: 10.1016/s0167-0115(98)00155-4. [DOI] [PubMed] [Google Scholar]
- 55.Ritzel U, Leonhardt U, Ottleben M, et al. A synthetic glucagon-like peptide-1 analog with improved plasma stability. Journal of Endocrinology. 1998;159(1):93–102. doi: 10.1677/joe.0.1590093. [DOI] [PubMed] [Google Scholar]
- 56.Deacon CF, Knudsen LB, Madsen K, Wiberg FC, Jacobsen O, Holst JJ. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia. 1998;41(3):271–278. doi: 10.1007/s001250050903. [DOI] [PubMed] [Google Scholar]
- 57.Goke R, Fehmann HC, Linn T, et al. Exendin-4 is a high potency agonist and truncated exendin-(9–39)-amide an antagonist at the glucagon-like peptide 1-(7–36)-amide receptor of insulin- secreting beta-cells. The Journal of Biological Chemistry. 1993;268(26):19650–19655. [PubMed] [Google Scholar]
- 58.Fehmann HC, Jiang J, Schweinfurth J, Wheeler MB, Boyd AE, Goke B. Stable expression of the rat GLP-I receptor in CHO cells: activation and binding characteristics utilizing GLP-I(7–36)-amide, oxyntomodulin, exendin- 4, and exendin(9–39) Peptides. 1994;15(3):453–456. doi: 10.1016/0196-9781(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 59.Fehmann HC, Jiang J, Schweinfurth J, et al. Ligand-specificity of the rat GLP-I receptor recombinantly expressed in Chinese hamster ovary (CHO-) cells. Zeitschrift fur Gastroenterologie. 1994;32(4):203–207. [PubMed] [Google Scholar]
- 60.Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. The Journal of Biological Chemistry. 1992;267(11):7402–7405. [PubMed] [Google Scholar]
- 61.Eng J, Andrews PC, Kleinman WA, Singh L, Raufman JP. Purification and structure of exendin-3, a new pancreatic secretagogue isolated from Heloderma horridum venom. The Journal of Biological Chemistry. 1990;265(33):20259–20262. [PubMed] [Google Scholar]
- 62.Russell FE, Bogert CM. Gila monster: its biology, venom and bite—a review. Toxicon. 1981;19(3):341–359. doi: 10.1016/0041-0101(81)90040-4. [DOI] [PubMed] [Google Scholar]
- 63.Thum A, Hupe-Sodmann K, Göke R, Voigt K, Göke B, McGregor GP. Endoproteolysis by isolated membrane peptidases reveal metabolic stability of glucagon-like peptide-1 analogs, exendins-3 and -4. Experimental and Clinical Endocrinology and Diabetes. 2002;110(3):113–118. doi: 10.1055/s-2002-29087. [DOI] [PubMed] [Google Scholar]
- 64.Edwards CM, Stanley SA, Davis R, et al. Exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers. American Journal of Physiology. 2001;281(1):E155–E161. doi: 10.1152/ajpendo.2001.281.1.E155. [DOI] [PubMed] [Google Scholar]
- 65.Simonsen L, Holst JJ, Deacon CF. Exendin-4, but not glucagon-like peptide-1, is cleared exclusively by glomerular filtration in anaesthetised pigs. Diabetologia. 2006;49(4):706–712. doi: 10.1007/s00125-005-0128-9. [DOI] [PubMed] [Google Scholar]
- 66.Greig NH, Holloway HW, De Ore KA, et al. Once daily injection of exendin-4 to diabetic mice achieves long-term beneficial effects on blood glucose concentrations. Diabetologia. 1999;42(1):45–50. doi: 10.1007/s001250051111. [DOI] [PubMed] [Google Scholar]
- 67.Szayna M, Doyle ME, Betkey JA, et al. Exendin-4 decelerates food intake, weight gain, and fat deposition in Zucker rats. Endocrinology. 2000;141(6):1936–1941. doi: 10.1210/endo.141.6.7490. [DOI] [PubMed] [Google Scholar]
- 68.Young AA, Gedulin BR, Bhavsar S, et al. Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta) Diabetes. 1999;48(5):1026–1034. doi: 10.2337/diabetes.48.5.1026. [DOI] [PubMed] [Google Scholar]
- 69.Kolterman OG, Kim DD, Shen L, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. American Journal of Health-System Pharmacy. 2005;62(2):173–181. doi: 10.1093/ajhp/62.2.173. [DOI] [PubMed] [Google Scholar]
- 70.Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. Journal of Medicinal Chemistry. 2000;43(9):1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 71.Agerso H, Jensen LB, Elbrønd B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45(2):195–202. doi: 10.1007/s00125-001-0719-z. [DOI] [PubMed] [Google Scholar]
- 72.Elbrond B, Jakobsen G, Larsen S, et al. Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diabetes Care. 2002;25(8):1398–1404. doi: 10.2337/diacare.25.8.1398. [DOI] [PubMed] [Google Scholar]
- 73.Neidigh JW, Fesinmeyer RM, Prickett KS, Andersen NH. Exendin-4 and glucagon-like-peptide-1: NMR structural comparisons in the solution and micelle-associated states. Biochemistry. 2001;40(44):13188–13200. doi: 10.1021/bi010902s. [DOI] [PubMed] [Google Scholar]
- 74.Underwood CR, Garibay P, Knudsen LB, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. The Journal of Biological Chemistry. 2010;285(1):723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Runge S, Thøgersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. The Journal of Biological Chemistry. 2008;283(17):11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- 76.Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga JP. Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(44):16084–16089. doi: 10.1073/pnas.0503942102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Al-Sabah S, Donnelly D. A model for receptor-peptide binding at the glucagon-like peptide-1 (GLP-1) receptor through the analysis of truncated ligands and receptors. British Journal of Pharmacology. 2003;140(2):339–346. doi: 10.1038/sj.bjp.0705453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Runge S, Wulff BS, Madsen K, Bräuner-Osborne H, Knudsen LB. Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. British Journal of Pharmacology. 2003;138(5):787–794. doi: 10.1038/sj.bjp.0705120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perrin MH, Grace CRR, Riek R, Vale WW. The three-dimensional structure of the n-terminal domain of corticotropin-releasing factor receptors sushi domains and the B1 family of G protein-coupled receptors. Annals of the New York Academy of Sciences. 2006;1070:105–119. doi: 10.1196/annals.1317.065. [DOI] [PubMed] [Google Scholar]
- 80.Pioszak AA, Xu HE. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(13):5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bazarsuren A, Grauschopf U, Wozny M, et al. In vitro folding, functional characterization, and disulfide pattern of the extracellular domain of human GLP-1 receptor. Biophysical Chemistry. 2002;96(2-3):305–318. doi: 10.1016/s0301-4622(02)00023-6. [DOI] [PubMed] [Google Scholar]
- 82.Hoare SR. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discovery Today. 2005;10(6):417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- 83.Jin L, Briggs SL, Chandrasekhar S, et al. Crystal structure of human parathyroid hormone 1-34 at 0.9−A resolution. The Journal of Biological Chemistry. 2000;275(35):27238–27244. doi: 10.1074/jbc.M001134200. [DOI] [PubMed] [Google Scholar]
- 84.Thornton K, Gorenstein DG. Structure of glucagon-like peptide(7–36) amide in a dodecylphosphocholine micelle as determined by 2D NMR. Biochemistry. 1994;33(12):3532–3539. doi: 10.1021/bi00178a009. [DOI] [PubMed] [Google Scholar]
- 85.Andersen NH, Brodsky Y, Neidigh JW, Prickett KS. Medium-dependence of the secondary structure of exendin-4 and glucagon-like-peptide-1. Bioorganic and Medicinal Chemistry. 2002;10(1):79–85. doi: 10.1016/s0968-0896(01)00263-2. [DOI] [PubMed] [Google Scholar]
- 86.Leech CA, Chepurny OG, Holz GG. Epac2-Dependent Rap1 Activation and the Control of Islet Insulin Secretion by Glucagon-Like Peptide-1. Vitamins and Hormones. 2010;84(C):279–302. doi: 10.1016/B978-0-12-381517-0.00010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wheeler MB, Lu M, Dillon JS, Leng XH, Chen C, Boyd AE. Functional expression of the rat glucagon-like peptide-I receptor, evidence for coupling to both adenylyl cyclase and phospholipase-C. Endocrinology. 1993;133(1):57–62. doi: 10.1210/endo.133.1.8391428. [DOI] [PubMed] [Google Scholar]
- 88.Montrose-Rafizadeh C, Avdonin P, Garant MJ, et al. Pancreatic glucagon-like peptide-1 receptor couples to multiple G proteins and activates mitogen-activated protein kinase pathways in Chinese hamster ovary cells. Endocrinology. 1999;140(3):1132–1140. doi: 10.1210/endo.140.3.6550. [DOI] [PubMed] [Google Scholar]
- 89.Cullinan CA, Brady EJ, Saperstein R, Leibowitz MD. Glucose-dependent alterations of intracellular free calcium by glucagon-like peptide-1((7–36amide)) in individual ob/ob mouse beta-cells. Cell Calcium. 1994;15(5):391–400. doi: 10.1016/0143-4160(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 90.Koole C, Wootten D, Simms J, et al. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Molecular Pharmacology. 2010;78(3):456–465. doi: 10.1124/mol.110.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Coopman K, Huang Y, Johnston N, Bradley SJ, Wilkinson GF, Willars GB. Comparative effects of the endogenous agonist glucagon-like peptide-1 (GLP-1)-(7–36) amide and the small-molecule ago-allosteric agent "compound 2" at the GLP-1 receptor. Journal of Pharmacology and Experimental Therapeutics. 2010;334(3):795–808. doi: 10.1124/jpet.110.166009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sassmann A, Gier B, Gröne HJ, Drews G, Offermanns S, Wettschureck N. The Gq/G11-mediated signaling pathway is critical for autocrine potentiation of insulin secretion in mice. Journal of Clinical Investigation. 2010;120(6):2184–2193. doi: 10.1172/JCI41541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yada T, Itoh K, Nakata M. Glucagon-like peptide-1-(7–36)amide and a rise in cyclic adenosine 3′,5′- monophosphate increase cytosolic free Ca2+ in rat pancreatic beta-cells by enhancing Ca2+ activity. Endocrinology. 1993;133(4):1685–1692. doi: 10.1210/endo.133.4.8404610. [DOI] [PubMed] [Google Scholar]
- 94.Kenakin T. Efficacy in drug receptor theory: outdated concept or under-valued tool? Trends in Pharmacological Sciences. 1999;20(10):400–405. doi: 10.1016/s0165-6147(99)01361-9. [DOI] [PubMed] [Google Scholar]
- 95.Guo J, Liu M, Yang D, et al. Phospholipase C signaling via the parathyroid hormone (PTH)/PTH-related peptide receptor is essential for normal bone responses to PTH. Endocrinology. 2010;151(8):3502–3513. doi: 10.1210/en.2009-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Takasaki J, Saito T, Taniguchi M, et al. A novel Galphaq/11-selective inhibitor. The Journal of Biological Chemistry. 2004;279(46):47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- 97.Jorgensen R, Kubale V, Vrecl M, Schwartz TW, Elling CE. Oxyntomodulin differentially affects glucagon-like peptide-1 receptor beta-arrestin recruitment and signaling through Galpha(s) Journal of Pharmacology and Experimental Therapeutics. 2007;322(1):148–154. doi: 10.1124/jpet.107.120006. [DOI] [PubMed] [Google Scholar]
- 98.Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Progress in Hormone Research. 1996;51:319–351. discussion 352-353. [PubMed] [Google Scholar]
- 99.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nature Reviews Drug Discovery. 2010;9(5):373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Quoyer J, Longuet C, Broca C, et al. GLP-1 mediates antiapoptotic effect by phosphorylating bad through a beta-arrestin 1-mediated ERK1/2 activation in pancreatic beta-cells. The Journal of Biological Chemistry. 2010;285(3):1989–2002. doi: 10.1074/jbc.M109.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luan B, Zhao J, Wu H, et al. Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature. 2009;457(7233):1146–1149. doi: 10.1038/nature07617. [DOI] [PubMed] [Google Scholar]
- 102.Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drueker DJ. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. The Journal of Biological Chemistry. 2003;278(1):471–478. doi: 10.1074/jbc.M209423200. [DOI] [PubMed] [Google Scholar]
- 103.Wang Q, Brubaker P. Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia. 2002;45(9):1263–1273. doi: 10.1007/s00125-002-0828-3. [DOI] [PubMed] [Google Scholar]
- 104.Burcelin R, Dejager S. GLP-1: what is known, new and controversial in 2010? Diabetes and Metabolism. 2010;36(6, part 1):503–509. doi: 10.1016/j.diabet.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 105.Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144(12):5149–5158. doi: 10.1210/en.2003-0323. [DOI] [PubMed] [Google Scholar]
- 106.Tian L, Gao J, Weng G, et al. Comparison of exendin-4 on beta-cell replication in mouse and human islet grafts. Transplant International. 2011;24(8):856–864. doi: 10.1111/j.1432-2277.2011.01275.x. [DOI] [PubMed] [Google Scholar]
- 107.Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacology and Therapeutics. 2007;113(3):546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Donnelly D. The structure and function of the glucagon-like peptide-1 receptor and its ligands. doi: 10.1111/j.1476-5381.2011.01687.x. British Journal of Pharmacology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dong M, Pinon DI, Asmann YW, Miller LJ. Possible endogenous agonist mechanism for the activation of secretin family G protein-coupled receptors. Molecular Pharmacology. 2006;70(1):206–213. doi: 10.1124/mol.105.021840. [DOI] [PubMed] [Google Scholar]
- 110.Dong M, Gao F, Pinon DI, Miller LJ. Insights into the structural basis of endogenous agonist activation of family B G protein-coupled receptors. Molecular Endocrinology. 2008;22(6):1489–1499. doi: 10.1210/me.2008-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dong M, Pinon DI, Miller LJ. Site of action of a pentapeptide agonist at the glucagon-like peptide-1 receptor. Insight into a small molecule agonist-binding pocket. Bioorganic and Medicinal Chemistry Letters. 2012;22(1):638–641. doi: 10.1016/j.bmcl.2011.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(2):643–648. doi: 10.1073/pnas.022460899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Janz JM, Ren Y, Looby R, et al. Direct interaction between an allosteric agonist pepducin and the chemokine receptor CXCR4. Journal of the American Chemical Society. 2011;133(40):15878–15881. doi: 10.1021/ja206661w. [DOI] [PubMed] [Google Scholar]
- 114.Runge S, Schimmer S, Oschmann J, et al. Differential structural properties of GLP-1 and exendin-4 determine their relative affinity for the GLP-1 receptor N-terminal extracellular domain. Biochemistry. 2007;46(19):5830–5840. doi: 10.1021/bi062309m. [DOI] [PubMed] [Google Scholar]
- 115.de Maturana RL, Willshaw A, Kuntzsch A, Rudolph R, Donnelly D. The isolated N-terminal domain of the glucagon-like peptide-1 (GLP-1) receptor binds exendin peptides with much higher affinity than GLP-1. The Journal of Biological Chemistry. 2003;278(12):10195–10200. doi: 10.1074/jbc.M212147200. [DOI] [PubMed] [Google Scholar]
- 116.Mann RJ, Nasr NE, Sinfield JK, Paci E, Donnelly D. The major determinant of exendin-4/glucagon-like peptide 1 differential affinity at the rat glucagon-like peptide 1 receptor N-terminal domain is a hydrogen bond from SER-32 of exendin-4: research paper. British Journal of Pharmacology. 2010;160(8):1973–1984. doi: 10.1111/j.1476-5381.2010.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schroder-Tittmann K, Bosse-Doenecke E, Reedtz-Runge S, et al. Recombinant expression, in vitro refolding, and biophysical characterization of the human glucagon-like peptide-1 receptor. Biochemistry. 2010;49(36):7956–7965. doi: 10.1021/bi101159s. [DOI] [PubMed] [Google Scholar]
- 118.Bisello A, Adams AE, Mierke DF, et al. Parathyroid hormone-receptor interactions identified directly by photocross-linking and molecular modeling studies. The Journal of Biological Chemistry. 1998;273(35):22498–22505. doi: 10.1074/jbc.273.35.22498. [DOI] [PubMed] [Google Scholar]
- 119.Dong M, Li Z, Pinon DI, Lybrand TP, Miller LJ. Spatial approximation between the amino terminus of a peptide agonist and the top of the sixth transmembrane segment of the secretin receptor. The Journal of Biological Chemistry. 2004;279(4):2894–2903. doi: 10.1074/jbc.M310407200. [DOI] [PubMed] [Google Scholar]
- 120.Dong M, Pinon DI, Cox RF, Miller LJ. Molecular approximation between a residue in the amino-terminal region of calcitonin and the third extracellular loop of the class B G protein-coupled calcitonin receptor. The Journal of Biological Chemistry. 2004;279(30):31177–31182. doi: 10.1074/jbc.M404113200. [DOI] [PubMed] [Google Scholar]
- 121.Ji Z, Hadac EM, Henne RM, Patel SA, Lybrand TP, Miller LJ. Direct identification of a distinct site of interaction between the carboxyl-terminal residue of cholecystokinin and the type A cholecystokinin receptor using photoaffinity labeling. The Journal of Biological Chemistry. 1997;272(39):24393–24401. doi: 10.1074/jbc.272.39.24393. [DOI] [PubMed] [Google Scholar]
- 122.Chen Q, Pinon DI, Miller LJ, Dong M. Molecular basis of glucagon-like peptide 1 docking to its intact receptor studied with carboxyl-terminal photolabile probes. The Journal of Biological Chemistry. 2009;284(49):34135–34144. doi: 10.1074/jbc.M109.038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen Q, Pinon DI, Miller LJ, Dong M. Spatial approximations between residues 6 and 12 in the amino-terminal region of glucagon-like peptide 1 and its receptor: a region critical for biological activity. The Journal of Biological Chemistry. 2010;285(32):24508–24518. doi: 10.1074/jbc.M110.135749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Miller LJ, Chen Q, Lam PC-H, et al. Refinement of glucagon-like peptide 1 docking to its intact receptor using mid-region photolabile probes and molecular modeling. The Journal of Biological Chemistry. 2011;286(18):15895–15907. doi: 10.1074/jbc.M110.217901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Adelhorst K, Hedegaard BB, Knudsen LB, Kirk O. Structure-activity studies of glucagon-like peptide-1. The Journal of Biological Chemistry. 1994;269(9):6275–6278. [PubMed] [Google Scholar]
- 126.Englander SW, Sosnick TR, Englander JJ, Mayne L. Mechanisms and uses of hydrogen exchange. Current Opinion in Structural Biology. 1996;6(1):18–23. doi: 10.1016/s0959-440x(96)80090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Katta V, Chait BT. Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Communications in Mass Spectrometry. 1991;5(4):214–217. doi: 10.1002/rcm.1290050415. [DOI] [PubMed] [Google Scholar]
- 128.Zhang X, Chien EYT, Chalmers MJ, et al. Dynamics of the beta2-adrenergic G-protein coupled receptor revealed by hydrogen-deuterium exchange. Analytical Chemistry. 2010;82(3):1100–1108. doi: 10.1021/ac902484p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.West GM, Chien EYT, Katritch V, et al. Ligand-dependent perturbation of the conformational ensemble for the GPCR beta(2) adrenergic receptor revealed by HDX. Structure. 2011;19(10):1424–1432. doi: 10.1016/j.str.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kahsai AW, Xiao K, Rajagopal S, et al. Multiple ligand-specific conformations of the beta(2)-adrenergic receptor. Nature Chemical Biology. 2011;7(10):692–700. doi: 10.1038/nchembio.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 132.Salon JA, Lodowski DT, Palczewski K. The significance of G protein-coupled receptor crystallography for drug discovery. Pharmacological Reviews. 2011;63(4):901–937. doi: 10.1124/pr.110.003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rasmussen SG, Devree BT, Zou Y, et al. Crystal structure of the beta(2) adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chae PS, Rasmussen SGF, Rana RR, et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nature Methods. 2010;7(12):1003–1008. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rosenbaum DM, Cherezov V, Hanson MA, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318(5854):1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 136.Rasmussen SG, Choi H-J, Fung JJ, et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature. 2011;469(7329):175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Caffrey M. Crystallizing membrane proteins for structure-function studies using lipidic mesophases. Biochemical Society Transactions. 2011;39(3):725–732. doi: 10.1042/BST0390725. [DOI] [PMC free article] [PubMed] [Google Scholar]