

Abstract
Microglia are multifunctional immune cells in the central nervous system (CNS). In the neurodegenerative diseases such as Alzheimer's disease (AD), accumulation of glial cells, gliosis, occurs in the lesions. The role of accumulated microglia in the pathophysiology of AD is still controversial. When neuronal damage occurs, microglia exert diversified functions, including migration, phagocytosis, and production of various cytokines and chemokines. Among these, microglial phagocytosis of unwanted neuronal debris is critical to maintain the healthy neuronal networks. Microglia express many surface receptors implicated in phagocytosis. It has been suggested that the lack of microglial phagocytosis worsens pathology of AD and induces memory impairment. The present paper summarizes recent evidences on implication of microglial chemotaxis and phagocytosis in AD pathology and discusses the mechanisms related to chemotaxis toward injured neurons and phagocytosis of unnecessary debris.
1. Introduction
Microglia are macrophage-like resident immune cells in the central nervous system (CNS) and possess both neurotoxic and neuroprotective function. Microglia accumulate in the lesions of a variety of neurodegenerative disorders, such as Alzheimer's disease (AD), Parkinson's disease, and multiple sclerosis, and are thought to play both toxic and protective functions for neuronal survival [1]. Microglia are considered to be a first line defense and respond quickly to various stimuli. When activated, microglia undergo morphological changes to ameboid, proliferate, migrate toward injured areas, and release many soluble factors and phagocytosis of foreign substances or unwanted self-debris. Appropriate migration of microglia to damaged area is controlled by chemokines and nucleotide ATP [2, 3]. Phagocytosis seems to be important to prevent the senile plaque expansion in AD by removing amyloid β (Aβ) deposit [4]. Microglia not only engulf the Aβ protein but also phagocytose apoptotic cells and degenerated neuronal debris. Phagocytosis of apoptotic or degenerated neuronal debris is crucial to reduce inflammation and maintain healthy neuronal networks. Another type of phagocytosis, phagocytosis with inflammation, occurs in chronic inflammatory-related neurodegenerative disorders including Alzheimer disease [5–7].
Degenerated neurons releases several signaling molecules, including nucleotides, cytokines, and chemokines, to recruit microglia and enhance their activities [8, 9]. The phenomenon are now termed as find-me, eat-me, and help-me signals. In this paper, we focused on find-me, and eat-me signals from degenerated neurons to microglia. Most distinguished and examined eat-me signal is phosphatidylserine (PS), which is a component of cellular membrane and is everted on apoptotic cell membrane [10, 11]. Nucleotides are also considered as the eat-me signal lately; microglia expresse various P2X and P2Y receptors, nucleotide receptors, which regulate not only chemotaxis but also phagocytosis [8, 12].
Microglia express many other surface receptors, which have direct interaction with the target to initiate phagocytosis, including PS receptor [6], lipopolysaccharide (LPS) receptor CD14 [13], the scavenger receptor CD36 [14], the purine receptor P2Y6 [8], and the toll-like receptors (TLRs) [15] (Table 1, Figure 1). Another surface receptor, the CX3C chemokine fractalkine receptor CX3CR1, is almost exclusively expressed in microglia throughout the CNS, which is involved in progression of neurodegenerative disease by altering microglial activities [16, 17] (Figure 2). Deletion of CX3CR1 expression in microglia results in progressive neuronal cell death in an animal model of neurodegenerative disease, by inducing microglial dysfunction. It has been identified recently that neurons themselves produce cytokine and chemokine, such as fractalkine. As shown in Figure 2, we previously reported that interleukin-34 (IL-34), a newly discovered cytokine, is produced by neurons, and that its receptor, colony-stimulating factor 1 receptor, is primarily expressed on microglia [18]. Fractalkine and IL-34 might be important mediator between neurons and microglia, and it is important to clarify this cellular crosstalk signaling pathways for seeking future therapeutic target of neurodegenerative diseases including AD. In the following sections, we will discuss about recent advances of microglial chemotaxis and phagocytosis and their implications for AD therapy.
Table 1.
Various chemotaxis or phagocytosis-related receptors in microglia and its ligand(s) or interacted factors. Microglia are activated with various stimuli through the specific receptor of each stimuli. For detailed review of chemokines, pathogens, and factors associated with tissue damage recognized by microglia, refer to [19–21].
| Receptor type | Subtypes | Ligand(s)/interacted factors |
|---|---|---|
| CCR1 | CCL3 (MIP-1α), CCL5 (RANTES), CCL7 (MCP-3), CCL9 (MIP-1γ), CCL14 (HCC-1), CCL15 (HCC-2/leukotactin-1), CCL16 (HCC-4/LEC), CCL23 (MPIF-1) |
|
| Chemokine receptor | CCR2 | CCL2 (MCP-1), CCL7 (MCP-3), CCL8 (MCP-2), CCL13 (MCP-4), CCL16 (HCC-4/LEC) |
| CCR5 | CCL3 (MIP-1α), CCL4 (MIP-1β), CCL5 (RANTES) | |
| CXCR3 | CXCL9 (Mig), CXCL10 (IP-10), CXCL11 (I-TAC) | |
| CX3CR1 | CX3CL1 (Fractalkine) | |
|
| ||
| Purinergic receptor | P2X4, P2Y7, P2Y12 P2Y6 |
ATP, ADP UDP |
|
| ||
| TLR | TLR1 TLR2 TLR4 TLR6 TLR9 |
Triacyl lipopeptides Glycolipids, Hsp70, HMGB1, Aβ LPS, Hsp Diacyl lipopeptides CpG-DNA |
|
| ||
| Phosphatidylserine (PS) receptor | MFG-E8, Tim1, Tim4 RAGE |
PS of apoptotic cells Aβ, AGE, HMGB1 PS of apoptotic cells |
|
| ||
| Scavenger receptor (SR) | SR-AI/II (CD204), SR-BI, CD36 | Cellular debris, apoptotic cells, Aβ (CD36) |
|
| ||
| Immunoglobulin (Ig) receptor | FcγRI, FcγRIII | Ig-opsonized particles |
|
| ||
| Complement receptor (CR) | CR3 (MAC-1; CD11b/CD18) | Complement components, opsonized particles |
|
| ||
| Other phagocytosis-related receptor | CD14 CD47 CD200R TREM2 |
LPS, Aβ
SIRPα (CD172a) CD200 Hsp60, DAP12 |
Figure 1.

Chemotaxis or phagocytosis-involved receptors in microglia and correlation of the inflammatory or anti-inflammatory response. Many of the receptors correlated with microglial activities of chemotaxis (migration) or phagocytosis, respectively. Among these, some of the receptors possess not merely single function; CCR1 is the migration-inducing receptor that also possesses phagocytotic activity. CCR2 and CCR5 are also the migration-inducting receptors that lead to anti-inflammatory response. CX3CR1 contributes to migration, phagocytosis, and anti-inflammatory response. TREM2 and MFG-E8 induce phagocytosis and anti-inflammatory response. CD47 and CD200R usually induce phagocytosis under pathological condition, so that they indirectly contribute to anti-inflammatory status. TLR9 activates microglia to induce phagocytosis with producing proinflammatory and anti-inflammatory molecules. There are receptors inducing not only phagocytosis but also inflammatory response (CD14, CD36, RAGE, TLR1, TLR2, TLR4, and TLR6). Within these receptors that the synergistic signaling involved Aβ-triggering inflammatory response are CD14-TLR2-TLR4 and CD36-TLR2-TLR6.
Figure 2.
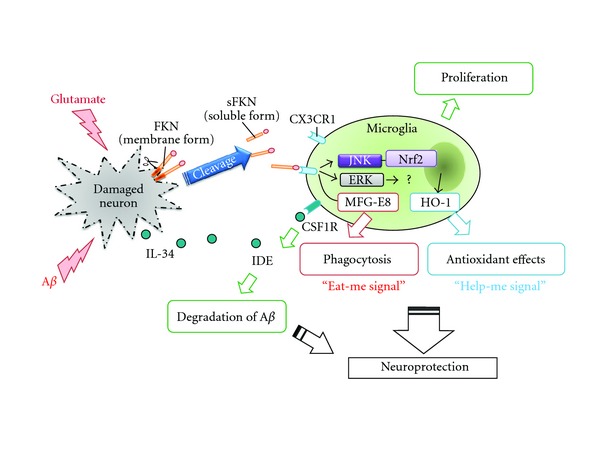
Model of the role of neuronal chemokine (FKN) and neuronal cytokine (IL-34) in microglial phagocytosis and neuroprotection. Neuronal cells primary produce chemokine fractalkine (CX3CL1; FKN) and cytokine IL-34. Microglia predominantly express its receptor, CX3CR1, and colony-stimulating factor 1 receptor (CSF1R). Soluble form of FKN (sFKN) is secreted from damaged neurons and promotes microglial phagocytosis of neuronal debris through the release of MFG-E8. sFKN also induces the expression of the antioxidant enzyme HO-1 in microglia via Nrf2 recruitment and activation of the JNK MAPK signaling pathway. The neuroprotective effects of sFKN are also mediated in part by activation of ERK MAPK, although the downstream signaling pathway has not yet been elucidated. IL-34 promoted microglial proliferation and clearance of Aβ which mediates insulin-degrading enzyme (IDE) expression. Therefore, sFKN and IL-34 may be an intrinsic neuroprotectant for damaged yet surviving neurons.
2. Microglial Chemotaxis in CNS Injury
Chemokine and its receptors are expressed in broad area of the CNS. The expression levels were increased under pathological conditions, which seem to facilitate the recruitment and trafficking of glial cells to the damaged area [22]. Microglia constitutively express several chemokine receptors (Table 1), which are implicated in the recruitment and accumulation microglia in AD lesions. When exposed to Aβ, microglia are induced to produce several chemokines, such as CCL2, CCL4, and CXCL12 [23]. CCR2, the receptor of CCL2, deficiency resulted in reduction of microglial accumulation and higher brain Aβ levels in mouse model of AD, which might be mediated via suppression of anti-inflammatory molecule, transforming growth factor β (TGF-β) [3]. However, there is a conflicting report showing increased TGF-β signaling in microglia surrounding Aβ plaques in CCR2 knockout in AD model mouse (APPSwe/PS1/CCR2−/−) [24]. The other chemokine receptor CX3CR1 expression in microglia was also increased in the mice. This AD model has been shown to have accumulation of oligomeric Aβ and memory impairment [24]. CX3CR1 is the sole receptor of CX3C chemokine CX3CL1 (fractalkine). The roles of CX3CL1-CX3CR1 signaling on AD pathology are discussed in next section. CCL2 expression level is also related to another neurodegenerative disorder, multiple sclerosis (MS); CCL2 level is downregulated in cerebrospinal fluid from MS patients [25].
CCL21 is a neuronal chemokine, expressed in neurons. Expression of CCL21 is upregulated in neurons undergoing degeneration [26, 27]. CCL21 triggers chemotaxis of microglia through CXCR3, but not CCR7 which implicated in peripheral lymphoid organs [28].
CXCR3-CXCL10 interaction is also implicated in microglial migration [29]. CXCR3 knockout mice reveal impairment of the microglial migration but no change in proliferation. CXCL10 is also expressed in neurons. CXCL10 and CCL21 synergistically induce microglial homing through the receptor CXCR3 [30]. CXCL10 inhibits CCL21-induced migration in microglia through CXCR3 [31]. Neuronal CCL21 upregulates P2X4 receptor, the nucleotide receptor, expressed in microglia [32]. This cascade is implicated in pathophysiology of tactile allodynia to cause chronic neuropathic pain.
CCR5 also plays a role in neuronal survival [33]. In ischemic stroke models, brain damage is severer by CCR5 deficiency [34].
Inter- and intracellular transmitter nucleotides can influence microglial migration and phagocytosis. Microglia express various nucleotide receptors, P2X and P2Y receptors (Table 1) [35]. Aβ-induced microglial activation is mediated through P2X7 receptor that is reported as proinflammatory response conductive receptor [36]. ATP predominantly induced microglial migration among nucleotides through P2Y receptors, especially P2Y12 [2, 37]. Following CNS injury, expression of P2Y12 in microglia drastically reduced after microglial activation, suggesting that P2Y12 is a primary and temporary receptor to induce microglial chemotaxis at early stages of the local CNS injury [37]. The other nucleotide UDP increases mainly microglial phagocytosis (uptake of microspheres) via P2Y6 receptor [8]. In the condition brain damage by kainic acid administration, the P2Y6 receptor is upregulated and can act as a sensor for phagocytosis [38].
3. CX3CL1-CX3CR1
The CX3C chemokine CX3CL1 (fractalkine, also called as neurotactin), which has been identified as two forms, soluble or membrane-anchored forms, plays a pivotal role in signaling between degenerating neurons and microglia [39]. CX3CL1 and its receptor CX3CR1 are highly expressed in brain tissue, particularly in neurons and microglia [40–42]. CX3CL1 directly induces various microglial functions including migration [41], proliferation [43], inhibition of Fas-ligand-induced cell death [44] and glutamate-induced neurotoxicity [45, 46], and inhibition of proinflammatory cytokine production [42, 47]. Recently, we have shown that soluble form of CX3CL1 also directly enhances microglial clearance of degenerated neuronal debris, which is mediated through phosphatidylserine (PS) receptor and production of Milk fat globule-EGF factor 8 protein (MFG-E8) [46] (Figure 2). The source of soluble form of CX3CL1 is neuron. The membrane-anchored CX3CL1 is cleaved by several proteases including a disintegrin and metalloprotease (ADAM) family, ADAM-10 and ADAM-17 [48–50], and cathepsin S [51]. When neurons are injured or exposed to glutamate, shedding of CX3CL1 occurs immediately [41, 52]. However, little is known about direct connection with Aβ-induced neuronal toxicity and the CX3CL1-shedding.
Another important function of ADAM family enzyme, except for CX3CL1 shedding, is an α-secretase. It cleaves APP in the centre of the Aβ domain, and generated α-APP is considered to have neurotrophic function [53–56]. The ADAMs include ADAM-9, ADAM-10, and ADAM-17 [57, 58]. Cathepsin S is expressed predominantly in microglia and implicated in microglial activation of neuropathic pain [51]. Therefore, these CX3CL1-shedding proteases may regulate the microglial phagocytosis directly or indirectly through CX3CL1 expression, and may also play a role on pathogenesis of AD.
Microglia respond to CX3CL1 through CX3CR1. In a previous study, we have shown that CX3CL1 functions neuroprotective against activated microglia-induced neurotoxicity [46]. There are some reports showing that CCL2 activates CX3CR1 expression, and the induction of CX3CR1 expression by CCL2-CCR2 axis is mediated through p38 MAPK activation [59]. CX3CR1 deficiency increases susceptibility to neurotoxicity in mouse models of Parkinson's disease, amyotrophic lateral sclerosis, and systemic LPS administration [16]. CX3CL1-induced neuroprotection in a rat model of Parkinson's disease has also been reported recently [60]. In addition, there are some reports showing that pathologic features of AD mouse model are worsened by knockout of CX3CR1 [17, 61].
4. Microglial Receptors Involving Phagocytosis with or without Inflammation: Possible Implication to AD Pathology
Microglial phagocytosis is classified into two categories, with or without inflammation [62]. Some receptors are involved in both types of phagocytosis (Table 1). The receptor leading to inflammatory status includes CD14, CD36, the receptor for advanced glycation end products (RAGEs), and toll-like receptor (TLR) 1, TLR2, TLR4, and TLR6. The receptor leading to anti-inflammatory status includes triggering receptor expressed on myeloid cells 2 (TREM2) and PS receptor (PSR), MFG-E8. Microglia also express many other phagocytosis-related receptors which are not yet unclear to the correlation of inflammatory status.
CR3 —
Microglia express classical phagocytosis-related receptor, the α M β 2 integrin complement-receptor-3 (CR3; MAC-1; CD11b/CD18), and scavenger-receptor (SR)-AI/II (SR-AI/II; CD204). CR3 synergistically activated SR-AI/II-mediated myelin phagocytosis by microglia [63, 64]. CR3 is implicated in clearance of bacteria through induction of major histocompatibility complex class II (MHC II) antigens [65]. CR3 is involved in endocytosis in normal conditions and MHC II antigens in an inflammatory state. Microglia express SR-AI/II and SR-BI during neonatal period, while in adult they lack the expression. However SR-AI/II expression in microglia is upregulated in AD [66].
CD14 —
CD14 is the LPS receptor which is also considered as classical phagocytosis-related receptor in macrophage and microglia [67]. CD14-mediated phagocytosis of apoptotic cells occurred in both normal and inflammatory conditions [67]. CD14-mediated phagocytosis does not require expression of PS receptor and possibly induces inflammatory conditions through activation of CD14 signaling [68]. CD14 also contributes phagocytosis of Aβ by microglia [13]. However, deletion of CD14 reportedly attenuates pathological features of AD mouse model. The authors suggested that it might be due to reduction of insoluble, but not soluble, Aβ [69].
FcγReceptor —
Exposure of fibrillar Aβ to microglia induces phagocytosis through the receptors distinct from those used by the classical phagocytosis: immunoglobulin receptors (FcγRI and FcγRIII) or complement receptors [70].
CD36 and CD47 —
Aβ directly interacted with microglial cell surface receptor complex comprising the class B scavenger receptor CD36, α 6 β 1 integrin, and integrin-associated protein CD47, all of them involved in migration and phagocytosis of microglia [70–72]. CD36 is required for fibrillar Aβ-induced chemotaxis and proinflammatory molecules including reactive oxygen species (ROS), TNFα, IL-1β, and several chemokines in microglia [23]. Aβ activates microglial recruitment to amyloid deposition site through CD36-dependent signaling cascade involving the Src kinase family members, Lyn and Fyn, and the ERK1/2 [73]. CD47 is a membrane glycoprotein, broadly expressed in the various cell types in the CNS, including neurons and microglia. Signal regulatory protein-α (SIRPα; CD172a) is a receptor binding to CD47, which is also expressed on neurons and myeloid cells. SIRPα is an inhibitory molecule of CD47 that downregulates MAPK phosphorylation which is a downstream pathway of CD47 [74]. SIRPα interacts with the protein tyrosine phosphatases SHP-1 and SHP-2, which are predominantly expressed in neurons, dendritic cells, and macrophages [75]. Intact myelin expresses CD47 to suppress myelin phagocytosis by microglia via SIRPα-CD47 interaction [76]. Therefore, CD47 functions normally as a marker of “self” to protect intact body component [76, 77].
RAGE —
In has been reported that the direct interaction of Aβ peptide with the receptor for RAGE is important in AD pathophysiology. In AD brains, RAGE expression is increased, and RAGE directly induces neurotoxicity by production of oxidative stressors and indirectly by activating microglia [78]. RAGE increases macrophage-colony stimulating factor (M-CSF) from neurons via nuclear-factor-κB- (NF-κB-) dependent pathway and released M-CSF induced interaction of cognate receptor c-fms on microglia which prolongs survival of microglia [79]. RAGE recognizes multiple ligands other than Aβ peptide, such as advanced glycation end products (AGEs), PS, and high-mobility group box 1 protein (HMGB1) [80, 81]. These molecules act as an agonist of RAGE on microglia, by inducing proinflammatory molecules, such as NO, TNF-α, IL-1β, and IL-6, via MAP-kinase-kinase (MEK) and phosphatidylinositol 3-kinase (PI3K) pathways [81]. Activation of RAGE leaded to NF-κB and MAPK-mediated signaling to propagate and perpetuate inflammation status [82]. RAGE also mediates the transport of peripheral Aβ into the brain across the blood-brain barrier (BBB) [83].
CD200R —
CD200 is a transmembrane glycoprotein and is expressed on many different cell types including neurons, endothelial cells, lymphocytes, and dendritic cells [84]. The receptor of CD200, CD200R expression, is predominant in myeloid cells, macrophages, and microglia [84, 85]. As in the case of SIRPα-CD47, CD200 exerts inhibitory effect on CD200R, so that CD200-CD200R interaction can downregulate activity of microglia. In retina, it has been shown that activation of CD200R in microglia does not show direct effect on migration, but CD200-CD200R signaling restores LPS/IFNγ-induced loss of migration [86]. CD200 knockout leads to an expansion of the myeloid population in several tissues and increased expression of the activation markers in microglia, including the signaling adaptor protein DNAX-activating protein of 12 kDa (DAP12), CD11b, CD45, CD68, and inducible NO synthase (iNOS) [87]. Blocking of CD200R increases neurodegeneration in mouse model of Parkinson's disease [88]. These observations suggest that CD200-CD200R signaling leads to anti-inflammatory state and protection against neurotoxic stimuli. CD200 and CD200R expression levels (neurons and microglia, resp.) are decreased in AD hippocampus and inferior temporal gyrus, indicating that inhibition of CD200-CD200R axis contributes to AD pathology [89].
TREM2 —
TREM2, a recently identified innate immune receptor, and its adaptor protein DAP12 are expressed on microglia and cortical neurons, but not on hippocampal neurons, astrocytes, and oligodendrocytes. Their expression correlates with clearance of apoptotic neurons by microglia without inflammation [90–92]. However, endogenous ligand or specific agonist of TREM2 had not been identified until recently. Heat shock protein 60 (Hsp60) is a mitochondrial chaperone, which interacts with TREM2 directly. Hsp60-induced phagocytosis is only found in microglia which have robust expression of TREM2 [93]. In AD model mouse, TREM2 expression was highest in the outer zone in Aβ plaques, and the expression level correlated with the size of Aβ plaque [94]. Forced expression of TREM2 positively regulated microglial phagocytosis, the ability of microglia to stimulate CD4+ T-cell proliferation, TNF-α and CCL2 production, but not IFNγ production [94].
5. TLRs
TLRs are class of pattern-recognition receptors in the innate immune system to induce inflammatory responses. 13 TLRs have been identified in human and mouse to date, except for TLR10 which is expressed only in human [95, 96]. TLRs may also contribute to the microglial inflammatory response to promote AD pathogenesis [15].
CD11b, a marker of macrophages and microglia, has been shown to interact with TLR signaling. CD11b knockout mouse exhibited enhanced TLR-mediated responses and subsequent more susceptibility to endotoxin shock [97].
Among the TLRs, LPS receptor TLR4 potently activates microglia in various aspects, such as Aβ phagocytosis and proinflammatory molecules production [98, 99]. Activation of TLR1, TLR2, TLR3, and TLR9 by each selective agonist also increased phagocytosis and several cytokines and chemokine production [98, 100]. CD14 is a coreceptor of TLR4. The response of microglia to fibrillar Aβ is mediated via CD14, which act together with TLR4 and TLR2 to bind fibrillar Aβ and induced microglial activation through p38 MAPK [101]. Deficiency of CD200 induces the expression of TLR2 and TLR4 in glial cells and proliferation of CD11b+/MHCII+/CD40+ activated microglia [102]. These mice show memory impairment, suggesting that dual activation of TLR2 and TLR4 may induce an inflammatory phenotype of microglia which negatively regulate synaptic plasticity in the AD model. Aβ triggers the inflammatory status in microglia via heterodimer of TLR4 and TLR6, which is regulated by CD36 [103]. Therefore, CD14-TLR2-TLR4 and CD36-TLR4-TLR6 signaling are crucial to Aβ-induced inflammatory response, and also in microglial phagocytosis.
Bacterial DNA containing motifs of unmethylated CpG dinucleotides (CpG-DNA) is a ligand of TLR9, which is initially identified to activate microglia and strongly induces TNF-α and IL-12 production [104]. However, we have shown that CpG activated microglia to produce neuroprotective molecule, such as hemeoxygenase-1 (HO-1) and matrix metalloproteinase 9 (MMP-9) without producing neurotoxic molecules, such as TNF-α, glutamate and nitric oxide (NO), and enhanced Aβ clearance to protect memory disturbance in vivo [105]. There are the discrepancies about CpG effect between aforementioned two reports despite using the same origin cells (mouse primary microglia). It may be due to concentrations of this TLR9 ligand used: higher concentration (10 μM) in former report [104], whereas lower concentrations (1 to 100 nM) in latter report [105]. Moreover, the latter report revealed the difference on neuroprotective effect of CpG among synthetic oligodeoxynucleotides (ODNs) classes (A to C). Class A CpG did not activate microglia, but classes B and C CpGs increased microglial neuroprotective effect through induction of clearance of Aβ and production of neuroprotectant. These suggested that the CpG sequence-dependent microglial activation and responses are present.
CpG increased chemokine CCL9 and its receptor CCR1 expression in macrophages and microglia via TLR9/MyD88 signaling involving ERK, p38 MAPK, and PI3K pathways. Thus it can enhance microglial migration as well as phagocytosis [106].
6. PSR
Phagocytotic cells recognize apoptotic cells by several mechanisms, including recognition of PS expressed on the cells. PS is receiving much attention because it is responsible for phagocytosis without inducing inflammation [10]. The receptors of PS (PSRs) had not been clarified for a long time but have uncovered in recent years. These include MFG-E8 (the lactadherin homolog in humans) and T-cell immunoglobulin mucin domain 4 (Tim4) [107, 108]. These act as a bridge between PS-expressing apoptotic cells and PSR expressing phagocytes and trigger engulfment of cellular debris.
MFG-E8 is expressed on various macrophage subsets in the periphery and on microglia in the CNS. Recently, we have shown that CX3CL1 induces MFG-E8 expression in primary mouse microglia to lead to the microglial clearance of degenerated neuronal debris [46]. Others also reported the induction of MFG-E8 by CX3CL1 in macrophages and rat microglia [109, 110]. MFG-E8 bridges PS and integrins α v β 3 or α v β 5 on the surface of phagocytes [107, 111]. High-mobility group box 1 protein (HMGB1) is an intracellular protein that activates transcriptional factors, including p53 and NF-κB. HMGB1 reportedly suppresses the interaction between MFG-E8 and α v β 3 integrin in macrophage and inhibits the phagocytosis of apoptotic cells through ERK-mediated signaling pathway [112]. MFG-E8 may also be involved in Aβ phagocytosis, since its expression is reduced in AD [113]. We previously showed the neutralization of MFG-E8 progressed neuronal degeneration [46]. MFG-E8 reportedly induces anti-inflammation status in the periphery. Therefore, MFG-E8 may possibly lead to targeted clearance of unwanted molecules, such as Aβ, without inflammation.
The other well-studied PSR, Tim4, is expressed in MAC-1+ cells in various mouse tissues, including spleen, lymph nodes, and fetal liver [108]. Among the other Tim family members, Tim1, but neither Tim2 nor Tim3, also specifically binds to PS. Tim1-PS subsequently connects with exosomes to recognize and engulf apoptotic cells [114–116].
It has been shown recently that RAGE also recognizes PS and induces apoptotic cell clearance [80]. However as mentioned previously, RAGE-guided intracellular signaling pathway induces prolonged inflammatory status.
7. γ-Secretase and Phagocytosis in AD Pathology
γ-secretase is a protein complex of four essential membrane proteins: aph-1, pen-2, nicastrin, and presenilin. A recent study suggests that presenilin increases microglial phagocytosis of Aβ, and this γ-secretase has dual role for AD pathogenesis: one is cleavage of amyloid precursor protein (APP) to produce pathologic Aβ, and the other is reduction of microglial phagocytosis of Aβ by γ-secretase inhibitor [117]. Vertebrates have two presenilin genes, PSEN1 (located on chromosome 14 in humans; encoded presenilin 1) and PSEN2 (located on chromosome 1; encoded presenilin 2). There is a report that presenilin 2 is identified as the predominant γ-secretase in mouse microglia (but not presenilin 1), which repressed microglial activation via its function as a γ-secretase, and its expression is increased by inflammatory stimuli (IFN-γ) [118].
In order to explore the detailed mechanism how γ-secretase regulates microglial activity, further studies are needed since γ-secretase is a therapeutic target for AD. In traumatic injury of the brain, presenilin and nicastrin expressions are elevated in activated microglia and astrocytes [119]. γ-secretase mainly cleaves APP to lead to accumulation of Aβ 1-42, which then results in aggregation of Aβ protein to worsen AD pathology. However Aβ 1-42 is also a target of microglial phagocytosis.
8. Concluding Remarks
Microglia express a wide array of receptors characteristic to immune cell, such as CD molecules, integrins, chemokine receptors, and PSR (Figure 1). These receptors are involved in multiple functions of microglia. Chemokine receptors not only induce migration of microglia but also contribute directly to AD pathogenesis through regulation of phagocytosis and neuroprotective activity. PS acts as eat-me signal, and PSR-mediated phagocytosis so far is regarded as inducing anti-inflammatory responses. However, according to some recent reports, RAGE interacts with PSR and facilitates phagocytosis with robust inflammation status [80, 120]. Therefore, if the other phagocytosis-related receptors including TLRs interact with PSR, microglia would be activated to outbreak excessive phagocytosis with robust inflammation.
Microglia from old APP/PS1 mouse, but not from younger ones, show the reduction of SRA, CD36, RAGE, and the Aβ-degrading enzymes including insulysin, neprilysin, and MMP9 [121]. Dysfunction of microglia after progression of disease development may lead to neurodegeneration. Thus, it is important to consider the microglial status depending on the disease stage, to treat AD effectively. As shown in Figure 2, FKN and IL-34 may be an intrinsic neuroprotectant for damaged but still surviving neurons through activation of microglia. Therefore, it is important to elucidate neurons-microglia crosstalk in neurodegenerative condition.
References
- 1.Farfara D, Lifshitz V, Frenkel D. Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer’s disease. Journal of Cellular and Molecular Medicine. 2008;12(3):762–780. doi: 10.1111/j.1582-4934.2008.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honda S, Sasaki Y, Ohsawa K, et al. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. Journal of Neuroscience. 2001;21(6):1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends in Pharmacological Sciences. 2008;29(12):626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Medicine. 2000;6(8):916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 5.Hoarau JJ, Krejbich-Trotot P, Jaffar-Bandjee MC, et al. Activation and control of CNS innate immune responses in health and diseases: a balancing act finely tuned by neuroimmune regulators (NIReg) CNS & neurological disorders drug targets. 2011;10(1):25–43. doi: 10.2174/187152711794488601. [DOI] [PubMed] [Google Scholar]
- 6.Fuller AD, van Eldik LJ. MFG-E8 regulates microglial phagocytosis of apoptotic neurons. Journal of Neuroimmune Pharmacology. 2008;3(4):246–256. doi: 10.1007/s11481-008-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McArthur S, Cristante E, Paterno M, et al. Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. Journal of Immunology. 2010;185(10):6317–6328. doi: 10.4049/jimmunol.1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446(7139):1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biber K, Neumann H, Inoue K, Boddeke HWGM. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends in Neurosciences. 2007;30(11):596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Green DR, Beere HM. Apoptosis: gone but not forgotten. Nature. 2000;405(6782):28–29. doi: 10.1038/35011175. [DOI] [PubMed] [Google Scholar]
- 11.Sambrano GR, Steinberg D. Recognition of oxidatively damaged and apoptotic cells by an oxidized low density lipoprotein receptor on mouse peritoneal macrophages: role of membrane phosphatidylserine. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(5):1396–1400. doi: 10.1073/pnas.92.5.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvath RJ, DeLeo JA. Morphine enhances microglial migration through modulation of P2X4 receptor signaling. Journal of Neuroscience. 2009;29(4):998–1005. doi: 10.1523/JNEUROSCI.4595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Walter S, Stagi M, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128:1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- 14.Stolzing A, Grune T. Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. The FASEB journal. 2004;18(6):743–745. doi: 10.1096/fj.03-0374fje. [DOI] [PubMed] [Google Scholar]
- 15.Landreth GE, Reed-Geaghan EG. Toll-like receptors in Alzheimer’s disease. Current Topics in Microbiology and Immunology. 2009;336(1):137–153. doi: 10.1007/978-3-642-00549-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nature Neuroscience. 2006;9(7):917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 17.Lee S, Varvel NH, Konerth ME, et al. CX3CR1 deficiency alters microglial activation and reduces β-amyloid deposition in two Alzheimer’s disease mouse models. American Journal of Pathology. 2010;177(5):2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizuno T, Doi Y, Mizoguchi H, et al. Interleukin-34 selectively enhances the neuroprotective effects of microglia to attenuate oligomeric amyloid-β neurotoxicity. The American Journal of Pathology. 2011;179(4):2016–2027. doi: 10.1016/j.ajpath.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 20.Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64(1):110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature Neuroscience. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 22.Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Research Reviews. 2005;48(1):16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 23.El Khoury JB, Moore KJ, Means TK, et al. CD36 mediates the innate host response to β-amyloid. Journal of Experimental Medicine. 2003;197(12):1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naert G, Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Journal of Neuroscience. 2011;31(16):6208–6220. doi: 10.1523/JNEUROSCI.0299-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4(4):590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Jong EK, Vinet J, Stanulovic VS, et al. Expression, transport, and axonal sorting of neuronal CCL21 in large dense-core vesicles. The FASEB Journal. 2008;22(12):4136–4145. doi: 10.1096/fj.07-101907. [DOI] [PubMed] [Google Scholar]
- 27.de Jong EK, Dijkstra IM, Hensens M, et al. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. Journal of Neuroscience. 2005;25(33):7548–7557. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rappert A, Biber K, Nolte C, et al. Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. Journal of Immunology. 2002;168(7):3221–3226. doi: 10.4049/jimmunol.168.7.3221. [DOI] [PubMed] [Google Scholar]
- 29.Rappert A, Bechmann I, Pivneva T, et al. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. Journal of Neuroscience. 2004;24(39):8500–8509. doi: 10.1523/JNEUROSCI.2451-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vinet J, de Jong EK, Boddeke HWGM, et al. Expression of CXCL10 in cultured cortical neurons. Journal of Neurochemistry. 2010;112(3):703–714. doi: 10.1111/j.1471-4159.2009.06495.x. [DOI] [PubMed] [Google Scholar]
- 31.Dijkstra IM, Hulshof S, van der Valk P, Boddeke HWGM, Biber K. Cutting edge: activity of human adult microglia in response to CC chemokine ligand 21. Journal of Immunology. 2004;172(5):2744–2747. doi: 10.4049/jimmunol.172.5.2744. [DOI] [PubMed] [Google Scholar]
- 32.Biber K, Tsuda M, Tozaki-Saitoh H, et al. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. The EMBO Journal. 2011;30(9):1864–1873. doi: 10.1038/emboj.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sorce S, Myburgh R, Krause KH. The chemokine receptor CCR5 in the central nervous system. Progress in Neurobiology. 2011;93(2):297–311. doi: 10.1016/j.pneurobio.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Sorce S, Bonnefont J, Julien S, et al. Increased brain damage after ischaemic stroke in mice lacking the chemokine receptor CCR5. British Journal of Pharmacology. 2010;160(2):311–321. doi: 10.1111/j.1476-5381.2010.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crain JM, Nikodemova M, Watters JJ. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. Journal of Neuroinflammation. 2009;6, article no. 1742:p. 24. doi: 10.1186/1742-2094-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanz JM, Chiozzi P, Ferrari D, et al. Activation of microglia by amyloid β requires P2X7 receptor expression. Journal of Immunology. 2009;182(7):4378–4385. doi: 10.4049/jimmunol.0803612. [DOI] [PubMed] [Google Scholar]
- 37.Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nature Neuroscience. 2006;9(12):1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- 38.Inoue K, Koizumi S, Kataoka A, Tozaki-Saitoh H, Tsuda M. Chapter 12 P2Y6-evoked microglial phagocytosis. International Review of Neurobiology. 2009;85:159–163. doi: 10.1016/S0074-7742(09)85012-5. [DOI] [PubMed] [Google Scholar]
- 39.Pan Y, Lloyd C, Zhou H, et al. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature. 1997;387(6633):611–617. doi: 10.1038/42491. [DOI] [PubMed] [Google Scholar]
- 40.Nishiyori A, Minami M, Ohtani Y, et al. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? The FEBS Letters. 1998;429(2):167–172. doi: 10.1016/s0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- 41.Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Research. 2003;979(1-2):65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- 43.Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. Journal of Neuroscience Research. 2002;69(3):418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- 44.Boehme SA, Lio FM, Maciejewski-Lenoir D, Bacon KB, Conlon PJ. The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. Journal of Immunology. 2000;165(1):397–403. doi: 10.4049/jimmunol.165.1.397. [DOI] [PubMed] [Google Scholar]
- 45.Lauro C, Angelantonio SD, Cipriani R, et al. Activity of adenosine receptors type 1 is required for CX3CL1-mediated neuroprotection and neuromodulation in hippocampal neurons. Journal of Immunology. 2008;180(11):7590–7596. doi: 10.4049/jimmunol.180.11.7590. [DOI] [PubMed] [Google Scholar]
- 46.Noda M, Doi Y, Liang J, et al. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. Journal of Biological Chemistry. 2011;286(3):2308–2319. doi: 10.1074/jbc.M110.169839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zujovic V, Benavides J, Vige X, et al. Fractalkine modulates TNF-α secretion and neurotoxicity induced by microglial activation. Glia. 2000;29(4):305–315. [PubMed] [Google Scholar]
- 48.Garton KJ, Gough PJ, Blobel CP, et al. Tumor necrosis factor-α-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) Journal of Biological Chemistry. 2001;276(41):37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- 49.Hundhausen C, Misztela D, Berkhout TA, et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102(4):1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 50.Tsou CL, Haskell CA, Charo IF. Tumor necrosis factor-α-converting enzyme mediates the inducible cleavage of fractalkine. Journal of Biological Chemistry. 2001;276(48):44622–44626. doi: 10.1074/jbc.M107327200. [DOI] [PubMed] [Google Scholar]
- 51.Clark AK, Yip PK, Grist J, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(25):10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. Journal of Neuroscience. 2000;20(15):p. RC87. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fahrenholz F. α-secretase as a therapeutic target. Current Alzheimer Research. 2007;4(4):412–417. doi: 10.2174/156720507781788837. [DOI] [PubMed] [Google Scholar]
- 54.Postina R, Schroeder A, Dewachter I, et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. Journal of Clinical Investigation. 2004;113(10):1456–1464. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milward EA, Papadopoulos R, Fuller SJ, et al. The amyloid protein precursor of Alzheimer’s disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9(1):129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- 56.Jin LW, Ninomiya H, Roch JM, et al. Peptides containing the RERMS sequence of amyloid β/A4 protein precursor bind cell surface and promote neurite extension. Journal of Neuroscience. 1994;14(9):5461–5470. doi: 10.1523/JNEUROSCI.14-09-05461.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walter J, Kaether C, Steiner H, Haass C. The cell biology of Alzheimer’s disease: uncovering the secrets of secretases. Current Opinion in Neurobiology. 2001;11(5):585–590. doi: 10.1016/s0959-4388(00)00253-1. [DOI] [PubMed] [Google Scholar]
- 58.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120(4):545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 59.Green SR, Han KH, Chen Y, et al. The CC chemokine MCP-1 stimulates surface expression of CX3CR1 and enhances the adhesion of monocytes to fractalkine/CX3CL1 via p38 MAPK. Journal of Immunology. 2006;176(12):7412–7420. doi: 10.4049/jimmunol.176.12.7412. [DOI] [PubMed] [Google Scholar]
- 60.Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. Journal of Neuroinflammation. 2011;8, article 9 doi: 10.1186/1742-2094-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fuhrmann M, Bittner T, Jung CKE, et al. Microglial CX3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nature Neuroscience. 2010;13(4):411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neumann H, Kotter MR, Franklin RJM. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reichert F, Rotshenker S. Complement-receptor-3 and scavenger-receptor-AI/II mediated myelin phagocytosis in microglia and macrophages. Neurobiology of Disease. 2003;12(1):65–72. doi: 10.1016/s0969-9961(02)00008-6. [DOI] [PubMed] [Google Scholar]
- 64.Rotshenker S. Microglia and macrophage activation and the regulation of complement-receptor-3 (CR3/MAC-1)-mediated myelin phagocytosis in injury and disease. Journal of Molecular Neuroscience. 2003;21(1):65–72. doi: 10.1385/JMN:21:1:65. [DOI] [PubMed] [Google Scholar]
- 65.Kaur C, Too HF, Ling EA. Phagocytosis of Escherichia coli by amoeboid microglial cells in the developing brain. Acta Neuropathologica. 2004;107(3):204–208. doi: 10.1007/s00401-003-0798-7. [DOI] [PubMed] [Google Scholar]
- 66.Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 2002;40(2):195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]
- 67.Schlegel RA, Krahling S, Callahan MK, Williamson P. CD14 is a component of multiple recognition systems used by macrophages to phagocytose apoptotic lymphocytes. Cell Death and Differentiation. 1999;6(6):583–592. doi: 10.1038/sj.cdd.4400529. [DOI] [PubMed] [Google Scholar]
- 68.Devitt A, Pierce S, Oldreive C, Shingler WH, Gregory CD. CD14-dependent clearance of apoptotic cells by human macrophages: the role of phosphatidylserine. Cell Death and Differentiation. 2003;10(3):371–382. doi: 10.1038/sj.cdd.4401168. [DOI] [PubMed] [Google Scholar]
- 69.Reed-Geaghan EG, Reed QW, Cramer PE, Landreth GE. Deletion of CD14 attenuates Alzheimer’s disease pathology by influencing the brain’s inflammatory milieu. Journal of Neuroscience. 2010;30(46):15369–15373. doi: 10.1523/JNEUROSCI.2637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. Journal of Neuroscience. 2004;24(44):9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. Journal of Neuroscience. 2003;23(7):2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Persaud-Sawin DA, Banach L, Harry GJ. Raft aggregation with specific receptor recruitment is required for microglial phagocytosis of Aβ42. Glia. 2009;57(3):320–335. doi: 10.1002/glia.20759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moore KJ, El Khoury J, Medeiros LA, et al. A CD36-initiated signaling cascade mediates inflammatory effects of β-amyloid. Journal of Biological Chemistry. 2002;277(49):47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- 74.Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature. 1997;386(6621):181–186. doi: 10.1038/386181a0. [DOI] [PubMed] [Google Scholar]
- 75.Matozaki T, Murata Y, Okazawa H, Ohnishi H. Functions and molecular mechanisms of the CD47-SIRPα signalling pathway. Trends in Cell Biology. 2009;19(2):72–80. doi: 10.1016/j.tcb.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 76.Gitik M, Liraz-Zaltsman S, Oldenborg PA, Reichert F, Rotshenker S. Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPα (signal regulatory protein-α) on phagocytes. Journal of Neuroinflammation. 2011;8, article 24 doi: 10.1186/1742-2094-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288(5473):2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 78.Yan SD, Chen X, Fu J, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382(6593):685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 79.Yan SD, Zhu H, Fu J, et al. Amyloid-β peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(10):5296–5301. doi: 10.1073/pnas.94.10.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Friggeri A, Banerjee S, Biswas S, et al. Participation of the receptor for advanced glycation end products in efferocytosis. Journal of Immunology. 2011;186(11):6191–6198. doi: 10.4049/jimmunol.1004134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dukic-Stefanovic S, Gasic-Milenkovic J, Deuther-Conrad W, Münch G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs) Journal of Neurochemistry. 2003;87(1):44–55. doi: 10.1046/j.1471-4159.2003.01988.x. [DOI] [PubMed] [Google Scholar]
- 82.Kang R, Tang D, Schapiro NE, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death and Differentiation. 2010;17(4):666–676. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dearie R, Sagare A, Zlokovic BV. The role of the cell surface LRP and soluble LRP in blood-brain barrier Aβ clearance in Alzheimer’s disease. Current Pharmaceutical Design. 2008;14(16):1601–1605. doi: 10.2174/138161208784705487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koning N, Swaab DF, Hoek RM, Huitinga I. Distribution of the immune inhibitory molecules CD200 and CD200R in the normal central nervous system and multiple sclerosis lesions suggests neuron-glia and glia-glia interactions. Journal of Neuropathology and Experimental Neurology. 2009;68(2):159–167. doi: 10.1097/NEN.0b013e3181964113. [DOI] [PubMed] [Google Scholar]
- 85.Matsumoto H, Kumon Y, Watanabe H, et al. Expression of CD200 by macrophage-like cells in ischemic core of rat brain after transient middle cerebral artery occlusion. Neuroscience Letters. 2007;418(1):44–48. doi: 10.1016/j.neulet.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 86.Carter DA, Dick AD. CD200 maintains microglial potential to migrate in adult human retinal explant model. Current Eye Research. 2004;28(6):427–436. doi: 10.1080/02713680490503778. [DOI] [PubMed] [Google Scholar]
- 87.Langmann T. Microglia activation in retinal degeneration. Journal of Leukocyte Biology. 2007;81(6):1345–1351. doi: 10.1189/jlb.0207114. [DOI] [PubMed] [Google Scholar]
- 88.Zhang S, Wang XJ, Tian LP, et al. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. Journal of Neuroinflammation. 2011;8(1):p. 154. doi: 10.1186/1742-2094-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Experimental Neurology. 2009;215(1):5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takahashi K, Rochford CDP, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. Journal of Experimental Medicine. 2005;201(4):647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sessa G, Podini P, Mariani M, et al. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. European Journal of Neuroscience. 2004;20(10):2617–2628. doi: 10.1111/j.1460-9568.2004.03729.x. [DOI] [PubMed] [Google Scholar]
- 92.Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Medicine. 2007;4(4):p. e124. doi: 10.1371/journal.pmed.0040124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stefano L, Racchetti G, Bianco F, et al. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. Journal of Neurochemistry. 2009;110(1):284–294. doi: 10.1111/j.1471-4159.2009.06130.x. [DOI] [PubMed] [Google Scholar]
- 94.Melchior B, Garcia AE, Hsiung BK, et al. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: Implications for vaccine-based therapies for Alzheimer’s disease. ASN Neuro. 2010;2(3) doi: 10.1042/AN20100010. Article ID e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mishra BB, Gundra UM, Teale JM. Expression and distribution of Toll-like receptors 11–13 in the brain during murine neurocysticercosis. Journal of Neuroinflammation. 2008;5, article 53 doi: 10.1186/1742-2094-5-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shi Z, Cai Z, Sanchez A, et al. A novel toll-like receptor that recognizes vesicular stomatitis virus. Journal of Biological Chemistry. 2011;286(6):4517–4524. doi: 10.1074/jbc.M110.159590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nature Immunology. 2010;11(8):734–742. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 98.Ribes S, Ebert S, Regen T, et al. Toll-like receptor stimulation enhances phagocytosis and intracellular killing of nonencapsulated and encapsulated Streptococcus pneumoniae by murine microglia. Infection and Immunity. 2010;78(2):865–871. doi: 10.1128/IAI.01110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song M, Jin J, Lim JE, et al. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Journal of Neuroinflammation. 2011;8:p. 92. doi: 10.1186/1742-2094-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ribes S, Adam N, Ebert S, et al. The viral TLR3 agonist poly(I:C) stimulates phagocytosis and intracellular killing of Escherichia coli by microglial cells. Neuroscience Letters. 2010;482(1):17–20. doi: 10.1016/j.neulet.2010.06.078. [DOI] [PubMed] [Google Scholar]
- 101.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. Journal of Neuroscience. 2009;29(38):11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Costello DA, Lyons A, Denieffe S, et al. Long term potentiation is impaired in membrane glycoprotein CD200-deficient mice: a role for Toll-like receptor activation. The Journal of Biological Chemistry. 2011;286(40):34722–34732. doi: 10.1074/jbc.M111.280826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nature Immunology. 2010;11(2):155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dalpke AH, Schafer MK, Frey M, et al. Immunostimulatory CpG-DNA activates murine microglia. The Journal of Immunology. 2002;168(10):4854–4863. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- 105.Doi Y, Mizuno T, Maki Y, et al. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid β neurotoxicity in in vitro and in vivo models of Alzheimer’s disease. American Journal of Pathology. 2009;175(5):2121–2132. doi: 10.2353/ajpath.2009.090418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ravindran C, Cheng YC, Liang SM. CpG-ODNs induces up-regulated expression of chemokine CCL9 in mouse macrophages and microglia. Cellular Immunology. 2010;260(2):113–118. doi: 10.1016/j.cellimm.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 107.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417(6885):182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 108.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 109.Leonardi-Essmann F, Emig M, Kitamura Y, Spanagel R, Gebicke-Haerter PJ. Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. Journal of Neuroimmunology. 2005;160(1-2):92–101. doi: 10.1016/j.jneuroim.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 110.Miksa M, Amin D, Wu R, Dong W, Ravikumar TS, Wang P. Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Molecular Medicine. 2007;13(11-12):553–560. doi: 10.2119/2007-00019.Miksa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Akakura S, Singh S, Spataro M, et al. The opsonin MFG-E8 is a ligand for the αvβ5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Experimental Cell Research. 2004;292(2):403–416. doi: 10.1016/j.yexcr.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 112.Friggeri A, Yang Y, Banerjee S, Park YJ, Liu G, Abraham E. HMGB1 inhibits macrophage activity in efferocytosis through binding to the αvβ3-integrin. American Journal of Physiology. 2010;299(6):C1267–C1276. doi: 10.1152/ajpcell.00152.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boddaert J, Kinugawa K, Lambert JC, et al. Evidence of a role for lactadherin in Alzheimer’s disease. American Journal of Pathology. 2007;170(3):921–929. doi: 10.2353/ajpath.2007.060664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Feng D, Zhao WL, Ye YY, et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11(5):675–687. doi: 10.1111/j.1600-0854.2010.01041.x. [DOI] [PubMed] [Google Scholar]
- 115.Martinez J, Almendinger J, Oberst A, et al. Microtubule-associated protein 1 light chain 3 α (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proceedings of the National Academy of Sciences of the United States. 2011;108(42):17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yamanishi Y, Kitaura J, Izawa K, et al. TIM1 is an endogenous ligand for LMIR5/CD300b: LMIR5 deficiency ameliorates mouse kidney ischemia/reperfusion injury. Journal of Experimental Medicine. 2010;207(7):1501–1511. doi: 10.1084/jem.20090581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Farfara D, Trudler D, Segev-Amzaleg N, Galron R, Stein R, Frenkel D. γ-secretase component presenilin is important for microglia β-amyloid clearance. Annals of Neurology. 2011;69(1):170–180. doi: 10.1002/ana.22191. [DOI] [PubMed] [Google Scholar]
- 118.Jayadev S, Case A, Eastman AJ, et al. Presenilin 2 is the predominant γ-secretase in microglia and modulates cytokine release. PLoS One. 2010;5(12) doi: 10.1371/journal.pone.0015743. Article ID e15743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nadler Y, Alexandrovich A, Grigoriadis N, et al. Increased expression of the γ-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia. 2008;56(5):552–567. doi: 10.1002/glia.20638. [DOI] [PubMed] [Google Scholar]
- 120.He M, Kubo H, Morimoto K, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. The EMBO Reports. 2011;12(4):358–364. doi: 10.1038/embor.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging alzheimer’s disease mice. Journal of Neuroscience. 2008;28(33):8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]