

Abstract
Systemic lupus erythematosus (SLE) is a complex autoimmune disorder, known to have a strong genetic component. Concordance between monozygotic twins is approximately 30–40%, which is 8–20 times higher than that of dizygotic twins. In the last decade, genome-wide approaches to understanding SLE have yielded many candidate genes, which are important to understanding the pathophysiology of the disease and potential targets for pharmaceutical intervention. In this paper, we focus on the role of cytokines and examine how genome-wide association studies, copy number variation studies, and next-generation sequencing are being employed to understand the etiology of SLE. Prominent genes identified by these approaches include BLK, FCγR3B, and TREX1. Our goal is to present a brief overview of genomic approaches to SLE and to introduce some of the key discussion points pertinent to the field.
1. Introduction
Systemic lupus erythematosus (SLE) is a highly heterogeneous autoimmune disorder characterized by the prevalence of autoantibodies directed against double-stranded DNA. Small nuclear RNA-binding proteins, including anti-Ro, anti-La, and anti-RNP are also found in many patients. Worldwide, the prevalence is approximately 52 per 100,000 and may be highest among individuals of Afro-Caribbean descent at 159 per 100,000 (derived from a UK sample) [1]. In the United States, SLE is 2.6 times more common in individuals of African as opposed to European descent (19.5 versus 7.4 per 100,000), reflecting a disproportionate ethnic disease burden. For adult-onset SLE, the female : male ratio is 9 : 1 [2].
SLE is underscored by a range of environmental and genetic risk factors that can affect any part of the organ system, including cardiovascular, hematological, integumentary, musculoskeletal, nervous, renal, and respiratory. In this paper, we focus on three major approaches that are being used to uncover genetic correlates of the disease, genome-wide association studies (GWAS), copy number variation (CNV) studies, and next-generation sequencing (NGS). Our goal is to provide a brief overview of the genomic landscape and to introduce some of the key discussion points in terms of how these approaches inform our understanding of SLE, particularly in relation to cytokines. To date, more than 40 relevant loci have been identified and replicated; many of which directly or indirectly involve cytokines regulation.
Cytokines are important components of immune response and regulation and play an active role in activating, differentiating, and maturing immune cells [6]. An imbalance between pro- and anti-inflammatory cytokines is a well known characteristic of SLE [7]. They are heavily integrated in T-Cell and B-Cell signaling systems and abnormal cytokine levels, particularly interleukins, interferons, and tumor necrosis factors (TNFs), are important hallmarks of SLE.
2. Genome-Wide Association Studies
Genome-wide association studies began to proliferate approximately 6 years ago, coinciding with rapid developments in genomic hardware and software and falling costs of relevant technologies. GWAS use microarrays to tag up to several million single nucleotide polymorphisms (SNPs) at once, which gives broad coverage of exonic and intronic regions (though coverage is weighted toward the former). When a difference in SNP-frequency is observed between cases and controls, we can infer a difference in the underlying genomic locus, which may affect gene expression or regulation. Because of the large number of comparisons being made, most GWAS require very large numbers of patients and controls and sample sizes in excess of several thousand are the norm.
Table 1 lists the ~40 loci that have been associated with SLE by GWAS. A number of these genes, particularly in the human leukocyte antigen (HLA) complex, were identified before the genome era. Since the publication of the first GWA study into SLE in 2007, this list has expanded rapidly and at least nine GWAS have been published [8–16]. Prominent replicated genes are discussed below. A major benefit of the GWA approach is its applicability to complex disease.
Table 1.
Loci associated with SLE from GWAS. Table adapted from http://www.genome.gov/gwastudies/. A number of these genes were associated with SLE prior to GWAS and have now been replicated, while others await replication. Intergenic regions are listed in brackets. It is important to note that many genes associated with SLE in individuals of European ancestry are not associated with the disease in Asian populations, with the converse also holding true. Similar differences in susceptibility are certain to be found between other populations for whom the requisite GWAS have not yet been conducted. As Bustamante et al. [3] and others have pointed out, individuals of European ancestry are heavily overrepresented in GWAS, and many more comparison studies are needed to address this sampling bias.
| Region | Reported gene(s) | Reported OR Population | |
|---|---|---|---|
| 1p31.1 | NEGR1 | EU7 | |
| 1p32.3 | SLC1A7 | 1.32 | EU1 |
| 1q25.1 | TNFSF4 | 1.22–1.46 | AS4, EU6 |
| 1q25.3 | LAMC2 | 1.33 | EU1 |
| 1q31.1 | IL10 [RPS3AP9-FAM5C] | 1.19–1.3 | EU1,9 |
| 2p16.3 | [FSHR-RPL7P13] | NR | EU5 |
| 2p22.3 | RASGRP3 | 1.43 | AS4 |
| 2q31.3 | [CWC22-FTHL20] | 1.72 | EU1 |
| 2q32.3 | STAT4 | 1.41–1.77 | AS4, EU1,5,7 |
| 3p14.3 | PXK | 1.25 | EU6 |
| 3q26.31 | NAALADL2 | 1.37 | EU1 |
| 3q26.32 | [ASS1P7-KCNMB2] | 1.56 | EU1 |
| 4q24 | BANK1 | 1.38 | EU8 |
| 4q25 | COL25A1 | 1.33 | EU1 |
| 4q28.3 | [PCDH18-SLC7A11] | EU5 | |
| 5p13.1 | GHR | EU7 | |
| 5q33.1 | TNIP1 | 1.23–1.27 | AS4, EU9 |
| 5q33.3 | PTTG1 | 1.32 | EU1 |
| 6p21.31 | UHRF1BP1 | 1.17 | EU9 |
| 6p21.32 | HLA region HLA-DQA1 HLA-DQA2 HLA-DR3, HLA-DRB1 NOTCH4 C6orf10 TNXB |
1.38–2.36 | AS1, EU1,6,7 |
| 6q21 |
PRDM1
ATG5 |
1.20–1.25 | AS4, EU9 |
| 6q23.3 | TNFAIP3 | 1.72–2.28 | AS4, EU5 |
| 7p12.2 | IKZF1 | 1.39 | AS4 |
| 7p15.1 | JAZF1 | 1.19 | EU9 |
| 7q32.1 |
IRF5
TNPO3 |
1.43–1.92 | AS4, EU1,6,7 |
| 8p23.1 |
BLK
FAM167A-BLK C8orf13 |
1.29–1.45 | AS4, EU1,5,7 |
| 9q34.13 | NTNG2 | EU7 | |
| 10q11.23 |
LRRC18
WDFY4 c10orf64 |
1.24–1.30 | AS3,4, EU5 |
| 11p15.5 | KIAA1542 | 1.28–1.33 | EU1,6 |
| 11q11 | OR4A15 | 1.92 | EU1 |
| 11q24.3 | ETS1 | 1.29–1.37 | AS3,4 |
| 12q21.2 | RPL7AP59 | 1.28 | EU1 |
| 12q24.33 | SLC15A4 | 1.26 | AS4 |
| 13q14.11 | ELF1 | 1.26 | AS2 |
| 16p11.2 |
ITGAM
ITGAX |
1.33–1.80 | EU1,5,6,7 |
| 17p12 | [HS3ST3A1-CDRT15P] | EU5 | |
| 18q22.3 | SOCS6 | EU7 | |
| 20p13 | TMC2 | 1.37 | EU1 |
| 22q11.21 |
HIC2
UBE2L3 |
1.28–1.38 | AS4, EU1 |
1Chung et al. [9] (NOTE: reported associations are for anti ds-DNA autoantibody production); 2Yang et al. [10]; 3 Yang et al. [11]; 4Han et al. [12]; 5Graham et al. [13]; 6Harley et al. [14]; 7Hom et al. [15]; 8Kozyrev et al. [16]; 9Gateva et al. [24]. A study by Li et al. (2011) did not report any significant associations. NOTE: The study by Gateva et al. [24] (9) was not genome wide, having used SNPs from 2,466 regions that showed nominal evidence of association to SLE (P < 0.05).
3. HLA Regulation
The HLA system/histocompatibility complex (MHC) has long been associated with SLE. Because of its long-standing importance to immune function [17], the complete HLA/MHC was one of the first multigenic regions of the human genome to be sequenced [18]. It is densely packed with genes that regulate immune functions and all of the GWAS of SLE converge upon the region as the strongest predictor of genetic risk [12–16]. HLA genes are classified as types I, II, and III, and the region also includes a number of genes that encode tumor necrosis factor α (TNF-α) cytokines, which are discussed separately below. The class II and class III regions have been most closely associated with SLE.
3.1. HLA Class II
Genes belonging to HLA Class II are prominent candidates for SLE susceptibility and are known to play a major role in T-cell immunity. Graham et al. [19] differentiated three haplotypes in the region that are associated with SLE risk. More specifically, the DR beta 1 gene (HLA-DRB1) has been shown to associate with SLE across a range of ethnic populations, though the specific risk to each population varies by haplotype and serotype [20, 21]. The DR2 (DRB1*1501) serotype [22] as well as DR3 (DRB1*0301) holds the strongest evidence of association [23]. Recently, GWAS have confirmed these associations in European and Asian populations [11, 24]. HLA Class II genes including HLA-DRB1 are also a major component of T-Cell signaling networks, and they present relevant peptides for recognition by T-Cells. Comparing anti-dsDNA-negative SLE versus healthy controls, Chung et al. [9] found a significant association for a SNP, rs2301271, 9 kb downstream from HLA-DQA2 (P = 2 × 10−12). The authors point out that extensive linkage disequilibrium in this region may attribute the association to the HLA-DRB1 locus. The same study found a strong association between anti-dsDNA and HLA-DR3 at rs2187668. Further, the association with SLE was found to be far stronger in anti-dsDNA-positive SLE versus either anti-dsDNA-negative or the SLE phenotype. This important finding suggests that the HLADR3 allele is most closely aligned to the production of autoantibodies per se, rather than SLE in general. This was also true of observed associations with STAT4, IRF5, and ITGAM, which are discussed further below.
3.2. HLA Class III and the Complement System
The strongest single genetic risk factors of SLE are complement defects, particularly a complete deficiency of the C1q immune complex which, when present, is associated with a 93% risk of SLE [25]. Related complement defects in C4, C1r/s, and (to a lesser extent) C2, and C3 are firmly established as risk factors, with a prevalence of 75%, 57%, 32%, and 10%, respectively [26, 27]. These deficiencies are different from most candidate variants in that they are strong predictors if present. However, they are collectively rare, found in only 1-2% of cases [28]. Dunckley et al. [29] reported a significant increase in the C4A null allele (C4AQ0) in Chinese, European, and Japanese populations compared to controls. In a study of Japanese SLE patients, two SNPs in the C3 gene, rs7951 and rs2230201, have also been significantly associated with increased disease risk, with the former correlating with lower C3 serum levels [30].
The Class III gene, mutS homolog 5 (MSH5) gene, has been associated with SLE and was in fact the strongest association in the GWAS by Harley et al. [14]. MSH5 facilitates DNA rearrangements, which cause immunoglobulin class switching [31]. Super viralicidic activity 2-like (SKIV2L) is another Class III gene previously identified as an SLE candidate, and a study of 314 trios from the United Kingdom [32] implicated this locus independent of class II variants. SKIV2L is a putative RNA helicase and is expressed in T cells, B cells, and dendritic cells [32]. The integrin alpha M gene (ITGAM) has also been associated with SLE by a number of studies [9, 11, 14, 15]. Along with the β2 chain (ITGB2) protein, ITGAM forms a leukocyte integrin, complement receptor 3 (CC3), that facilitates adhesion of neutrophils and monocytes to the stimulated endothelium. ITGAM is also a receptor for the degraded CC3—iC3b [33]. Similarly, missense mutations in Fcγ receptors (see below) have been shown to disrupt influence immune complex processing [34].
4. Tumor Necrosis Factor α
The tumor necrosis factor α (TNF-α) family of cytokines have been repeatedly associated with SLE, though the specific role of TNF is still debated. As discussed further below, the interleukin, IL-10 is known to suppress TNF-α and the theoretical relationship between the two is illustrated in Figure 1. TNF-α plays an important role in autoimmune regulation and is a primary mediator of the response to infectious organisms. TNF-α is primarily activated by mononuclear phagocytes but is also secreted by Nκ cells, T cells, and mast cells [35]. Although TNF-α plays a prominent role in fighting infection, excessive levels of this cytokine have been associated with a number of autoimmune diseases including Crohn's disease, multiple sclerosis, and rheumatoid arthritis [36]. Treatment with anti-TNF medication has been proposed as a treatment for SLE [37] and is an active therapy for other autoimmune diseases, presumably by regulating the production of IFN-α by pDC [38].
Figure 1.
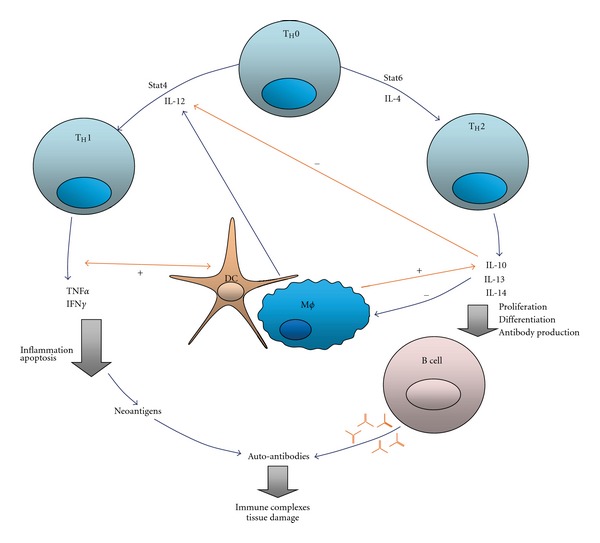
Feedback loops between IL10 and TNFα-simplified model (from López et al. [4]). Th1 cells produce TNFα, which activates dendritic cells, other antigen presenting cells (APCs) and induces IL-10 production. TNFα also promotes inflammation and apoptosis, generating neoantigens that can produce autoantibodies. Conversely, IL-10 antagonizes Th1 differentiation and inhibits APCs and T cells. IL-10 strongly stimulates B-cell proliferation, differentiation, and antibody production. As such, B-cell activation in the presence of neoantigens may cause autoantibodies to be secreted and lead to tissue damage. Stat is a signal transducer and transcription activator.
4.1. TNFSF4
TNF ligand superfamily 4 as well and its receptor, TNFRSF4, are primarily expressed on activated antigen-presenting cells [39] and activated CD4+ T cells [40], respectively. Graham et al. [41] used family-based and case-control approaches to identify a risk allele upstream of TNFS4 that predisposes to SLE and is correlated with increased TNFSF4 expression. The authors hypothesized that increased expression of TNFSF4 enhances interactions with either antigen-presenting cells or modulating T-cell activation. Recent GWAS [12, 24] have confirmed the association in European and Chinese samples. A recent study by Sanchez et al. [42] which targeted this and 15 other SLE-associated loci found that SLE renal disorder was significantly correlated with risk alleles in ITGAM and TNFSF4. Similar associations were found for ITGAM-discoid rash, STAT4-protection from oral ulcers, and IL21- hematological disorder and suggest ways in which genetic profiling may be used to predict SLE etiology.
4.2. TNFAIP3
Graham et al. [13] used whole-genome association to identify a significant relationship between tumor necrosis factor, alpha-induced protein 3 (TNFAIP3), and SLE, with the SNP rs5029939 the primary correlate. Subsequent GWAS [12, 24] replicated this association in Asian populations. A number of polymorphisms in TNFAIP3 have been associated with increased susceptibility to SLE [43, 44]. Recently, Adrianto et al. [45] identified a deletion (T) followed by a transversion (T to A) that resulted in reduced mRNA and A20 expression. By resequencing TNFAIP3 in affected European and Korean samples, the authors were able to compare nine risk variants at a signal locus. They found that only one (the TT to A polymorphism) showed major overlap with regulatory elements, which was therefore established as the functional polymorphism driving the association between TNFAIP3 and SLE. This is a prime example of how GWAS and NGS approaches can be used in combination to gain a deeper perspective on pathogenesis and is discussed further below.
4.3. TNIP1
TNFAIP3-interacting protein 1 (TNIP1) is a closely related gene, also identified by GWAS as significantly associated with SLE [12, 46, 47] in both European and Chinese populations. Both TNFAIP3 and TNIP1 regulate the NF-κB signaling pathway, which is critical to the immune response and is activated by TNF-α as well as other diverse stimuli [48]. The mechanisms by which TNIP1 and TNFAIP3 regulate NF-κB signaling are not fully understood, though Heyninck and Beyaert [49] found that sequential deubiquitination and ubiquitination of the TNF receptor-interacting protein (RIP) may mediate this process. Gregersen and Olsson [50] discuss a common pathway linking TNFAIP3, TRAF1, TRAF2, CD40, and c-Rel in autoimmunity.
5. Interferon Cytokines
Interferons (IFNs) derive their name from their ability to “interfere” with the host cells of viruses during the infection process and have been heavily associated with the pathogenesis of SLE. Type I interferons include IFN-α (produced by leukocytes) and IFN-β (produced by fibroblasts), both of which signal through the same receptor. IFN-α links the innate and adaptive immune systems and is therefore a critical component of the autoimmunity. A number of genes involved in IFN regulation have been associated with SLE, and individuals who have received treatment for other disorders using IFN-α supplementation have been known to develop SLE [51], which has also been found to resolve following discontinuation of IFN-α [52]. A recent study [53] of African American (n = 387), European American (n = 516), and Hispanic American (n = 186) patients found a strong association between SLE-associated autoantibodies and high IFN activity for all ancestral backgrounds, though non-European Americans have higher serum IFN activity.
5.1. IRF5
Interferon regulatory factor 5 (IRF5) is one of a family of nine IRFs, at least three of which have been associated with SLE. IRF5 is a particularly strong candidate because it can induce transcription of IFN-α mRNA [54]. Graham et al. [55] identified an association between the rs2004640 SNP and SLE, and this locus has subsequently been replicated by GWAS in individuals of European, African and Asian ancestry [46, 56–62]. Many functional variants have been identified, including SNPs at the first intron (rs2004640) and at the 3′ untranslated region (rs10954213 [46]), a five base-pair insertion-deletion proximal to the 5′ prime UTR [63] and a 30 bp [51] in the sixth exon. Haplotypes derived from combining these variants are associated with varying degrees of SLE risk, although linkage disequilibrium makes it difficult to disentangle the functional impact of specific variants. Niewold et al. [62] examined a risk haplotype containing a number of functional elements at this locus and found evidence of an increased risk for SLE, which is also associated with high-serum IFN-α. The study also found that the increased serum IFN-α was contingent upon the presence of anti-dsDNA and anti-RNA-binding protein autoantibodies, which may support a “gene + autoantibody = high IFN-α” model. Similar to their findings in relation the MHC locus, Chung et al. [9] showed that GWA threshold levels for anti-dsDNA-negative SLE had lower odds ratios and higher P values compared to associations with anti-dsDNA-positive SLE. While IRF5 is not an autoantibody susceptibility locus per se, the authors conclude that this locus may have a stronger effect in anti-dsDNA-positive SLE. Löfgren et al. [64] recently genotyped IRF5 using the CGGGG insertion/deletion and the rs2004640, rs2070197, and rs10954213 SNPs in 1488 SLE patients and 1466 healthy controls. This showed that the rs10954213 is the main SNP responsible for altered IRF5 expression in PBMC peripheral blood mononuclear cells (PBMC).
5.2. IRF7
The interferon regulatory factor 7 (IRF7) gene can also stimulate induction of IFN-α RNA [65]. Harley et al. [14] identified a significant association between SLE risk and locus between IRF7 and the PHRF1 (PHD and ring finger domains 1) gene in a European population. Similar to the approach taken with the IRF5 haplotype above, the Niewold group [66] examined the relationship between this locus, the presence of serum IFN-α, and the presence of autoantibodies. Again, IFN-α was only observed in the presence of specific autoantibodies. Furthermore, when the risk IRF5 and IRF7 genotypes were examined together, an additive effect was observed upon IFN-α that was not found in the autoantibody-negative group.
5.3. IRF8
The GWA replication study by Gateva et al. [24] highlighted a third member of the interferon regulatory factor family, IRF8, which is also an associated risk factor for SLE. A different variant at the same locus has also been associated with multiple sclerosis [67], adding further weight to this region as a potentially important region in relation to autoimmune susceptibility. Hikami et al. [68] found that a functional polymorphism in the 3′-untranslated region of SPI1, known to regulate expression of IRF2, IRF4, and IRF8 [69, 70] is associated with increased risk of SLE.
5.4. STAT4
Kariuki et al. [71] identified an association between a signal transducer and activator of transcription 4 (STAT4) risk allele (the T allele at rs7574865), which is associated with lower serum IFN-α activity and increased sensitivity to IFN-α signaling. Remmers et al. [72] reported an association between signal transducer and activator of transcription 4 (STAT4) and SLE (as well as rheumatoid arthritis). Again, the association with SLE has since been replicated in a number of GWAS in European and Asian populations [12–14, 24]. It represents one of six primary members of the STAT family, all of which play important roles in cytokine signaling. STAT4 is integral to IL-12 signaling in both T and Nκ Cells and increases the production of IFN-γ and differentiation of CD4 T Cells [73]. After it binds to the IL-12 receptor, phosphorylated STAT4 forms homodimers, which translocate to the nucleus and initiate transcription of genetic targets, IFN-γ is amongst them [74]. The SNP rs7574865 in the third intron of STAT4 that is the most strongly associated SNP and is also associated with an alternate SLE phenotype that is more severe has a younger age of onset (<30 years) and is characterized by characterized by double-stranded DNA autoantibodies [75]. This SNP also correlates with increased sensitivity to IFN-α [71]. The GWA study by Chung et al. [9] confirms an association between rs7574865 and anti-dsDNA-positive autoantibody production in SLE (P = 2 × 10−20). Indeed associations between STAT4 and this phenotype were found to be stronger than with SLE per se. As such, the authors suggest that STAT4 be considered an autoantibody-propensity locus as opposed to and SLE-susceptibility locus. This claim is also made for the ITGAM and HLA-DR3 loci.
Namjou et al. [75] identified three major STAT4 haplotypes that were highly significant in Europeans and moderately significant in Korean and Hispanic samples. Interestingly, Sigurdsson et al. [76] report that the STAT4 SNP, rs7582694, correlates with the production of anti-double-stranded DNA antibodies and have a multiplicative risk effect of 1.82 with two independent IRF5 risk alleles. This provides a strong indication that interactions between the two genes contribute to the pathogenesis of SLE. However, a study using 30 tagged SNPs by Abelson et al. [77] revealed no significant interaction effects between SNPs in STAT4 and IRF5 and suggests that an additive model may most closely describe their combined contribution to SLE risk.
6. Interleukins
As a major component of the immune system, interleukins are strongly linked to the pathophysiology of SLE, and genes that encode interleukin proteins have been widely examined as possible SLE susceptibility candidates. Interleukin (IL)-10, which is an important immunoregulator that inhibits T-cell function and suppresses proinflammatory cytokines such as TNFα, IL-1, IL-6, IL-8, and IL-12 [78, 79] is particularly important in this regard.
6.1. IL10
A GWA replication study by Gateva et al. [24] confirmed an association between a SNP (rs3024505) on IL10 and SLE in individuals of European ancestry (P = 3.95 × 10−8). This followsup on a number of smaller scale studies that have previously identified an association between SLE and polymorphisms in IL10 [80, 81] (n = 58 and 158 resp.). It is also consistent with a large-scale association study in an Asian sample (n = 554) [82], which used six IL-10 promoter SNPs to identify six haplotypes. The authors found an association between one of the haplotypes and decreased IL-10 production and also observed a dose-dependent effect of the microsatellite IL10. G as a significant risk factor. IL10 is also known to promote B-cell functions by facilitating antibody production, differentiation, and proliferation [83]. Increased production of IL-10 by peripheral B cells has been shown to correlate with SLE severity [84]. Relevant polymorphisms include the SNP rs1800896, which is characterized by a glycine/alanine substitution at position −1082, rs1800871 (−819C/T), and rs1800872 (−592C/A) [4], all of which have been shown to affect IL-10 production [85–87]. Summers et al. [88] showed that the ATA haplotype, specifically the −592A allele, was more frequent in 23 cases of sudden infant death syndrome, suggesting that irregular lower/higher IL-10 production may affect production of inflammatory cytokines.
A number of other genes that encode interleukin proteins have also been associated with SLE. IL18 that has long been touted as an SLE candidate, though a recent dense mapping of the locus failed to find any evidence of a common variant association [89]. A recent CNV study of 938 SLE patients and 1,017 healthy controls by [90] found a higher proportion of copy number amplifications of IL17F, IL21, and IL22 in cases (see below). Elevated expression of IL21 (4q26-q27), which shows homology with genes that encode IL2, IL4, and IL15, has been demonstrated in the sera of SLE cases and mouse models [91]. Further work is needed to clarify the roles of these and other interleukin coding genes in SLE. A thorough review of SLE/interleukins is provided by López et al. [4].
6.2. IRAK1/MECP2
IL-1 receptor-associated kinase 1 (IRAK1) is part of the Toll/interleukin-1 receptor and nuclear NFκ signaling pathway and has been associated with both adult- and pediatric-onset SLE [92]. However, the neighboring locus, methyl-CpG-binding protein 2 (MECP2), which is in strong linkage disequilibrium with IRAK1 and is involved in regulating methylation-sensitive loci [93], has also been proposed as the source of this signal [94], though the two are not necessarily mutually exclusive. GWA by Graham et al. [13] identified the IRAK1-MECP2 locus as significantly associated with SLE risk in Europeans, replicating a previous finding by Sawalha et al. [95].
7. B-Cell Signaling
Although B cells have been strongly associated with SLE, a direct relationship between candidate genes and susceptibility has been difficult to determine. However, GWAS have facilitated the identification of a number of variants involved in B-cell signaling that may predispose to SLE. The most prominent is B lymphocyte kinase (BLK), which is located at 8p23.1. GWAS by Han et al. [12], Harley et al. [14], and a replication study by Gateva et al. [24] identified this locus as strongly associated with SLE risk in European and Asian populations. BLK is a tyrosine kinase that transduces signals downstream of the B-cell receptor and can phosphorylate inhibitory Fc receptors on B cells [52]. A meta-analysis by Fan et al. [96] utilizing 11,000+ cases and 20,000+ controls from European and Asian populations examined the risk alleles of rs13277113 and rs2248932. For the former, BLK mRNA expression was ~50% lower in A homozygotes than G allele homozygotes. Similarly, at rs2248932, the C allele was associated with lower levels of BLK mRNA, with C homozygotes having ~30% lower expression levels than T allele homozygotes. Both the A allele of rs13277113 and the T allele of rs2248932 were confirmed as significant risk factors for SLE. Similar to findings in relation to the MHC, STAT4, ITGAM, and IRF5, the GWA study of anti-dsDNA autoantibody production by Chung et al. [9] found evidence of an association between this phenotype and BLK but did not meet threshold criteria for genome-wide significance.
7.1. BANK1
A GWA study by Kozyrev et al. [16] identified a nonsynonymous SNP among a Swedish population in the B-cell scaffold protein with ankyrin repeats 1 (BANK1) gene at rs10516487 (R61H), which was replicated in four independent case-control sets. A study from our group confirmed this association in both European (n = 178) and African American (n = 148) populations [97]. Although a number of GWAS have not found a significant association with this locus, a large study of 1,724 SLE patients and 2,024 healthy controls of African American descent did replicate BANK1 as an SLE candidate in this cohort (as well as C8orf13-BLK, TNFSF4, KIAA1542, and CTLA4) [98]. Kozyrev et al. [16] identified two other nonsynonymous SNPs at rs17266594 (intronic) and rs3733197 (A383T), which have yet to be replicated as confirmed SLE risk factors. BANK1 has also been associated anti-dsDNA-positive but not anti-dsDNA-negative autoantibody production in SLE [9].
8. T-Cell Signaling: Candidate Genes
Cytokines are usually grouped in accordance with their functional capacity as T helper (Th) Th1, Th2, and Th17. Overproduction of Th1 and Th17 most often results in T-cell hyperactivity, whereas the overproduction of Th2 is linked to hyperactive B Cells and humoral responses [35].
8.1. PTPN22
Protein phosphatase nonreceptor type 22 (PTPN22) is known to inhibit T-Cell activation [99]. It has been associated with a wide range of autoimmune disorders and initially came to prominence as a candidate gene for type 1 diabetes [100]. Indeed, as Gregersen and Olsson [50] point out, the association between PTPN22 and a wide range of phenotypes (including Graves' disease [101–103], Hashimoto thyroiditis [104], myasthenia gravis [105], systemic sclerosis [106], generalized vitiligo [107], Addison's disease [108], alopecia areata [109], juvenile idiopathic arthritis [110–112], and SLE [14, 15]) provided one of the earliest indications of a shared pathophysiology for many autoimmune disorders. In spite of this, however, it is notable that PTPN22 risk alleles are not a universal feature of all autoimmune diseases, and indeed the 1858 T autoimmune risk allele is associated with protection against Crohn's disease [113]. Kariuki and Niewold [114] point out that this same risk allele is associated with TNF-α-related diseases but not with multiple sclerosis (treated with a related interferon, IFN-β). This suggests a possible relationship between PTPN22 and cytokine profile, albeit at a secondary level. The rs2476601 SNP polymorphism (cysteine to threonine, C1858T) in the Lyp protein is significantly associated with the underactivation of both T and B Cells and deregulated cytokine production [115, 116].
GWAS by Harley et al. [14] and replication by Gateva et al. [24] identified a positive association between the PTPN22 SNP, rs2476601, and SLE in European populations, but a similar association has not been observed in the Asian GWAS reviewed here. Deng and Tsao [117] point out that this difference may be attributable to more variability in European populations that may be between 2–15% [50]. PTPN22 has also been associated with anti-dsDNA-positive but not anti-dsDNA-negative autoantibody production in SLE [9].
8.2. PPP2R2B
Although not specifically identified by the GWAS listed in Table 1, protein phosphatase 2, regulatory subunit Bβ (PPP2R2B) may be important to SLE pathogenesis. A recent study by Crispín et al. [118] found that the regulatory Bβ that is expressed in resting human T cells is downregulated during T-cell activation and is upregulated by interleuikin-2 (IL-2). In a study of SLE patients, the group found that levels of PP2A Bβ were not increased by IL-2 deprivation in 7 of the 14 cases, and this phenomenon was paralleled by resistance to apoptosis. Furthermore, following IL-2 withdrawal in T cells, levels of Bβ in these patients remained unchanged. This contrasts markedly with responses from healthy controls as well as the remaining SLE (nonapoptosis-resistant) patients, who demonstrated an approximate threefold increase in Bβ. It would therefore appear that, at least in a subset of SLE patients, Bβ may be a primary cause of apoptosis resistance in T cells.
9. Fcγ Receptors
The fragment crystallizable (Fc) region is found at the tail of antibody proteins. Fc receptors are involved in clearing the immune complex and include Fcα, Fcε, and Fcγ. The latter has been most closely linked to the SLE susceptibility, with a number of low-affinity Fc receptors for immunoglobulin G (IgC) identified as SLE candidates. These include FCγR2A, FCγR2B, FCγR3A, and FCγR3B, all of which are regulated by cytokines, including IL-4, IL-10 or TGF-β [119]. Cytokine-mediated regulation of FcR expression is cell type-specific; however, IL-4 upregulates FCγR2B expression in myeloid cells but downregulates it in activated B cells [120]. One should bear in mind that, particularly with earlier studies, low coverage of Fcγ receptors by commercial manufacturers (i.e., Affymetrix and Illumina) has been an issue. This is mainly due to the presence of homologous sequence paralogs at this locus [121].
9.1. FCγR2A
The nonsynonymous SNP, rs1801274 in the FCγR2A (Fc fragment of IgG, low affinity IIa, receptor) gene has been associated with reduced clearance of immune complexes, where the (C) allele encodes arginine and the (T) allele encodes the variant histidine (H) [122]. Karassa et al. [123] conducted a meta-analysis of this polymorphism across European, African, and Asian populations and confirmed a positive risk to SLE. In general; however, the relationship has been inconsistent, and a majority of GWAS have not found significant evidence of an association. The exception is the 2008 GWA by Harley et al. [14] which identified an association between rs1801274 but only among European women.
9.2. FCγR2B
FCγR2B is involved in antibody production and macrophage activation. Tsuchiya et al. [124] examined a nonsynonymous SNP within the transmembrane domain, which was significantly associated with SLE in Chinese, Japanese, and Thai populations but was found to be rare in Europeans. The SNP (rs1050501) led to an aminoacid substitution within the transmembrane domain at position 232, isoleucine to tryptophan (I232T). The same group found that an FCγR2B promoter polymorphism that has been associated with SLE in Europeans [125] was largely absent in Asians. In a human B-cell line lacking endogenous FCγR2B, Kono et al. [126] demonstrated that I232T was less effective than wild-type at inhibiting B-cell receptor (BCR-) mediated signaling, and that distribution of FCγR2B to detergent-insoluble lipid rafts was disrupted. This is supported by Floto et al. [127], who showed that the lack of inhibition of activatory receptors resulted in unopposed proinflammatory signaling.
9.3. FCγR3A
Nonsynonymous SNPs in FCγR3A have been found to alter binding affinities in the four immunoglobulin G subclasses. At the SNP rs396991, the (T) allele encodes phenylalanine (F), and the (G) allele encodes the valine variant (F158V) [117]. The low-affinity phenylalanine allele is associated with disrupted immune complex clearance [128], while the high-affinity valine allele was a strong predictor of end-stage renal disease [129]. A copy number variation study by Niederer et al. [130] found significant differences in FCγR3A and FCγR3B CNV profiles between European, East Asian, and Kenyan populations. Reduced copy number and homozygosity of FCγR3B were strongly predictors of SLE susceptibility, but the same association was not reported in relation to FCγR3A.
9.4. FCγR3B
Three allotypic variants have been identified for FCγR3B—NA1, NA2, and SH and are defined by six SNPs. Hatta et al. [131] report an association between NA2 and SLE, but this has not been replicated (see Yuan et al. [132]). Nevertheless a number of copy number variation studies have converged upon the FCγR3B gene as a potentially important locus for SLE susceptibility. These are reviewed separately below.
10. Interpreting SLE GWAS
All of the discovery SNPs used to identify the genes listed in Table 1 have published odds ratios (ORs) between 1.2 and 2.4, with the majority ranking toward the lower end of this range. This is comparable with GWAS in other autoimmune disorders including Crohn's disease [133], rheumatoid arthritis [134], and psoriasis [135] and indeed the GWAS field as a whole. While it is important not to downplay their significance, it should be noted that the predictive value of such ratios is relatively low [136] and explains less than 15% [137] of the risk for SLE (as would be expected by GWAS variants all of which are designed to tag LD blocks showing association). We know from twin and family studies that the heritability of SLE is approximately 44–69% [138–140], which means we must consider the problem of missing heritability. There are a number of possible explanations between the discordance between the two figures: (1) SLE is underscored by an even larger number of genes each contributing smaller and smaller proportions of risk variance, (2) that the variants identified by GWAS lose significant power in the process of tagging the causative variants, and/or (3) that the broader SLE phenotype may consist of a several distinct and rare subtypes. It is also likely that epigenetic factors are important elements of the missing heritability, though this is not explored in the current paper.
(1) Gene Networks and Pathway Analysis —
The first of these conclusions, that an increasingly larger pool of genes is required to account for the heritability of SLE, is logical but complex. The common disease common variant model is predicated upon the conclusion that the complex diseases are caused by the interactions of a large network of genes, and the number of possible causal loci is only constrained by the number of genes and gene regulators in the human genome. However, as more and more genes are implicated in this network, we run into the law of diminishing returns. Pathway analysis, which leverages existing biological knowledge about gene function to examine how causal factors may interact, is an attractive mechanism for navigating this law. Pathway-based approaches typically examine whether test statistics for a predefined gene-set have concordant (albeit moderate) deviation from chance. Analyses are based on pathway association approaches in gene expression microarray analysis, where examination of groups of related genes has yielded major insights into functional capacity [141, 142].
A recent study from our group [5] adopted the pathways approach to GWAS of Crohn's disease (CD), which is known to share certain pathophysiological properties with SLE [37]. The study examined enrichment of association signals for genes previously identified as belonging to certain gene pathway networks, as defined by gene ontology, biocarta and KEGG, with careful adjustment for gene size, number of SNPs per each genes, and pathway size. The pathway that most significantly enriched for association signals was the interleukin-12 gene pathway that harbors the cytokines, interleukin 12 and 23 (IL-12/IL-23). These share one cellular receptor subunit and numerous intracellular signaling components previously shown to associate with CD [113], together with multiple other genes associating with CD, for the first time [5]. Indeed, only three genes (IL12B, IL23R, and IL12RB2) at two loci (5q23, 1p31) showed genome-wide signals in previous studies [113]. However, three further genes in the IL-12-IL-23 pathway (JAK2, CCR6, and STAT3) were confirmed as candidate genes in replication studies [109], and six more genes (including STAT4 and a number of interleukin-coding genes) supported by association in this pathway have been previously reported as CD susceptibility genes in other association and functional studies [5, 142–147]. Thus, since only three genes in this pathway have surpassed the threshold for genome-wide significance, we begin to develop a much richer picture of the pathophysiology of the disease through the pathway-based approach. This includes related variants that have remained above significance criteria but collectively contribute significantly to the risk variance. Furthermore, given that the strongest candidate gene is sometimes not the most suitable drug target, the pathway approach also opens up possible alternatives for targeted intervention. These interactions are outlined in more detail in Figure 2. Given the shared etiology between many of the autoimmune diseases, it is likely that a similar approach would be productive in extrapolating gene networks in SLE.
Figure 2.
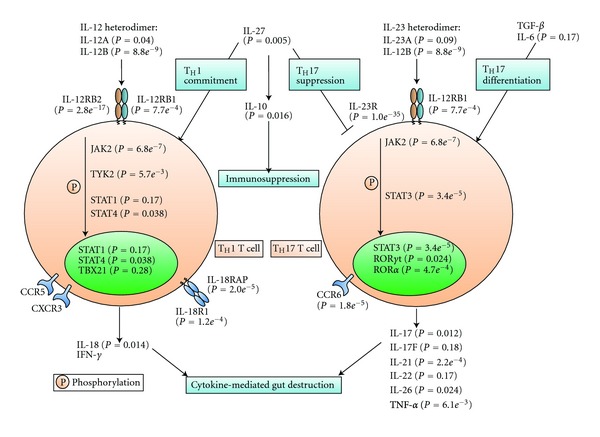
Linking pathways to disease: Crohn's disease (from Wang et al. [5]). As an example of how biological pathways are involved in disease pathogenesis, the authors illustrate a manually compiled pathway centered on IL-12 and IL-23. For each gene, the most significant P value among SNPs close to the gene (based on GWAS) was annotated. Only three genes at two loci (IL12B at 5q33 and IL23R-IL12RB2 at 1p31) showed genome-wide significant signals, but three further genes (JAK2, CCR6, and STAT3) in the pathway were confirmed as susceptibility candidates in replication studies. Six further genes in this pathway were reported as Crohn's disease susceptibility genes by other association and functional studies.
(2) Causative Variants and the GWAS Signal —
A study from our group substantiates the second conclusion, that at least some of the variants tagged by GWAS may actually be tagging rarer variants that may be responsible for the GWA signal. This phenomenon is known as synthetic association and was confirmed in a recent study by Wang et al. [148], who used sequencing to examine NOD2 as a candidate gene for CD. Three rare variants (nonsynonymous SNPs) in NOD2, rs2066844, rs2066845, and rs2066847, had previously been associated with susceptibility to CD [149, 150], which had additionally been identified by functional assays as potentially causal [151]. Although NOD2 had not previously been shown to harbor common causal variants, a 2007 study by the Wellcome Trust Case Control Consortium (WTCCC) [152] did implicate a common tag SNP (rs17221417, MAF = 29). From HapMap, we know that the first two of the rare NOD2 variants are in complete linkage disequilibrium with rs17221417 (the third variant was not listed). A large-scale meta-analysis of NOD2 estimates the allelic odds ratio (OR) at between 2.2 to 4.1, which, respectively, explain 0.54%, 1.2%, and 3.4% of genetic risk (5.1% in total). For the common tag SNP, on the other hand, the OR is 1.37 and explains on 0.69% of genetic risk for CD. In other words, the GWA signal dramatically underestimates the proportion of explained risk at this locus.
A long range haplotype analysis of the GWA data for all the genes identified above is therefore recommended. Our group previously showed that synthetic associations can cover intervals as long as 2.5-Mb, and include numerous “blocks” of associated variants [136]. As such, followup and interpretation of GWA data merit careful deliberation. Namely, this approach may enrich sample sets for individuals with rare-causative variants, who should be filtered out from the cohort and subsequently sequenced for confirmation [148]. Anderson et al. [153] make the point that certain GWAS signals are more amenable to the synthetic association effect than others. This includes signals that are found inconsistently between different populations; signals that are not universally common are more likely to have arisen recently or been unequally selected for through population history. A number of the SLE candidate genes reviewed above fall into this category, including ETS1, ITGAM, and BANK1.
(3) Rare Variants —
Another source of hidden variance is the existence of rare phenotypes subsumed under the broader SLE umbrella but with a separable genetic basis. The heterogeneity of the SLE phenotype lends itself to this possibility, which is also supported by a number of genome sequencing studies that have emerged in recent years. As mentioned above, complement defects are present in 1-2% of SLE cases and represent the strongest single genetic risk factors of SLE. Additional rare variants associated with SLE include mutations in the three prime repair exonuclease 1 (TREX1) gene, which is the most common known cause of monogenic (i.e., single gene defect) SLE [154, 155] and may be an instructive vehicle by which to explore the application of sequencing technologies to the disease. Located on chromosome 3p21, TREX1 can metabolize reverse-transcribed DNA and encodes 3′ repair exonuclease 1, the primary 3′ to 5′ exonuclease in humans. It is a regulator of the IFN-stimulatory DNA (ISD) response [156], and mutations of TREX1 can cause Aicardi-Goutières syndrome (AGS) [157], a severe neurological brain disease that is sometimes comorbid with pediatric SLE [158–161]. It is also linked to several other diseases accelerated atherosclerosis, antiphospholipid syndrome, and fetal loss [162].
TREX1 mutations in AGS are predominantly recessive and reduce exonuclease activity. This is the case with a nonsynonymous SNP at position 114 that results in an arginine to histidine substitution (R114H) [163, 164]. A large-scale genotyping study of 40 TREX1 SNPs in ~8370 SLE patients and ~7490 controls by Namjou et al. [165] identified nine European patients with heterozygous mutations at this locus, which was also found in five European controls. Among Asians included in the study, two SLE cases had heterozygote mutations at this locus, but none were found in controls. Moreover, one Asian case, a male with early-onset SLE, was found to have a homozygous R114H mutation. The patient, who also had positive anti-dsDNA antibody, was negative for neurological manifestations, which are found in AGS children with the R114H mutation. A number of other mutations were observed in SLE patients but not respective controls. Among Europeans cases, five heterozygous mutations were detected at Y305C but none in controls. Y305C is a missense coding mutation located outside of the catalytic domain. Among Africans, five cases had a mutation at E266G, but none were found in controls. Interestingly, this mutation was also present in Europeans but did not differentiate cases from controls. For the SLE group as a whole, the coding mutation frequency was approximately 0.5%.
Importantly, a case-control association study for common SNPs did not identify significant associations between the affected and the unaffected either for the group as a whole, or for respective racial groups. This underscores the point that the association approach is limited in terms of probing rare variants. However, as discussed above in relation to CD, GWA signals at the site of a common tag SNP can prime the locus for followup by sequencing and genotyping. Thus, in the Namajou et al. study, common SNPs (defined by a minor allele frequency of 10% or greater) in Europeans characterized a seizure-associated risk haplotype that was present in 58% of cases, compared to 45% in controls (P = 0.0008, OR = 1.73, 95% CI = 1.25–2.39).
Although untested, one may speculate that many of the loci identified by GWAS may similarly harbor rare variants. Monogenic subtypes of cutaneous lupus erythematosus (a rare cutaneous form of lupus erythematosus) have been observed at the TREX1 locus [166]. Similarly, Qari et al. [167] describe seven Saudi families, where pedigree data is consistent with autosomal recessive Mendelian inheritance. In the last two years, the application of NGS to monogenic and oligogenic disorders has accelerated rapidly, reflecting the increased availability of relevant technologies. In our own group, we have used NGS to indentify the causal variants in a number of Mendelian phenotypes including rare forms of Glycogen Storage Disease, familial forms of epilepsy, hemolytic anemia [168], and Ogden syndrome [169].
It remains to be seen what proportion of rare variants will account for hidden variance in SLE. Regardless, their study will continue to play an important role in explicating the pathogenesis of the disorder. Because rare mutations carry large effects, they make resolution of underlying networks distinctly less complex and are also amenable to modeling in other systems. The widespread application of sequencing technologies in the clinic will also help characterize and differentiate SLE subphenotypes. In this vein, De Vries et al. [170] used direct sequencing of exonic TREX1 in 60 patients with neuropsychiatric SLE and identified a novel heterozygous substitution at position 128 (arginine histidine) in one case. Because TREX1 mutations are also linked to AGS, which is characterized by a number of neurological abnormalities, it is possible that this form of SLE shares a common pathogenic mechanism.
11. SLE and Copy Number Variants
The role that rare variants in SLE has is also becoming apparent from a series of copy number variation (CNV) studies, in the past several years. CNVs are insertions, deletions, or inversions in the genome that are universal in the general population and vary in length from many megabases to 1 kilobase or smaller. Although CNVs per se are not associated with any observable phenotype, their presence in genic regions has been associated with a number of major diseases, including autism [171], schizophrenia [172, 173], neuroblastoma [174], and many others. Arguably the most widely known CNV occurs in down syndrome, which is characterized by an extra copy of chromosome 21. The origin of most CNVs is unknown, but causal mechanisms can include replication errors, meiotic recombination, and homologous/nonhomologous repair of double-strand breaks [175].
Recent studies by the 1000 Genomes Consortium [176] and Conrad et al. [177] report that common CNVs are well covered by SNPs in existing arrays and many likely have been indirectly examined in a range of GWAS. The impact of rare CNVs, on the other hand, may be substantial. Pang et al. [178] reexamined data from the Venter genome and identified over 12,000 structural variants spanning more than 40 Mb of sequence. These variants were found in 4,867 genes, which are often large and under negative selection. Because rarer alleles are more likely to have a higher penetrance, these results strongly support the role of CNVs as causal factors in genetic diseases. The study also showed that 24% of CNVs would not be imputed from SNP association alone, which stresses the point that CNVs may be more accurately detected using the NGS approach.
A number of CNV studies have highlighted FCγR3B as an important locus for CNV differences between SLE cases and controls. As outlined above, Fcγ receptors for immunoglobulin G (IgC) are involved in clearing the immune complex, and many members of the Fcγ have been proposed and replicated as SLE candidates. As far back as 2006, Aitman et al. [179] reported an association between low copy number in FCγR3B and autoimmune glomerulonephritis in a subsample of 30 individuals with SLE. The same group [180] replicated this association in 161 Europeans with SLE-associated glomerulonephritis (control n = 312), as well as an Afro-Caribbean cohort of 134 patients [181]. This second study also examined the broader autoimmune disorder phenotype (n = 1.279) and found that 25 (2%) have no copies of FCγR3B. The same analysis of 862 controls indentified only one individual with no FCγR3B copies. Interestingly, the group did not observe an association between Graves' or Addison's diseases, both of which are organ-specific autoimmune disorders.
Niederer et al. [130] also described a strong link between FCγR3B copy number and identified a linkage disequilibrium between FCγR3B and an FCγR2B variant (I232T). Morris et al. [182] confirmed the association between FCγR3B low copy number, which was contingent upon allotype; SLE risk was greater for NA1 deletion, than deletion of NA2. The authors also reported a significant correlation between FCγR3B copy number and neutrophil expression in healthy and control participants. FCγR3B is expressed by neutrophils and eosinophils. Neutrophils, which mediate inflammation responses to host injury [183] are attracted by cytokines at the early stages of infection and also release cytokines as part of the inflammatory response [184]. Eosinophils, also an important part of the inflammatory response, are activated by several cytokines, namely, IL-3, IL-5, and GM-CSF [185].
CNVs have been identified at other candidate SLE loci. In a study of 532 Asian patients with SLE and 576 controls, Yu et al. [186] identified CNVs at IL-12B and T-bet as significantly associated with SLE risk. For IL-12B, the frequency of copy number amplification was 63 versus 13 in controls. For T-bet, the respective values were 46 versus 7. The same group [187] identified CNVs at histamine H4 receptor in a cohort of 340 SLE patients (versus 392 controls). These correlated with the presence of antinuclear antibody abnormalities, as well as incidence of arthritis and proteinuria. The group also identified CNV enrichment in interleukins-17F, -21, and -22, in two Chinese SLE cohorts (Yu et al. [90]). Allele frequencies amplifications for cases versus controls were 107 versus 33, 166 versus 16, and 108 versus 19, respectively. Other studies have implicated copy number differences at the TLR7 locus in women with SLE [188] and at the HIN200 locus in UK SLE families and French males [189]. Although the majority of these CNVs are found in only a handful of cases, these results have broad implications for SLE as a whole and may collectively account for a relative large proportion of SLE cases.
12. Conclusions
Taken together, these studies show that SLE is highly heritable, and advances in gene-finding technology in the past decade have rapidly accelerated gene discovery. Over this period, a number of themes have begun to emerge: (1) an ever-increasing catalog of candidate genes that replicate across different studies, (2) a growing list of causal rare variants, and (3) the emergence of monogenic subtypes. Monogenic SLE is particularly interesting from a treatment perspective, as it provides a mechanism for studying the phenotype in model systems and is a more obvious target for drug intervention. In order to capitalize upon these findings, high-quality phenotype data is required. SLE is notoriously heterogeneous and is fractionated in terms of onset, symptoms, and trajectory. The systematic collection of clinical and biomedical data (e.g., cellular, serological, and mRNA transcripts) will dramatically increase our ability to generate testable hypothesis about the contributions of specific genes and gene networks to SLE pathophysiology. Conversely, knowledge of gene function can be used to target treatments and to predict onset.
Ultimately, the primary goal is not to determine the frequency of variation/mutation in cases versus controls but to determine the pathways that lead to disease pathology. This is no simple task, especially when we consider the other major risk factors such as epigenetics, RNA regulatory elements, and environmental exposures. While daunting, the elucidation of these elements will doubtlessly take us closer to developing more effective treatments for SLE targeting selective patients for interventions aimed at restoring the impact from variants within specific molecular pathways and gene networks.
References
- 1.Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15(5):308–318. doi: 10.1191/0961203306lu2305xx. [DOI] [PubMed] [Google Scholar]
- 2.Mina R, Brunner HI. Pediatric lupus-are there differences in presentation, genetics, response to therapy, and damage accrual compared with adult lupus? Rheumatic Disease Clinics of North America. 2010;36(1):53–80. doi: 10.1016/j.rdc.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bustamante CD, De La Vega FM, Burchard EG. Genomics for the world. Nature. 2011;475(7355):163–165. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.López P, Gutiérrez C, Suáre A. IL-10 and TNFα Genotypes in SLE. Journal of Biomedicine and Biotechnology. 2010;2010:11 pages. doi: 10.1155/2010/838390. Article ID 838390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nature Reviews Genetics. 2010;11(12):843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- 6.Yap DYH, Lai KN. Cytokines and their roles in the pathogenesis of systemic lupus erythematosus: from basics to recent advances. Journal of Biomedicine and Biotechnology. 2010;2010 doi: 10.1155/2010/365083. Article ID 365083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dean GS, Tyrrell-Price J, Crawley E, Isenberg DA. Cytokines and systemic lupus erythematosus. Annals of the Rheumatic Diseases. 2000;59(4):243–251. doi: 10.1136/ard.59.4.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature Cell Biology. 2010;12(9):814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung SA, Taylor KE, Graham RR, et al. Differential genetic associations for systemic lupus erythematosus based on anti-dsdna autoantibody production. Plos Genetics. 2011;7(3) doi: 10.1371/journal.pgen.1001323. Article ID e1001323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Yang W, Hirankarn N, et al. Elf1 is associated with systemic lupus erythematosus in asian populations. Human Molecular Genetics. 2011;20(3):601–607. doi: 10.1093/hmg/ddq474. Article ID ddq474. [DOI] [PubMed] [Google Scholar]
- 11.Yang W, Shen N, Ye DQ, et al. Genome-wide association study in asian populations identifies variants in ets1 and wdfy4 associated with systemic lupus erythematosus. Plos Genetics. 2010;6(2) doi: 10.1371/journal.pgen.1000841. Article ID e1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han JW, Zheng HF, Cui Y, et al. Genome-wide association study in a chinese han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nature Genetics. 2009;41(11):1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 13.Graham RR, Cotsapas C, Davies L, et al. Genetic variants near tnfaip3 on 6q23 are associated with systemic lupus erythematosus. Nature Genetics. 2008;40(9):1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in itgam, pxk, kiaa1542 and other loci. Nature Genetics. 2008;40(2):204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with c8orf13-blk and itgam-itgax. New England Journal of Medicine. 2008;358(9):900–909. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- 16.Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the b-cell gene bank1 are associated with systemic lupus erythematosus. Nature Genetics. 2008;40(2):211–216. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- 17.Grumet FC, Coukell A, Bodmer JG, Bodmer WF, McDevitt HO. Histocompatibility (hl-a) antigens associated with systemic lupus erythematosus. a possible genetic predisposition to disease. New England Journal of Medicine. 1971;285(4):193–196. doi: 10.1056/NEJM197107222850403. [DOI] [PubMed] [Google Scholar]
- 18.Beck S, Geraghty D, Inoko H, Rowen L. Complete sequence and gene map of a human major histocompatibility complex. Nature. 1999;401(6756):921–923. doi: 10.1038/44853. [DOI] [PubMed] [Google Scholar]
- 19.Graham RR, Ortmann WA, Langefeld CD, et al. Visualizing human leukocyte antigen class ii risk haplotypes in human systemic lupus erythematosus. American Journal of Human Genetics. 2002;71(3):543–553. doi: 10.1086/342290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doherty DG, Ireland R, Demaine AG, et al. Major histocompatibility complex genes and susceptibility to systemic lupus erythematosus in southern chinese. Arthritis and Rheumatism. 1992;35(6):641–646. doi: 10.1002/art.1780350607. [DOI] [PubMed] [Google Scholar]
- 21.Hong GH, Kim HY, Takeuchi F, et al. Association of complement c4 and hla-dr alleles with systemic lupus erythematosus in koreans. Journal of Rheumatology. 1994;21(3):442–447. [PubMed] [Google Scholar]
- 22.Hashimoto H, Tsuda H, Matsumoto T. Hla antigens associated with systemic lupus erythematosus in japan. Journal of Rheumatology. 1985;12(5):919–923. [PubMed] [Google Scholar]
- 23.Yuan YJ, Luo XB, Shen N. Current advances in lupus genetic and genomic studies in asia. Lupus. 2010;19(12):1374–1383. doi: 10.1177/0961203310376639. [DOI] [PubMed] [Google Scholar]
- 24.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies tnip1, prdm1, jazf1, uhrf1bp1 and il10 as risk loci for systemic lupus erythematosus. Nature Genetics. 2009;41(11):1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arason GJ, Jorgensen GH, Ludviksson BR. Primary immunodeficiency and autoimmunity: lessons from human diseases. Scandinavian Journal of Immunology. 2010;71(5):317–328. doi: 10.1111/j.1365-3083.2010.02386.x. [DOI] [PubMed] [Google Scholar]
- 26.Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Advances in Immunology. 2000;76:227–324. doi: 10.1016/s0065-2776(01)76021-x. [DOI] [PubMed] [Google Scholar]
- 27.Sullivan KE. Complement deficiency and autoimmunity. Current Opinion in Pediatrics. 1998;10(6):600–606. doi: 10.1097/00008480-199810060-00011. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan KE, Wisnieski JJ, Winkelstein JA, et al. Serum complement determinations in patients with quiescent systemic lupus erythematosus. Journal of Rheumatology. 1996;23(12):2063–2067. [PubMed] [Google Scholar]
- 29.Dunckley H, Gatenby PA, Hawkins B, Naito S, Serjeantson SW. Deficiency of c4a is a genetic determinant of systemic lupus erythematosus in three ethnic groups. Journal of Immunogenetics. 1987;14(4-5):209–218. doi: 10.1111/j.1744-313x.1987.tb00383.x. [DOI] [PubMed] [Google Scholar]
- 30.Miyagawa H, Yamai M, Sakaguchi D, et al. Association of polymorphisms in complement component c3 gene with susceptibility to systemic lupus erythematosus. Rheumatology. 2008;47(2):158–164. doi: 10.1093/rheumatology/kem321. [DOI] [PubMed] [Google Scholar]
- 31.Sekine H, Ferreira RC, Pan-Hammarström Q, et al. Role for msh5 in the regulation of ig class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(17):7193–7198. doi: 10.1073/pnas.0700815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernando MM, Stevens CR, Sabeti PC, et al. Identification of two independent risk factors for lupus within the MHC in United Kingdom families. Plos Genetics. 2007;3(11, article e192) doi: 10.1371/journal.pgen.0030192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annual Review of Immunology. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marnell L, Mold C, Du Clos TW. C-reactive protein: ligands, receptors and role in inflammation. Clinical Immunology. 2005;117(2):104–111. doi: 10.1016/j.clim.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Lourenço EV, La Cava A. Cytokines in systemic lupus erythematosus. Current Molecular Medicine. 2009;9(3):242–254. doi: 10.2174/156652409787847263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antoni C, Braun J. Side effects of anti-tnf therapy: current knowledge. Clinical and Experimental Rheumatology. 2002;20(6, supplement 28):S-152–S-157. [PubMed] [Google Scholar]
- 37.Zhu LJ, Yang X, Yu XQ. Anti-TNF-α therapies in systemic lupus erythematosus. Journal of Biomedicine and Biotechnology. 2010;2010:8 pages. doi: 10.1155/2010/465898. Article ID 465898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004;20(5):539–550. doi: 10.1016/s1074-7613(04)00108-6. [DOI] [PubMed] [Google Scholar]
- 39.Ohshima Y, Tanaka Y, Tozawa H, Takahashi Y, Maliszewski C, Delespesse G. Expression and function of ox40 ligand on human dendritic cells. Journal of Immunology. 1997;159(8):3838–3848. [PubMed] [Google Scholar]
- 40.Latza U, Durkop H, Schnittger S, et al. The human ox40 homolog: cdna structure, expression and chromosomal assignment of the act35 antigen. European Journal of Immunology. 1994;24(3):677–683. doi: 10.1002/eji.1830240329. [DOI] [PubMed] [Google Scholar]
- 41.Graham DSC, Graham RR, Manku H, et al. Polymorphism at the tnf superfamily gene tnfsf4 confers susceptibility to systemic lupus erythematosus. Nature Genetics. 2008;40(1):83–89. doi: 10.1038/ng.2007.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez E, Nadig A, Richardson BC, et al. Phenotypic associations of genetic susceptibility loci in systemic lupus erythematosus. Annals of the Rheumatic Diseases. 2011;70(10):1752–1757. doi: 10.1136/ard.2011.154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bates JS, Lessard CJ, Leon JM, et al. Meta-analysis and imputation identifies a 109kb risk haplotype spanning tnfaip3 associated with lupus nephritis and hematologic manifestations. Genes and Immunity. 2009;10(5):470–477. doi: 10.1038/gene.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the tnfaip3 region are independently associated with systemic lupus erythematosus. Nature Genetics. 2008;40(9):1062–1064. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adrianto I, Wen F, Templeton A, et al. Association of a functional variant downstream of tnfaip3 with systemic lupus erythematosus. Nature Genetics. 2011;43(3):253–258. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of ifn regulatory factor 5 (irf5) define risk and protective haplotypes for human lupus. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(16):6758–6763. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawasaki A, Ito S, Furukawa H, et al. Association of tnfaip3 interacting protein 1, tnip1 with systemic lupus erythematosus in a japanese population: a case-control association study. Arthritis Research & Therapy. 2010;12(5):p. R174. doi: 10.1186/ar3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basu S, Rosenzweig KR, Youmell M, Price BD. The DNA-dependent protein kinase participates in the activation of NF kappa B following DNA damage. Biochemical and Biophysical Research Communications. 1998;247(1):79–83. doi: 10.1006/bbrc.1998.8741. [DOI] [PubMed] [Google Scholar]
- 49.Heyninck K, Beyaert R. A20 inhibits nf-κb activation by dual ubiquitin-editing functions. Trends in Biochemical Sciences. 2005;30(1):1–4. doi: 10.1016/j.tibs.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 50.Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annual Review of Immunology. 2009;27:363–391. doi: 10.1146/annurev.immunol.021908.132653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ronnblom LE, Oberg KE, Alm GV. Possible induction of systemic lupus erythematosus by interferon α-treatment in a patient with a malignant carcinoid tumour. Journal of Internal Medicine. 1990;227(3):207–210. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 52.Ho V, McLean A, Terry S. Severe systemic lupus erythematosus induced by antiviral treatment for hepatitis C. Journal of Clinical Rheumatology. 2008;14(3):166–168. doi: 10.1097/RHU.0b013e3181775e80. [DOI] [PubMed] [Google Scholar]
- 53.Weckerle CE, Franek BS, Kelly JA, et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis and Rheumatism. 2011;63(4):1044–1053. doi: 10.1002/art.30187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, irf-5, results in the induction of distinct interferon α genes. Journal of Biological Chemistry. 2001;276(26):23382–23390. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 55.Graham RR, Kozyrev SV, Baechler EC, et al. A common haplotype of interferon regulatory factor 5 (irf5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nature Genetics. 2006;38(5):550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 56.Demirci FYK, Manzi S, Ramsey-Goldman R, et al. Association of a common interferon regulatory factor 5 (IRF5) variant with increased risk of systemic lupus erythematosus (SLE) Annals of Human Genetics. 2007;71(3):308–311. doi: 10.1111/j.1469-1809.2006.00336.x. [DOI] [PubMed] [Google Scholar]
- 57.Shin HD, Sung YK, Choi CB, Lee SO, Lee HW, Bae SC. Replication of the genetic effects of ifn regulatory factor 5 (irf5) on systemic lupus erythematosus in a korean population. Arthritis Research and Therapy. 2007;9, article no. R32 doi: 10.1186/ar2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawasaki A, Kyogoku C, Ohashi J, et al. Association of irf5 polymorphisms with systemic lupus erythematosus in a japanese population: support for a crucial role of intron 1 polymorphisms. Arthritis and Rheumatism. 2008;58(3):826–834. doi: 10.1002/art.23216. [DOI] [PubMed] [Google Scholar]
- 59.Siu HO, Yang W, Lau CS, et al. Association of a haplotype of irf5 gene with systemic lupus erythematosus in chinese. Journal of Rheumatology. 2008;35(2):360–362. [PubMed] [Google Scholar]
- 60.Niewold TB, Kelly JA, Kariuki SN, et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Annals of the Rheumatic Diseases. 2012;71(3):463–468. doi: 10.1136/annrheumdis-2011-200463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kelly JA, Kelley JM, Kaufman KM, et al. Interferon regulatory factor-5 is genetically associated with systemic lupus erythematosus in african americans. Genes and Immunity. 2008;9(3):187–194. doi: 10.1038/gene.2008.4. [DOI] [PubMed] [Google Scholar]
- 62.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the irf5 risk haplotype with high serum interferon-α activity in systemic lupus erythematosus patients. Arthritis and Rheumatism. 2008;58(8):2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sigurdsson S, Göring HHH, Kristjansdottir G, et al. Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 (irf5) reveals a novel 5 bp length polymorphism as strong risk factor for systemic lupus erythematosus. Human Molecular Genetics. 2008;17(6):872–881. doi: 10.1093/hmg/ddm359. [DOI] [PubMed] [Google Scholar]
- 64.Löfgren SE, Yin H, Delgado-Vega AM, et al. Promoter insertion/deletion in the irf5 gene is highly associated with susceptibility to systemic lupus erythematosus in distinct populations, but exerts a modest effect on gene expression in peripheral blood mononuclear cells. Journal of Rheumatology. 2010;37(3):574–578. doi: 10.3899/jrheum.090440. [DOI] [PubMed] [Google Scholar]
- 65.Zhang L, Pagano JS. Structure and function of irf-7. Journal of Interferon and Cytokine Research. 2002;22(1):95–101. doi: 10.1089/107999002753452700. [DOI] [PubMed] [Google Scholar]
- 66.Salloum R, Franek BS, Kariuki SN, et al. Genetic variation at the irf7/phrf1 locus is associated with autoantibody profile and serum interferon-α activity in lupus patients. Arthritis and Rheumatism. 2010;62(2):553–561. doi: 10.1002/art.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Jager PL, Jia X, Wang J, et al. Meta-analysis of genome scans and replication identify cd6, irf8 and tnfrsf1a as new multiple sclerosis susceptibility loci. Nature Genetics. 2009;41(7):776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hikami K, Kawasaki A, Ito I, et al. Association of a functional polymorphism in the 3’-untranslated region of spi1 with systemic lupus erythematosus. Arthritis and Rheumatism. 2011;63(3):755–763. doi: 10.1002/art.30188. [DOI] [PubMed] [Google Scholar]
- 69.Yee AA, Yin P, Siderovski DP, Mak TW, Litchfield DW, Arrowsmith CH. Cooperative interaction between the dna-binding domains of pu.1 and irf4. Journal of Molecular Biology. 1998;279(5):1075–1083. doi: 10.1006/jmbi.1998.1838. [DOI] [PubMed] [Google Scholar]
- 70.Huang W, Horvath E, Eklund EA. Pu.1, interferon regulatory factor (irf) 2, and the interferon consensus sequence-binding protein (icsbp/irf8) cooperate to activate nf1 transcription in differentiating myeloid cells. Journal of Biological Chemistry. 2007;282(9):6629–6643. doi: 10.1074/jbc.M607760200. [DOI] [PubMed] [Google Scholar]
- 71.Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB. Cutting edge: autoimmune disease risk variant of stat4 confers increased sensitivity to ifn-α in lupus patients in vivo. Journal of Immunology. 2009;182(1):34–38. doi: 10.4049/jimmunol.182.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Remmers EF, Plenge RM, Lee AT, et al. Stat4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. New England Journal of Medicine. 2007;357(10):977–986. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacobson NG, Szabo SJ, Weber-Nordt RM, et al. Interleukin 12 signaling in t helper type 1 (th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (stat)3 and stat4. Journal of Experimental Medicine. 1995;181(5):1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O’Shea JJ. Signaling by il-12 and il-23 and the immunoregulatory roles of stat4. Immunological Reviews. 2004;202:139–156. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- 75.Namjou B, Sestak AL, Armstrong DL, et al. High-density genotyping of stat4 reveals multiple haplotypic associations with systemic lupus erythematosus in different racial groups. Arthritis and Rheumatism. 2009;60(4):1085–1095. doi: 10.1002/art.24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sigurdsson S, Nordmark G, Garnier S, et al. A risk haplotype of stat4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsdna and shows additive effects with two risk alleles of irf5. Human Molecular Genetics. 2008;17(18):2868–2876. doi: 10.1093/hmg/ddn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abelson AK, Delgado-Vega AM, Kozyrev SV, et al. Stat4 associates with systemic lupus erythematosus through two independent effects that correlate with gene expression and act additively with irf5 to increase risk. Annals of the Rheumatic Diseases. 2009;68(11):1746–1753. doi: 10.1136/ard.2008.097642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Waal Malefyt R, Haanen J, Spits H, et al. Interleukin 10 (il-10) and viral il-10 strongly reduce antigen-specific human t cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class ii major histocompatibility complex expression. Journal of Experimental Medicine. 1991;174(4):915–924. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse t helper cell. iv. th2 clones secrete a factor that inhibits cytokine production by th1 clones. Journal of Experimental Medicine. 1989;170(6):2081–20095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eskdale J, Wordsworth P, Bowman S, Field M, Gallagher G. Association between polymorphisms at the human il-10 locus and systemic lupus erythematosus. Tissue Antigens. 1997;49(6):635–639. doi: 10.1111/j.1399-0039.1997.tb02812.x. [DOI] [PubMed] [Google Scholar]
- 81.Mehrian R, Quismorio FP, Jr., Strassmann G, et al. Synergistic effect between IL-10 and bcl-2 genotypes in determining susceptibility to systemic lupus erythematosus. Arthritis & Rheumatism. 1998;41(4):596–602. doi: 10.1002/1529-0131(199804)41:4<596::AID-ART6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 82.Chong WP, Ip WK, Wong WHS, Lau CS, Chan TM, Lau YL. Association of interleukin-10 promoter polymorphisms with systemic lupus erythematosus. Genes and Immunity. 2004;5(6):484–492. doi: 10.1038/sj.gene.6364119. [DOI] [PubMed] [Google Scholar]
- 83.Rousset F, Garcia E, Defrance T, et al. Interleukin 10 is a potent growth and differentiation factor for activated human b lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(5):1890–1893. doi: 10.1073/pnas.89.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hagiwara E, Gourley MF, Lee S, Klinman DM. Disease severity in patients with systemic lupus erythematosus correlates with an increased ratio of interleukin-10:interferon-γ-secreting cells in the peripheral blood. Arthritis and Rheumatism. 1996;39(3):379–385. doi: 10.1002/art.1780390305. [DOI] [PubMed] [Google Scholar]
- 85.Edwards-Smith CJ, Jonsson JR, Purdie DM, Bansal A, Shorthouse C, Powell EE. Interleukin-10 promoter polymorphism predicts initial response of chronic hepatitis c to interferon alfa. Hepatology. 1999;30(2):526–530. doi: 10.1002/hep.510300207. [DOI] [PubMed] [Google Scholar]
- 86.Eskdale J, Gallagher G, Verweij CL, Keijsers V, Westendorp RGJ, Huizinga TWJ. Interleukin 10 secretion in relation to human il-10 locus haplotypes. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9465–9470. doi: 10.1073/pnas.95.16.9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Turner DM, Williams DM, Sankaran D, Lazarus M, Sinnott PJ, Hutchinson IV. An investigation of polymorphism in the interleukin-10 gene promoter. European Journal of Immunogenetics. 1997;24(1):1–8. doi: 10.1111/j.1365-2370.1997.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 88.Summers AM, Summers CW, Drucker DB, Hajeer AH, Barson A, Hutchinson IV. Association of il-10 genotype with sudden infant death syndrome. Human Immunology. 2000;61(12):1270–1273. doi: 10.1016/s0198-8859(00)00183-x. [DOI] [PubMed] [Google Scholar]
- 89.Guerra SG, Morris DL, Gateva V, Graham RR, Vyse TJ, Graham DSC. Dense mapping of il18 shows no association in sle. Human Molecular Genetics. 2011;20(5):1026–1033. doi: 10.1093/hmg/ddq536. Article ID ddq536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu B, Guan M, Peng Y, et al. Copy number variations of interleukin-17F, interleukin-21, and interleukin-22 are associated with systemic lupus erythematosus. Arthritis Rheum. 2011;63(11):3487–3492. doi: 10.1002/art.30595. [DOI] [PubMed] [Google Scholar]
- 91.Li J, Pan HF, Cen H, et al. Interleukin-21 as a potential therapeutic target for systemic lupus erythematosus. Mol Biol Rep. 2011;38(6):4077–4081. doi: 10.1007/s11033-010-0527-y. [DOI] [PubMed] [Google Scholar]
- 92.Jacob CO, Zhu J, Armstrong DL, et al. Identification of irak1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(15):6256–6261. doi: 10.1073/pnas.0901181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Webb R, Wren JD, Jeffries M, et al. Variants within mecp2, a key transcription regulator, are associated with increased susceptibility to lupus and differential gene expression in patients with systemic lupus erythematosus. Arthritis and Rheumatism. 2009;60(4):1076–1084. doi: 10.1002/art.24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sestak AL, Furnrohr BG, Harley JB, Merrill JT, Namjou B. The genetics of systemic lupus erythematosus and implications for targeted therapy. Annals of the Rheumatic Diseases. 2011;70, supplement 1:37–43. doi: 10.1136/ard.2010.138057. [DOI] [PubMed] [Google Scholar]
- 95.Sawalha AH, Webb R, Han S, et al. Common variants within mecp2 confer risk of systemic lupus erythematosus. Plos One. 2008;3(3) doi: 10.1371/journal.pone.0001727. Article ID e1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fan Y, Tao JH, Zhang LP, Li LH, Ye DQ. Association of blk (rs13277113, rs2248932) polymorphism with systemic lupus erythematosus: a meta-analysis. Molecular Biology Reports. 2010;38(7):4445–4453. doi: 10.1007/s11033-010-0573-5. [DOI] [PubMed] [Google Scholar]
- 97.Grant SFA, Petri M, Bradfield JP, et al. Association of the BANK1 r61h variant with systemic lupus erythematosus in americans of european and african ancestry. Application of Clinical Genetics. 2008;2009, article 2:1–5. doi: 10.2147/tacg.s4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sanchez E, Comeau ME, Freedman BI, et al. Identification of novel genetic susceptibility loci in African American lupus patients in a candidate gene association study. Arthritis & Rheumatism. 2011;63(11):3493–3501. doi: 10.1002/art.30563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, lyp. Blood. 1999;93(6):2013–2024. [PubMed] [Google Scholar]
- 100.Bottini N, Musumeci L, Alonso A, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type i diabetes. Nature Genetics. 2004;36(4):337–338. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 101.Velaga MR, Wilson V, Jennings CE, et al. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (lyp) gene is a major determinant of graves’ disease. Journal of Clinical Endocrinology and Metabolism. 2004;89(11):5862–5865. doi: 10.1210/jc.2004-1108. [DOI] [PubMed] [Google Scholar]
- 102.Smyth D, Cooper JD, Collins JE, et al. Replication of an association between the lymphoid tyrosine phosphatase locus (lyp/ptpn22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes. 2004;53(11):3020–3023. doi: 10.2337/diabetes.53.11.3020. [DOI] [PubMed] [Google Scholar]
- 103.Skorka A, Bednarczuk T, Bar-Andziak E, Nauman J, Ploski R. Lymphoid tyrosine phosphatase (PTPN22/LYP) variant and Graves' disease in a Polish population: association and gene dose-dependent correlation with age of onset. Clinical Endocrinology. 2005;62(6):679–682. doi: 10.1111/j.1365-2265.2005.02279.x. [DOI] [PubMed] [Google Scholar]
- 104.Criswell LA, Pfeiffer KA, Lum RF, et al. Analysis of families in the multiple autoimmune disease genetics consortium (madgc) collection: the ptpn22 620w allele associates with multiple autoimmune phenotypes. American Journal of Human Genetics. 2005;76(4):561–571. doi: 10.1086/429096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vandiedonck C, Capdevielle C, Giraud M, et al. Association of the ptpn22∗r620w polymorphism with autoimmune myasthenia gravis. Annals of Neurology. 2006;59(2):404–407. doi: 10.1002/ana.20751. [DOI] [PubMed] [Google Scholar]
- 106.Dieude P, Guedj M, Wipff J, et al. The PTPN22 620W allele confers susceptibility to systemic sclerosis: findings of a large case-control study of European Caucasians and a meta-analysis. Arthritis & Rheumatism. 2008;58(7):2183–2188. doi: 10.1002/art.23601. [DOI] [PubMed] [Google Scholar]
- 107.LaBerge GS, Bennett DC, Fain PR, Spritz RA. PTPN22 is genetically associated with risk of generalized vitiligo, but CTLA4 is not. Journal of Investigative Dermatology. 2008;128(7):1757–1762. doi: 10.1038/sj.jid.5701233. [DOI] [PubMed] [Google Scholar]
- 108.Skinningsrud B, Husebye ES, Gervin K, et al. Mutation screening of ptpn22: association of the 1858t-allele with addison’s disease. European Journal of Human Genetics. 2008;16(8):977–982. doi: 10.1038/ejhg.2008.33. [DOI] [PubMed] [Google Scholar]
- 109.Betz RC, Konig K, Flaquer A, et al. The R620W polymorphism in PTPN22 confers general susceptibility for the development of alopecia areata. British Journal of Dermatology. 2008;158(2):389–391. doi: 10.1111/j.1365-2133.2007.08312.x. [DOI] [PubMed] [Google Scholar]
- 110.Hinks A, Barton A, John S, et al. Association between the ptpn22 gene and rheumatoid arthritis and juvenile idiopathic arthritis in a uk population: further support that ptpn22 is an autoimmunity gene. Arthritis and Rheumatism. 2005;52(6):1694–1699. doi: 10.1002/art.21049. [DOI] [PubMed] [Google Scholar]
- 111.Viken MK, Amundsen SS, Kvien TK, et al. Association analysis of the 1858c> t polymorphism in the ptpn22 gene in juvenile idiopathic arthritis and other autoimmune diseases. Genes and Immunity. 2005;6(3):271–273. doi: 10.1038/sj.gene.6364178. [DOI] [PubMed] [Google Scholar]
- 112.Seldin MF, Shigeta R, Laiho K, et al. Finnish case-control and family studies support ptpn22 r620w polymorphism as a risk factor in rheumatoid arthritis, but suggest only minimal or no effect in juvenile idiopathic arthritis. Genes and Immunity. 2005;6(8):720–722. doi: 10.1038/sj.gene.6364255. [DOI] [PubMed] [Google Scholar]
- 113.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for crohn’s disease. Nature Genetics. 2008;40(8):955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kariuki SN, Niewold TB. Genetic regulation of serum cytokines in systemic lupus erythematosus. Translational Research. 2010;155(3):109–117. doi: 10.1016/j.trsl.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vang T, Miletic AV, Bottini N, Mustelin T. Protein tyrosine phosphatase ptpn22 in human autoimmunity. Autoimmunity. 2007;40(6):453–461. doi: 10.1080/08916930701464897. [DOI] [PubMed] [Google Scholar]
- 116.Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in ptpn22 corresponds to altered function of t and b lymphocytes. Journal of Immunology. 2007;179(7):4704–4710. doi: 10.4049/jimmunol.179.7.4704. [DOI] [PubMed] [Google Scholar]
- 117.Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nature Reviews Rheumatology. 2010;6(12):683–692. doi: 10.1038/nrrheum.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Crispín JC, Apostolidis SA, Finnell MI, Tsokos GC. Induction of pp2a bβ, a regulator of il-2 deprivation-induced t-cell apoptosis, is deficient in systemic lupus erythematosus. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(30):12443–12448. doi: 10.1073/pnas.1103915108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nimmerjahn F, Ravetch JV. Fcγ receptors: old friends and new family members. Immunity. 2006;24(1):19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 120.Rudge EU, Cutler AJ, Pritchard NR, Smith KGC. Interleukin 4 reduces expression of inhibitory receptors on b cells and abolishes cd22 and fcγrii-mediated b cell suppression. Journal of Experimental Medicine. 2002;195(8):1079–1085. doi: 10.1084/jem.20011435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Robinson JI, Barrett JH, Taylor JC, et al. Dissection of the fcgr3a association with ra: increased association in men and with autoantibody positive disease. Annals of the Rheumatic Diseases. 2010;69(6):1054–1057. doi: 10.1136/ard.2009.110874. [DOI] [PubMed] [Google Scholar]
- 122.Bredius RG, de Vries CE, Troelstra A, et al. Phagocytosis of Staphylococcus aureus and Haemophilus influenzae type B opsonized with polyclonal human IgG1 and IgG2 antibodies. Functional hFc gamma RIIa polymorphism to IgG2. The Journal of Immunology. 1993;151(3):1463–1472. [PubMed] [Google Scholar]
- 123.Karassa FB, Bijl M, Davies KA, et al. Role of the Fcgamma receptor IIA polymorphism in the antiphospholipid syndrome: an international meta-analysis. Arthritis & Rheumatism. 2003;48(7):1930–1938. doi: 10.1002/art.11059. [DOI] [PubMed] [Google Scholar]
- 124.Tsuchiya N, Honda ZI, Tokunaga K. Role of b cell inhibitory receptor polymorphisms in systemic lupus erythematosus: a negative times a negative makes a positive. Journal of Human Genetics. 2006;51(9):741–750. doi: 10.1007/s10038-006-0030-4. [DOI] [PubMed] [Google Scholar]
- 125.Su K, Li X, Edberg JC, Wu J, Ferguson P, Kimberly RP. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. II. Differential binding of GATA4 and Yin-Yang1 transcription factors and correlated receptor expression and function. The Journal of Immunology. 2004;172(11):7192–7199. doi: 10.4049/jimmunol.172.11.7192. [DOI] [PubMed] [Google Scholar]
- 126.Kono H, Kyogoku C, Suzuki T, et al. Fcγriib ile232thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on b cell receptor signaling. Human Molecular Genetics. 2005;14(19):2881–2892. doi: 10.1093/hmg/ddi320. [DOI] [PubMed] [Google Scholar]
- 127.Floto RA, Clatworthy MR, Heilbronn KR, et al. Loss of function of a lupus-associated fcγriib polymorphism through exclusion from lipid rafts. Nature Medicine. 2005;11(10):1056–1058. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 128.Koene HR, Kleijer M, Swaak AJ, et al. The Fc gammaRIIIA-158F allele is a risk factor for systemic lupus erythematosus. Arthritis & Rheumatism. 1998;41(10):1813–1818. doi: 10.1002/1529-0131(199810)41:10<1813::AID-ART13>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 129.Alarcón GS, McGwin G, Petri M, et al. Time to renal disease and end-stage renal disease in profile: a multiethnic lupus cohort. Plos Medicine. 2006;3(10, article e396):1949–1956. doi: 10.1371/journal.pmed.0030396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Niederer HA, Willcocks LC, Rayner TF, et al. Copy number, linkage disequilibrium and disease association in the fcgr locus. Human Molecular Genetics. 2010;19(16):3282–3294. doi: 10.1093/hmg/ddq216. Article ID ddq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hatta Y, Tsuchiya N, Ohashi J, et al. Association of fcγ receptor iiib, but not of fcγ receptor iia and iiia, polymorphisms with systemic lupus erythematosus in japanese. Genes and Immunity. 1999;1(1):53–60. doi: 10.1038/sj.gene.6363639. [DOI] [PubMed] [Google Scholar]
- 132.Yuan H, Ni JD, Pan HF, Li LH, Feng JB, Ye DQ. Lack of association of FcgammaRIIIb polymorphisms with systemic lupus erythematosus: a meta-analysis. Rheumatology International. 2011;31(8):1017–1021. doi: 10.1007/s00296-010-1386-5. [DOI] [PubMed] [Google Scholar]
- 133.Franke A, McGovern DPB, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed crohn’s disease susceptibility loci. Nature Genetics. 2010;42(12):1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gregersen PK, Amos CI, Lee AT, et al. Rel, encoding a member of the nf-b family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nature Genetics. 2009;41(7):820–823. doi: 10.1038/ng.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stuart PE, Nair RP, Ellinghaus E, et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nature Genetics. 2010;42(11):1000–1004. doi: 10.1038/ng.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. Plos Biology. 2010;8(1, article 1000294) doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.So HC, Gui AH, Cherny SS, Sham PC. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genetic Epidemiology. 2011;35(5):310–317. doi: 10.1002/gepi.20579. [DOI] [PubMed] [Google Scholar]
- 138.Deapen D, Escalante A, Weinrib L, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis and Rheumatism. 1992;35(3):311–318. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 139.Alarcon-Segovia D, Alarcon-Riquelme ME, Cardiel MH, et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis & Rheumatism. 2005;52(4):1138–1147. doi: 10.1002/art.20999. [DOI] [PubMed] [Google Scholar]
- 140.Wang J, Yang S, Chen JJ, et al. Systemic lupus erythematosus: a genetic epidemiology study of 695 patients from china. Archives of Dermatological Research. 2007;298(10):485–491. doi: 10.1007/s00403-006-0719-4. [DOI] [PubMed] [Google Scholar]
- 141.Song S, Black MA. Microarray-based gene set analysis: a comparison of current methods. Bmc Bioinformatics. 2008;9, article no. 502 doi: 10.1186/1471-2105-9-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hedegaard J, Arce C, Bicciato S, et al. Methods for interpreting lists of affected genes obtained in a DNA microarray experiment. BMC Proceedings. 2009;3, supplement 4:p. S5. doi: 10.1186/1753-6561-3-s4-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Glas J, Seiderer J, Nagy M, et al. Evidence for stat4 as a common autoimmune gene: rs7574865 is associated with colonic crohn’s disease and early disease onset. Plos One. 2010;5(4) doi: 10.1371/journal.pone.0010373. Article ID e10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Martinez A, Varade J, Marquez A, et al. Association of the STAT4 gene with increased susceptibility for some immune-mediated diseases. Arthritis Rheum. 2008;58(9):2598–2602. doi: 10.1002/art.23792. [DOI] [PubMed] [Google Scholar]
- 145.Zhernakova A, Festen EM, Franke L, et al. Genetic analysis of innate immunity in crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring card9 and il18rap. American Journal of Human Genetics. 2008;82(5):1202–1210. doi: 10.1016/j.ajhg.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Leach ST, Messina I, Lemberg DA, Novick D, Rubenstein M, Day AS. Local and systemic interleukin-18 and interleukin-18-binding protein in children with inflammatory bowel disease. Inflammatory Bowel Diseases. 2008;14(1):68–74. doi: 10.1002/ibd.20272. [DOI] [PubMed] [Google Scholar]
- 147.Sato K, Shiota M, Fukuda S, et al. Strong evidence of a combination polymorphism of the tyrosine kinase 2 gene and the signal transducer and activator of transcription 3 gene as a dna-based biomarker for susceptibility to crohn’s disease in the japanese population. Journal of Clinical Immunology. 2009;29(6):815–825. doi: 10.1007/s10875-009-9320-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang K, Dickson SP, Stolle CA, Krantz ID, Goldstein DB, Hakonarson H. Interpretation of association signals and identification of causal variants from genome-wide association studies. American Journal of Human Genetics. 2010;86(5):730–742. doi: 10.1016/j.ajhg.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hugot JP, Chamaillard M, Zouali H, et al. Association of nod2 leucine-rich repeat variants with susceptibility to crohn’s disease. Nature. 2001;411(6837):599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 150.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in nod2 associated with susceptibility to crohn’s disease. Nature. 2001;411(6837):603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 151.Cho JH, Weaver CT. The genetics of inflammatory bowel disease. Gastroenterology. 2007;133(4):1327–1339. doi: 10.1053/j.gastro.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 152.Burton PR, Clayton DG, Cardon LR, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Anderson CA, Soranzo N, Zeggini E, Barrett JC. Synthetic associations are unlikely to account for many common disease genome-wide association signals. Plos Biology. 2011;9(1) doi: 10.1371/journal.pbio.1000580. Article ID e1000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee-Kirsch MA, Gong M, Chowdhury D, et al. Mutations in the gene encoding the 3′-5′ dna exonuclease trex1 are associated with systemic lupus erythematosus. Nature Genetics. 2007;39(9):1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 155.Crow YJ, Rehwinkel J. Aicardi-goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Human Molecular Genetics. 2009;18(2):R130–136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134(4):587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Crow YJ, Hayward BE, Parmar R, et al. Mutations in the gene encoding the 3′-5′ dna exonuclease trex1 cause aicardi-goutières syndrome at the ags1 locus. Nature Genetics. 2006;38(8):917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 158.Morita M, Stamp G, Robins P, et al. Gene-targeted mice lacking the trex1 (dnase iii) 3′→5′ dna exonuclease develop inflammatory myocarditis. Molecular and Cellular Biology. 2004;24(15):6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Goutières F, Aicardi J, Barth PG, Lebon P. Aicardi-goutieres syndrome: an update and results of interferon-α studies. Annals of Neurology. 1998;44(6):900–907. doi: 10.1002/ana.410440608. [DOI] [PubMed] [Google Scholar]
- 160.Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. Journal of the Neurological Sciences. 1988;84(2-3):201–208. doi: 10.1016/0022-510x(88)90125-6. [DOI] [PubMed] [Google Scholar]
- 161.Dale RC, Ping Tang S, Heckmatt JZ, Tatnall FM. Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics. 2000;31(3):155–158. doi: 10.1055/s-2000-7492. [DOI] [PubMed] [Google Scholar]
- 162.Crow MK. Developments in the clinical understanding of lupus. Arthritis Research and Therapy. 2009;11(5, article no. 245) doi: 10.1186/ar2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rice G, Patrick T, Parmar R, et al. Clinical and molecular phenotype of aicardi-goutières syndrome. American Journal of Human Genetics. 2007;81(4):713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.De Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, Hollis T. The crystal structure of trex1 explains the 3′ nucleotide specificity and reveals a polyproline ii helix for protein partnering. Journal of Biological Chemistry. 2007;282(14):10537–10543. doi: 10.1074/jbc.M700039200. [DOI] [PubMed] [Google Scholar]
- 165.Namjou B, Kothari PH, Kelly JA, et al. Evaluation of the trex1 gene in a large multi-ancestral lupus cohort. Genes and Immunity. 2011;12(4):270–279. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lee-Kirsch MA, Gong M, Schulz H, et al. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. American Journal of Human Genetics. 2006;79(4):731–737. doi: 10.1086/507848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Qari A, Al-Mayouf S, Al-Owain M. Mode of inheritance in systemic lupus erythematosus in saudi multiplex families. Genetic Counseling. 2009;20(3):215–223. [PubMed] [Google Scholar]
- 168.Lyon GJ, Jiang T, Van Wijk R, et al. Exome sequencing and unrelated findings in the context of complex disease research: ethical and clinical implications. Discovery Medicine. 2011;12(62):41–55. [PMC free article] [PubMed] [Google Scholar]
- 169.Rope A, Wang K, Evjenth R, et al. Using vaast to identify an x-linked disorder resulting in lethality in male infants due to n-terminal acetyltransferase deficiency. American Journal of Human Genetics. 2011 doi: 10.1016/j.ajhg.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.De Vries B, Steup-Beekman GM, Haan J, et al. Trex1 gene variant in neuropsychiatric systemic lupus erythematosus. Annals of the Rheumatic Diseases. 2010;69(10):1886–1887. doi: 10.1136/ard.2009.114157. [DOI] [PubMed] [Google Scholar]
- 171.Glessner JT, Wang K, Cai G, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–572. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Glessner JT, Reilly MP, Kim CE, et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(23):10584–10589. doi: 10.1073/pnas.1000274107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Levinson DF, Duan J, Oh S, et al. Copy number variants in schizophrenia: confirmation of five previous finding sand new evidence for 3q29 microdeletions and vipr2 duplications. American Journal of Psychiatry. 2011;168(3):302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Diskin SJ, Hou C, Glessner JT, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009;459(7249):987–991. doi: 10.1038/nature08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1(1):p. 4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Altshuler DL, Durbin RM, Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Conrad DF, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pang AW, MacDonald JR, Pinto D, et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biology. 2010;11(5, article no. R52) doi: 10.1186/gb-2010-11-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439(7078):851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 180.Fanciulli M, Norsworthy PJ, Petretto E, et al. Fcgr3b copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nature Genetics. 2007;39(6):721–723. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Molokhia M, Fanciulli M, Petretto E, et al. Fcgr3b copy number variation is associated with systemic lupus erythematosus risk in afro-caribbeans. Rheumatology. 2011;50(7):1206–1210. doi: 10.1093/rheumatology/keq456. Article ID keq456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Morris DL, Roberts AL, Witherden AS, et al. Evidence for both copy number and allelic (NA1/NA2) risk at the fcgr3b locus in systemic lupus erythematosus. European Journal of Human Genetics. 2010;18(9):1027–1031. doi: 10.1038/ejhg.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Seely AJE, Pascual JL, Christou NV. Science review: cell membrane expression (connectivity) regulates neutrophil delivery, function and clearance. Critical Care. 2003;7(4):291–307. doi: 10.1186/cc1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yamashiro S, Kamohara H, Wang JM, Yang D, Gong WH, Yoshimura T. Phenotypic and functional change of cytokine-activated neutrophils: inflammatory neutrophils are heterogeneous and enhance adaptive immune responses. Journal of Leukocyte Biology. 2001;69(5):698–704. [PubMed] [Google Scholar]
- 185.Gregory B, Kirchem A, Phipps S, et al. Differential regulation of human eosinophil IL-3, IL-5, and GM-CSF receptor alpha-chain expression by cytokines: IL-3, IL-5, and GM-CSF down-regulate IL-5 receptor alpha expression with loss of IL-5 responsiveness, but up-regulate IL-3 receptor alpha expression. The Journal of Immunology. 2003;170(11):5359–5366. doi: 10.4049/jimmunol.170.11.5359. [DOI] [PubMed] [Google Scholar]
- 186.Yu B, Shao Y, Yue X, et al. Copy number variations of interleukin-12b and t-bet are associated with systemic lupus erythematosus. Rheumatology. 2011;50(7):1201–1205. doi: 10.1093/rheumatology/keq439. Article ID keq439. [DOI] [PubMed] [Google Scholar]
- 187.Yu B, Shao Y, Li P, et al. Copy number variations of the human histamine h4 receptor gene are associated with systemic lupus erythematosus. British Journal of Dermatology. 2010;163(5):935–940. doi: 10.1111/j.1365-2133.2010.09928.x. [DOI] [PubMed] [Google Scholar]
- 188.Garcia-Ortiz H, Velazquez-Cruz R, Espinosa-Rosales F, Jimenez-Morales S, Baca V, Orozco L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Annals of the Rheumatic Diseases. 2010;69(10):1861–1865. doi: 10.1136/ard.2009.124313. [DOI] [PubMed] [Google Scholar]
- 189.Fernando MMA, de Smith AJ, Coin L, et al. Investigation of the hin200 locus in uk sle families identifies novel copy number variants. Annals of Human Genetics. 2011;75(3):383–397. doi: 10.1111/j.1469-1809.2011.00641.x. [DOI] [PubMed] [Google Scholar]