

Abstract
Bcl-2, a prosurvival protein, regulates programmed cell death during development and repair processes, and can be oncogenic when cell proliferation is deregulated. The present study investigated what factors modulate Bcl-2 expression in airway epithelial cells and identified the pathways involved. Microarray analysis of mRNA from airway epithelial cells captured by laser microdissection showed that increased expression of IL-1β and IGF-1 coincided with induced Bcl-2 expression compared to controls. Treatment of cultured airway epithelial cells with IL-1β and IGF-1 induced Bcl-2 expression by increasing Bcl-2 mRNA stability with no discernible changes in promoter activity. Silencing the IGF-1 expression using shRNA showed that intracellular (IC)-IGF-1 was increasing Bcl-2 expression. Blocking EGFR or IGF-1R activation also suppressed IC-IGF-1, and abolished the Bcl-2 induction. Induced expression and co-localization of IC-IGF-1 and Bcl-2 were observed in airway epithelial cells of mice exposed to LPS or cigarette smoke and of patients with cystic fibrosis and chronic bronchitis but not in the respective controls. These studies demonstrate that IC-IGF-1 induces Bcl-2 expression in epithelial cells via IGF-1R and EGFR pathways, and targeting IC-IGF-1 could be beneficial to treat chronic airway diseases.
Introduction
Bcl-2, a prosurvival protein, was first identified in the most common translocation in human B cell follicular lymphoma (1, 2) and was found to have oncogenic properties by inhibiting cell death rather than by promoting cell proliferation. Bcl-2 frequently overexpressed in cancer cells is associated with resistance of cytotoxic anticancer drugs and poor clinical outcome (3), and downregulation of Bcl-2 restores the intrinsic apoptotic pathways with antitumor effects (4). Bcl-2 plays a key role in normal cellular homeostasis by regulating the integrity of the mitochondrial and endoplasmic reticulum membranes and is involved in an array of apoptotic and autophagic programs during development and repair processes (5, 6). Particularly, Bcl-2 is essential for developmentally programmed cell death of renal epithelial progenitors, and of mature B and T lymphocytes (5) and for epithelial injury and repair processes (7) that can lead to chronic inflammatory diseases. Pulmonary inflammation induced by viral or bacterial infections or by chronic exposure to environmental pollutants initiates airway epithelial cell proliferation and induced expression of Bcl-2 to sustain proliferating epithelial cells (8, 9). Gain- and loss-of-function studies showed that Bcl-2 expression sustains hyperplastic epithelial cells, and Bcl-2 expression is elevated in airway mucous cells of subjects with cystic fibrosis (7) and in airway epithelium of asthmatics (10). Therefore, identification of inflammatory mediators that regulate Bcl-2 expression in airway epithelial cells may provide useful targets for developing novel intervention strategies.
Several cytokines including TNF receptor-associated factors link transmembrane receptors to the NF-κB pathway to increase Bcl-2 expression (11–13). Both, IL-6 and IL-7 up-regulate Bcl-2 to increase survival in T lymphocytes by activating STAT3 (14) or viability and proliferation of T cell acute lymphoblastic leukemia cells, respectively (15). IL-3, IL-4, and IGF-I increase Bcl-2 expression to protect promyeloid cells from apoptosis (16). In contrast, IL-2 limits continued T-cell expansion by reducing Bcl-2 expression and rendering cells susceptible to apoptotic cell death (17) and TGF-β controls effector CD8+ T cell numbers by lowering Bcl-2 expression and selectively promoting apoptosis of short-lived effector cells (18). While these studies establish the role of cytokines and growth factors in hematopoietic cells, the inflammatory mediator(s) that regulate Bcl-2 expression in airway epithelial cells are unknown.
Recently, EGFR-dependent pathway was associated with epithelial cell survival and shown to be critical for remodeling of airway epithelium toward chronic mucous hypersecretory phenotype, but the mechanism of epithelial cell survival was not identified (19). Thus, this study also investigated whether Bcl-2 expression is involved in the EGFR-mediated airway epithelial cell survival mechanisms.
Materials and Methods
Human subjects and lung tissue analysis
Lung tissue specimens from subjects with cystic fibrosis were obtained under protocols approved by the University of North Carolina at Chapel Hill Office of Human Research Ethics Institutional Review Board. The lung specimens with the CFTR mutation were rapidly procured from individuals undergoing lung transplantation for end stage lung disease and specimens from non-CF controls were excess portions of donor lungs that were trimmed for size matching. Demographics of these subjects are shown in Table 1. Specimens were fixed by immersion in neutral buffered formalin and conventionally processed for paraffin embedding and sectioning. The lung tissue specimens from subjects diagnosed with chronic bronchitis and non-diseased controls (Table 2) were obtained from Lung Tissue Research Consortium (LTRC) that is sponsored by the National Heart Lung and Blood Institute.
Table 1.
Demographics of subjects with and without cystic fibrosis
| No CF (n=3) |
CF (n=4) |
|
|---|---|---|
| Age* | 35.3 ± 21.7 | 29.3 ± 9.5 |
| Gender, M/F | 3/0 | 4/0 |
| Diagnosis | N/A | 3 DF/DF 1, DF/621+1G>T |
Mean ± SD; CF = cystic fibrosis; M = male; F = female; DF= deletion of phenylalanine; G > T = transversion mutation
Table 2.
Demographics of former smokers with or without chronic bronchitis
| No CB (n=5) |
CB (n=4) |
|
|---|---|---|
| Age* | 62.4 ± 4.3 | 59.5 ± 10.2 |
| Gender, M/F | 2/3 | 4/0 |
| Smoking packs/y* | 36.8 ± 11.9 | 51.8 ± 28.8 |
| Stop Smoking (Y)* | 13.0 ± 6.0 | 10.5 ± 10.0 |
Mean ± SD; CB = chronic bronchitis; M = male; F = female; PY = Pack per Year; Y = years
Laboratory animals
Specific pathogen-free F344/NCrR male rats of 6–8 wk of age were obtained from NCI (Frederick, MD) and were housed until 8–10 weeks of age. The rats were housed in pairs and were provided food and water ad libitum, a 12-h light/dark cycle at 22.2°C, and 30–40% humidity. Rats were weighed and randomly assigned to each experimental group. Pathogen-free wild-type C57BL/6J mice were purchased from The Jackson Laboratory. Mice were housed in isolated cages under specific pathogen-free conditions. All experiments were approved by the Institutional Animal Care and Use Committee and were conducted at Lovelace Respiratory Research Institute, a facility approved by the Association for the Assessment and Accreditation for Laboratory Animal Care International.
Rat intratracheal instillation
For intratracheal instillations, rats were briefly anesthetized with 5% isoflurane in oxygen and instilled intratracheally with 1000 µg of LPS (Pseudomonas aeruginosa serotype 10, lot 31K4122, 3,000,000 LPS units (EU)/mg, Sigma, St. Louis, MO) in 0.5 ml of 0.9% pyrogen-free saline. Control rats were instilled with 0.5 ml of 0.9% pyrogen-free saline.
LCM and microarray analyses
The right lungs of F344/NCrR male rats at 0 and 2 d post LPS instillation were snap-frozen, and stored at −80°C after inflating with diluted (1:4 in PBS) Tissue-Tek O.C.T. (EMS Biosciences, Hatfeild, PA). Frozen tissue sections (8 µm thick) were fixed, dehydrated, air-dried, and epithelial cells from five large airways of each rat were captured using the laser onto Arcturus® CapSure® HS LCM Caps (Applied Biosystems, Foster City, CA). Total RNA was extracted from the cellular lysate in the cap using the PicoPure® RNA Isolation Kit (Applied Biosystems) and mRNA were amplified using the RiboAmp RNA amplification kit (Applied Biosystems) as recommended by the manufacturer’s instructions. Epithelia microdissected from eight tissue sections resulted in an average 1000 ng of amplified RNA. A second round of amplification was carried out and the biotinylated cRNA probes were then prepared. Microarray analysis was performed using a rat microarray chip RG-U34A (Affymetrix Inc., Sunnyvale, CA). All datasets have been deposited at National Center for Biotechnology Information/Gene Expression Omnibus under accession number GSE36174 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36174).
Cell culture treatments and quantitative RT-PCR
Immortalized rat airway epithelial cells, SPOC-1 cells were maintained as described (20, 21). The human airway epithelial cells, AALEB cells (22) and primary human airway epithelial cells (HAECs, Clontech, Walkersville, MD) were maintained in bronchial epithelial growth medium (BEGM, Lonza, Walkersville, MD). The SPOC-1 cells were treated with rat IL-1β (5 ng/ml), IL-6 (1 ng/ml), IL-9 (0.01 ng/ml), VEGF (5 ng/ml), TNFα (1 ng/ml), IGF-1 (100 ng/ml) or left untreated. The AALEBs and HAECs were treated with human IGF-1 and human IL-1β (R&D systems, Minneapolis, MN). Cells were also pre-treated with Picropodophyllin or PPP (Santa Cruz Biotechnology, Santa Cruz, CA), an inhibitor of IGF-1R activation or various inhibitors of EGFR tyrosine kinase activation, AG1478 (Santa Cruz Biotechnology, Santa Cruz, CA), EKB-569 or PD153035 (Selleck Chemicals, Houston, TX) for 2 h with no effect on cell viability prior to the treatment with IGF-1 or IL-1β. RNA was isolated from the snap-frozen right lungs of animals using TRIzol as described previously (22) whereas RNA from cultured cells was extracted using the RNeasy kit (Qiagen, Valencia, CA). The primer/probe sets for IL-1b, IGF-1, Bcl-2, MUC5AC and CDKN1B were obtained from Applied Biosystems (Foster City, CA) and were amplified by quantitative real-time PCR using RT-PCR Master Mix (Applied Biosystems, Foster City, CA) in the ABIPRISM 7900HT Real-Time PCR System. Relative quantities were calculated by normalizing averaged CT values to CDKN1B or 18S to obtain ΔCt, and the relative standard curve method was used for determining the fold change as described previously (23).
Western blot analysis
Protein was extracted from the right lungs of LPS instilled rats by homogenization in RIPA buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, and 5 mM EDTA) supplemented with a protease inhibitor cocktail (Sigma Chemical Co., St. Louis, MO) at 1:100 final concentration. Protein concentration was determined using the BCA kit (Pierce, Thermo Fisher Scientific, Rockford, IL) and 100–150 µg of protein lysate was analyzed by Western blotting. The Bcl-2, IGF-1, EGFR and β-actin were detected using appropriate peroxidase-conjugated secondary antibodies and visualized by chemiluminescence (Perkin Elmer, Waltham, MA) using the FujiFilm Image Reader LAS-4000 (Valhalla, NY).
Immunofluorescent staining and image analysis
Lung sections were deparaffinized, hydrated in graded ethanol and deionized water, then washed in 0.05% v/v Brij-35 in Dulbecco's PBS (pH 7.4). The antigens were unmasked by treating with Digest-All kit (Zymed Laboratories, San Francisco, CA) at a 1:3 dilution of trypsin to diluent at 37°C for 10 min. Sections were then blocked using 0.2% Triton X-100 with 0.2% Saponin in a blocking solution containing 3% IgG-free BSA, 1% Gelatin and 2% normal donkey serum followed by incubation with anti-Bcl-2 (N19, Santa Cruz Biotech, CA), anti-IGF-1 (H70, Santa Cruz Biotech, CA), or isotype controls at 1:200 dilution. The immunolabeled cells were detected using F(ab)2-fragments of respective secondary antibodies conjugated to either DylightTM-549 or DylightTM-649 (Jackson Immunoresearch, West Grove, PA) at 1:1000 dilution and mounted with 4',6-diamidino-2-phenylindole (DAPI) containing Fluormount-GTM (SouthernBiotech, Birmingham, AL) for nuclear staining. For cytometry, cells were grown on Lab-Tek-II 8-chamber slides (Nalge Nunc International, Rochester, NY) and treated with 100 ng/ml of IGF-1 or 5ng/ml of IL-1β or were left untreated and were fixed using 3% paraformaldehyde with 3% sucrose in PBS and processed for immunostaining as described above. Quantification of Bcl-2-positive and IGF-1-positve cells per mm of basal lamina was performed using the VisioMorph system (Visiopham A/S, Horsholm, Denmark).
mRNA half-life analysis
Cells were treated with the RNA polymerase inhibitor 5,6-dichloro-1-beta-D-ribobenzimidazole (DRB) (Sigma-Aldrich, St. Louis, MO) at a final concentration of 100ng/ml to block RNA polymerase activity and harvested at 0, 0.5, 1, 2, 4, 6, 8, 12, and 24 h post-treatment. The relative mRNA abundance was calculated using the ∆∆Ct method, and mRNA half-life was calculated using the Greenberg formula (24).
Bcl-2 promoter constructs and transfection
The generation of the P1–P2, P1, and P2 luciferase constructs has been previously described (25). The P1 promoter region is the main driving force for transcribing the Bcl-2 gene and P1 activity is suppressed by the P2 region (25). The P2 region (681 bp) was cloned in pCR2.1 vector to obtain P2-TA construct and various deletion mutants were generated by using standard techniques. The full-length mutant P2 construct (mut-P2) was generated by purine-pyrimidine substitutions (Mutagenex Inc., NJ) to fully destroy the consensus Brn3a-interacting region at residues 93–99. All transient transfections were carried out in 24 well-plates (USA Scientific, Orlando, FL) using Fugene 6.0 reagent (Roche Applied Biosystems, Indianapolis, IN) as described previously (25).
LPS instillation
Male C57BL/6 mice at 8–10 wks of age were briefly anesthetized with 5% isoflurane in oxygen and instilled intranasally with 60 µg of LPS (Pseudomonas aeruginosa serotype 10, lot 31K4122, 3,000,000 LPS units (EU)/mg, Sigma, St. Louis, MO) in 0.05 ml of 0.9% pyrogen-free saline. Control mice were instilled with 0.05 ml of 0.9% pyrogen-free saline. Mice were sacrificed 3 d post-instillation and lung tissues were processed and analyzed.
Exposure to cigarette smoke
Male C57BL/6 mice at 8–10 wks of age were exposed whole-body to mainstream CS or filtered air (FA) as described previously (26). Mice were acclimated to the CS exposure at 100 mg/m3 of total particulate material (TPM) for the first week and then exposed to CS concentration at 250 mg/m3 TPM for the following 22 wk. CS was generated from 2R4F filtered research cigarettes (University of Kentucky Tobacco Research and Development Center, Lexington, KY) using the AMESA Type 1300 smoking machine. Mice were sacrificed post-exposure and lung tissues were processed and analyzed.
IGF-1 gene silencing by shRNA transfection
AALEB cells were transfected with IGF-1 shRNA containing retroviral vectors or control vectors (Origene Technologies, Inc., Rockville, MD) as per manufacturer’s instructions. After infection with IGF-1 or control shRNAs, cells were treated with 20 ng/ml human IL-1β, and 48 h later cells were assessed for IGF-1 expression.
Statistical analysis
Grouped results were expressed as means ± SEM. Data were analyzed using GraphPad Prism Software (GraphPad Software, Inc., San Diego, CA). Grouped results were analyzed using two-way analysis of variance. When significant main effects were detected (P < 0.05), Fishers least significant difference test was used to determine differences between groups.
Results
Microarray analysis of laser-capture microdissected airway epithelia
Previously, we observed increased number of Bcl-2-positive epithelial cells in rats exposed to LPS, specifically on day 2 post LPS challenge (7). Thus, to identify the mediators involved in upregulation of Bcl-2 expression, we microdissected airway epithelial cells by laser-capture microscopy (Fig. 1A) from lungs of non-instilled rats or 2 d post LPS-exposure. Microarray profiling of the mRNA isolated from the dissected epithelial cells on rat RGU34A chips revealed 800 differentially expressed genes (p<0.05), and the heat map of the differentially expressed genes displayed consistent gene expression patterns among individual rats in the same treatment group (Fig. 1B). Among the 800 genes, there were 137 growth factors, 51 cytokines and 44 apoptosis-related genes (Fig. 1C). The most significantly induced genes (2–6-fold) were IGF-1, IL-1α, IL-1β, eotaxin, MCP-1, MIP-2, and MIP-3α; however, IGFBP3 levels were downregulated by 3-fold in LPS-treated rats. Highly induced expression of IGF-1 and IL-1β along with that of elevated MUC5AC levels was validated by qRT-PCR (Fig. 1D). Consistent with previous observations, Bcl-2 mRNA was also found to be 1.9-fold higher in LPS-exposed rats compared to non-instilled controls (Fig. 1D).
FIGURE 1. Microarray analysis of laser-capture microdissected (LCM) rat airway epithelia.

(A) Light micrographs of the rat large airways before and after laser-capture microdissection of epithelial cells. (B) Venn-diagram clustering of genes expression pattern obtained by microarray analysis of LCM-captured epithelial cells from LPS-instilled and control rats. (C) Heat-map displaying differential gene expression pattern (p<0.05) of microdissected airway epithelia mRNA from rats instilled with LPS or non-instilled controls (n=3 rats per group). (D) Relative quantities of Bcl-2, MUC5AC, IGF-1, and IL-1β mRNA levels in LCM-captured epithelial cells from LPS-instilled rats compared to control rats as quantified by qRT-PCR.
IL-1β and IGF-1 induce Bcl-2 expression in airway epithelial cells
We first tested the effect of the candidate inflammatory factors, IL-1β and IGF-1, on Bcl-2 expression by treating AALEB cells, a human airway epithelial cell line, with recombinant human IL-1β and IGF-1. A significant induction of Bcl-2 mRNA levels was consistently observed following IGF-1 (Fig. 2A) and IL-1β (Fig. 2B) treatments. Similar results were observed in SPOC-1 cells, a rat airway epithelial cell line, following treatment with recombinant rat IL-1β and IGF-1 (Fig. S1). Accordingly, 100 ng/ml of IGF-1 and 5 ng/ml of IL-1β were chosen for further analysis and resulted in increased Bcl-2 protein levels as analyzed by Western blot (Fig. 2C) and immunofluorescent staining (Fig. 2D) analyses.
FIGURE 2. IL-1β and IGF-1 induce Bcl-2 expression in airway epithelial cells.
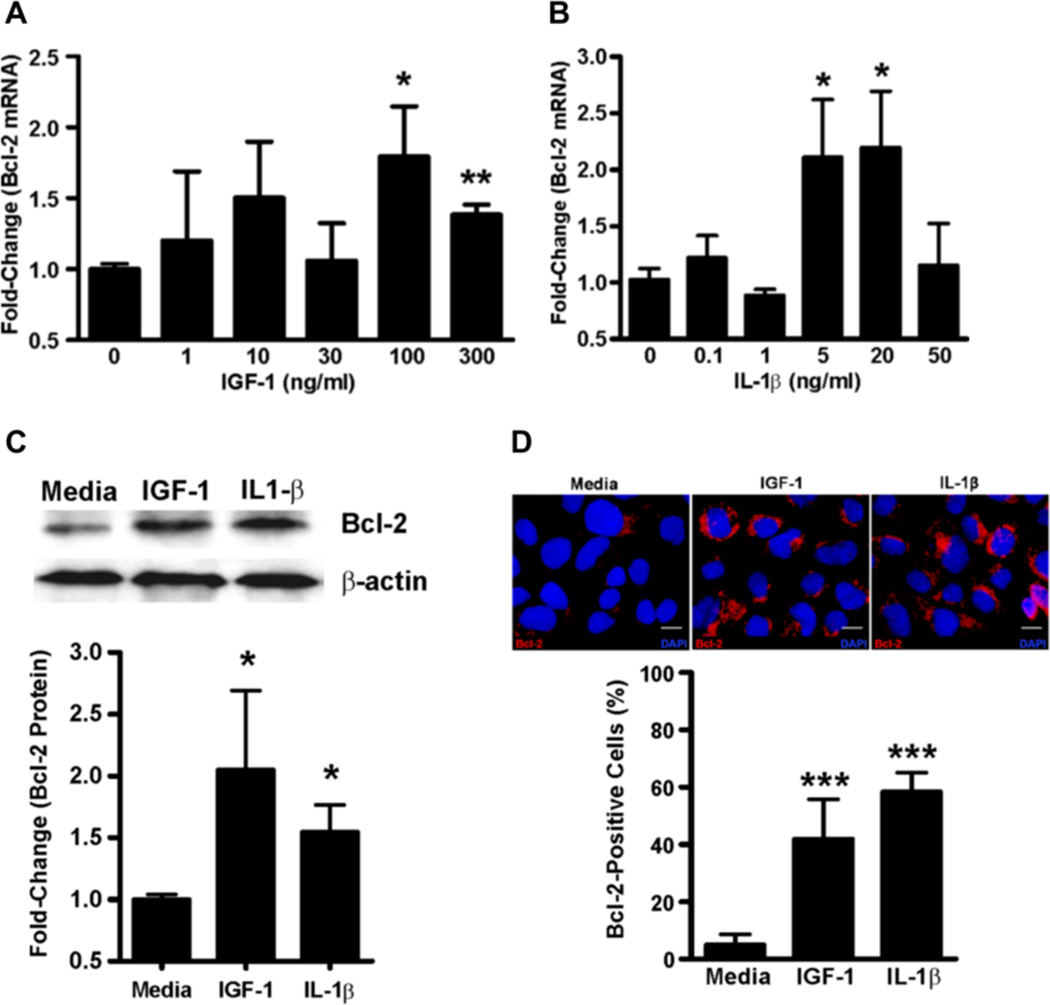
Bcl-2 mRNA levels in AALEB cells 24 h after treatment with rhIGF-1 (A) or rhIL-1β (B) as quantified by qRT-PCR. (C) Bcl-2 protein levels in AALEBs following treatment with IGF-1 (100 ng/ml) and IL-1β (5 ng/ml) compared to the non-treated cells and normalized to the β-actin levels (n=3). The upper panel shows a representative immunoblot. (D) In-situ analysis of Bcl-2 expression in AALEB cells by immunofluorescence following IGF-1 (100 ng/ml) and IL-1β (5 ng/ml) treatment. The Bcl-2-positive and total cells were counted 24 h after treatment and data expressed as the percent of Bcl-2 positive cells (n=3), results shown as mean ± SEM. * p<0.05, ** p<0.01, *** p<0.001. Scale bars, 10 µm.
Intracellular IGF-1 mediates Bcl-2 upregulation via EGFR and IGF-1R pathways
EGFR pathway has been shown to be essential in cell survival and proliferation pathways, therefore we tested whether IL-1β- and IGF-1-medaited induction of Bcl-2 requires EGFR pathway. For this purpose, AALEB cells were treated with IGF-1 or IL-1β in the presence and absence of a specific EGFR kinase inhibitor, AG1478. The pretreatment of cells with AG1478 abolished the IGF-1- and IL-1β-mediated increase in Bcl-2 mRNA levels (Fig. 3A). Pretreatment with other EGFR kinase inhibitors, PD153035 and EKB-569, a potent irreversible inhibitor, also showed identical results (Fig. S2). Because IL-1β was previously shown to induce IGF-1 (27), we hypothesized that IL-1β mediates IGF-1 and Bcl-2 expression. To test this hypothesis, AALEB cells were pretreated with specific IGF-1R tyrosine kinase inhibitor, PPP followed by IGF-1 or IL-1β treatment. Both IGF-1 and IL-1β-medaited increase in Bcl-2 mRNA levels was abolished with PPP pretreatment (Fig. 3A).
FIGURE 3. Intracellular IGF-1 mediates IL-1β- and IGF-1-induced Bcl-2 expression using both EGFR and IGF-1R pathways.

(A) Bcl-2 mRNA levels in AALEBs treated with IGF-1 and IL-1β in the presence or absence of PPP (300 nM) or AG1478 (1 µM), tyrosine kinase inhibitors for IGF-1R and EGFR, respectively (n=4). (B) Immunofluorescence analysis of intracellular-IGF-1-positive (IC-IGF-1 pos) and Bcl-2-positive (Bcl-2 pos) AALEB cells following treatments with IGF-1 or IL-1β in the presence or absence of PPP or AG1478 (n=4). Representative micrographs showing AALEB cells expressing IC-IGF-1 (left panels, in red), Bcl-2 (middle panels, in green), and merged image (right panels) denoting co-expression of IC-IGF-1 and Bcl-2 following IGF-1(upper panels) and IL-1β (lower panels) treatments. (C) Changes in the percentage of Bcl-2-positive cells in IGF-1 or IL-1β treated AALEB cells at 1, 2, 3, 4, 8 and 12 h (n=4). (D) Analysis of IGF-1 and Bcl-2 expression following IL-1β treatment in AALEBs with blocked IGF-1 expression (n=4). The AALEB cells transfected with shIGF-1 (shRNA specific for IGF-1) and shCon (control shRNA) constructs were treated with IL-1β (5 ng/ml) for 48 h before co-immunostaining for IGF-1 (red) and Bcl-2 (green). The percent of IC-IGF-1- and Bcl-2-positive cells infected with shCon and shIGF-1 retroviral vectors were quantified. (E) In-vivo analysis of Bcl-2 expression in rat airway epithelial cells following intratracheal instillation of rat IGF-1. The micrographs show bronchial epithelium of proximal airway (G-5) from vehicle-instilled (left panel) and IGF-1-instilled (right panel) rat with arrow indicating Bcl-2-positive (red) cell. The numbers of Bcl-2 positive cells per mm of basal lamina were counted (n=8 per group). Results shown as mean±SEM; * p<0.05; ** p<0.01; *** p<0.001; # p<0.05 compared to IGF-1-treated group; ψ p<0.05 compared to IL-1β-treated group; scale = 10µ.
Furthermore, both IGF-1 and IL-1β treatment increased the immunopositivity for intracellular (IC)-IGF-1 and Bcl-2 by 4–5-fold and IC-IGF-1 and Bcl-2 were co-localized in the same cells (Fig. 3B). Bcl-2 was absent in cells lacking IC-IGF-1 expression and pretreatment with either AG1478 or PPP suppressed this effect (Fig. 3B). Similar results were obtained when IGF-1R and EGFR were inhibited in SPOC-1 cells treated with rat IGF-1 and rat IL-1β (Fig. S3).
In order to understand the kinetics of Bcl-2 induction, we determined the expression of Bcl-2 in HAECs by immunofluorescence staining of cells treated with IGF-1 or IL-1β for 1, 2, 3, 4, 8 and 12 h. Compared to untreated cells, both IGF-1 and IL-1β treatments showed increase in Bcl-2-posiitve cells already at 1 h post-treatment with further increases in Bcl-2-positive cells at later time-points (Fig. 3C). However, there were no significant differences between the two treatments. The role of IC-IGF-1 in inducing Bcl2 expression was investigated by blocking its expression using shRNA specific for IGF-1 (shIGF-1). The IL-1β-induced expression of both IC-IGF-1 and Bcl-2 were completely suppressed compared to control shRNA transfected cells (Fig. 3D).
Because IC-IGF-1 appeared to be central in increasing Bcl-2 expression, we examined whether IGF-1 induces Bcl-2 expression in airway epithelial cells in vivo, by instilling rats with IGF-1 intratracheally and harvesting lungs at 24h for immunostaining. Bcl-2-positivity of airway epithelial cells was increased by 2-fold compared to vehicle-instilled group (Fig. 3E). Thus, IGF-1 induced Bcl-2 expression in airway epithelial cells both in-vitro and in-vivo.
Collectively, these data suggest that Bcl-2 expression is upregulated by IC-IGF-1 via convergence of EGFR and IGF-1R pathways.
IGF-1 increases Bcl-2 mRNA stability
We tested whether IGF-1 induces Bcl-2 promoter activity by transfecting AALEB cells with various Bcl-2 promoter luciferase reporter constructs described earlier (25). However, no changes in luciferase activity were observed following IGF-1 treatment (Fig. S4 A), suggesting that transcriptional activity was not affected. However, IGF-1 increased Bcl-2 mRNA half-life by ~4.6-fold in AALEB cells (Fig. 4A). Because the Bcl-2 mRNA contains the P2 region upstream of the transcriptional start site (25), we investigated whether merely transfection with plasmids containing this upstream region may compete for mRNA degrading factors and thereby affect Bcl-2 mRNA levels in IGF-1-treated cells. Interestingly, cells transfected with the constructs containing only the P2 promoter region consistently showed enhanced increase in Bcl-2 mRNA levels compared to those transfected with empty vector when treated with IGF-1 (Fig. 4B). Similar results were obtained when cells transfected with P2 promoter constructs were treated with IL-1β (Fig. S4 B). These highly reproducible findings suggested that P2 region may interact with mRNA destabilizing factors that facilitate the IGF-1-mediated stability of Bcl-2 mRNA.
FIGURE 4. IGF-1 induces Bcl-2 expression by increasing Bcl-2 mRNA stability in a mechanism that involves the Bcl-2 P2 promoter region.

(A) The calculated half-life of Bcl-2 mRNA in AALEB cells in the presence and absence of rhIGF-1 (n=3) as described in Material and Methods. (B) Bcl-2 mRNA levels in AALEB cells treated with nothing as control or with IGF-1 and transfected with nothing, empty vector, or a vector carrying the P2 construct (681 bp). Transfected and non-transfected (NT) cells were treated with IGF-1 for 4 h and Bcl-2 mRNA levels were analyzed by qRT-PCR and were compared to the media-treated NT cells (denoted by empty bar) (n=4). (C) Bcl-2 mRNA levels following IGF-1 treatment of AALEB cells transfected with various deletion-constructs of P2-promoter region and a mutant P2 construct (mutP2). Left panel illustrates the full-length P2 construct, various deletion-constructs, and the mutant P2 construct (mutP2); the detailed description of constructs is in Material and Methods. All cells were treated with rhIGF-1 for 4 h and Bcl-2 mRNA determined by qRT-PCR was normalized to empty TA-cloning vector-transfected cells (n=4). Data shown as mean±SEM; * p<0.05, ** p<0.01.
In order to identify the minimum possible sub-region within P2 that was responsible for facilitating IGF-1-induced Bcl-2 mRNA stability, we generated various P2 deletion-constructs and a mutant P2 (mut-P2) construct with substitutions at 93–99 region that would destroy the Brn3a-interacting site (28) (Fig. 4C). Following IGF-1 treatment, the cells transfected with D1 (1–356) and D2 (1–126) deletion constructs showed IGF-1-induced increase in Bcl-2 mRNA while cells transfected with D3 deletion construct (1–106) and mut-P2 construct failed to show any change (Fig. 4C). These findings suggest that the region 91–108 previously shown to interact with Brn3a (29) is involved with IGF-1-induced stabilization of Bcl-2 mRNA.
IC-IGF-1 and Bcl-2 are co-expressed in epithelial cells of chronic inflammatory airways
We next tested whether IC-IGF-1 and Bcl-2 are expressed in animal models, and more importantly, in subjects with chronic airway diseases. As reported earlier (7–9), Bcl-2 mRNA levels were increased significantly in LPS- compared to saline-instilled rats (Fig. 5A). The airway epithelial cells when analyzed for IC-IGF-1 and Bcl-2 immunopositivity showed a 9- and 11-fold increase, respectively, in LPS-instilled mice compared to saline-instilled controls (Fig. 5B). In addition, in cystic fibrosis (CF) the percentage of Bcl-2 positive epithelial cells is significantly increased compared to non-diseased controls (7). Because the present study shows that IC-IGF-1 is crucial for inducing Bcl-2 expression, we immunostained airway tissues from CF and controls with antibodies to IC-IGF-1 and Bcl-2. We found that in subjects with CF the number of IC-IGF-1- and Bcl-2-positive AECs/mm BL was increased 3- and 7-fold compared to normal subjects, respectively (Fig. 5C).
FIGURE 5. IC-IGF-1- and Bcl-2-positivity in airway epithelia of lungs with chronic inflammation.
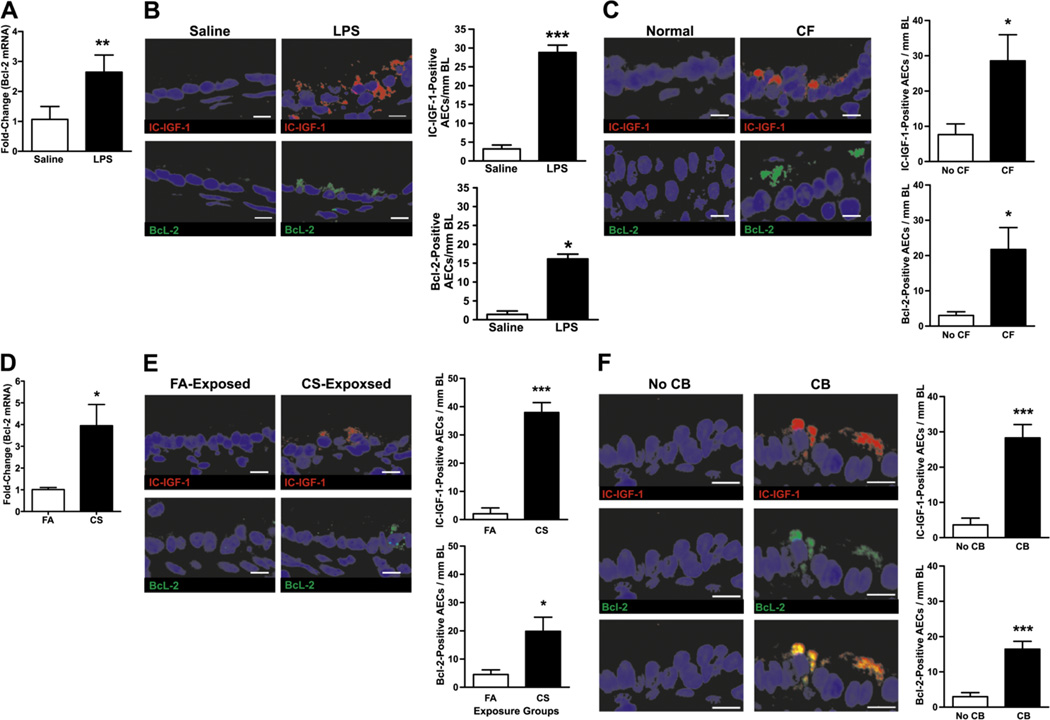
(A) Bcl-2 mRNA levels in lung tissues of mice instilled with LPS or saline as quantified by qRT-PCR. (B) Analysis of IC-IGF-1- and Bcl-2-positive airway epithelial cells in lung tissues of mice instilled with LPS or saline. Mouse lung tissues harvested 3 d post LPS or saline instillation. (C) IC-IGF-1- and Bcl-2-positive epithelial cells in airways of subjects with cystic fibrosis (CF) or no lung disease. (D) Bcl-2 mRNA levels in lung tissues of mice exposed to cigarette smoke (CS) or filtered air (FA) controls for 22 wks. (E) IC-IGF-1 and Bcl-2-positive airway epithelial cells in lung tissues of mice exposed to CS or FA. (F) IGF-1- and Bcl-2-positive airway epithelial cells in lung tissue from former-smokers with chronic bronchitis (CB) compared to former-smokers with no CB. Lung tissue sections from former-smokers with no CB (left panels) and with CB (right panels) were co-immunostained for IGF-1 (top-panels) and Bcl-2 (middle panels). The bottom panels illustrate the merged image showing the co-expression of IGF-1 and Bcl-2 in airway epithelial cells of CB (n=4 subjects/group). For B, C, E and F, lung tissue sections were immunostained for IC-IGF-1 (top-panels) and Bcl-2 (bottom panels) and number of positive-cells per mm basal lamina were quantified (n=4 /group). Results shown as mean±SEM. * p<0.05; *** p<0.0001; scale = 10µ.
In a mouse model of chronic cigarette smoke (CS) exposure we observed a 4-fold increase in Bcl-2 mRNA levels in CS-exposed mice compared to filtered-air (FA) controls (Fig. 5D). When airway tissues were analyzed by immunofluorescence we observed a 7- and 4-fold increase in the IC-IGF-1- and Bcl-2-positive cells in CS-exposed mice compared to FA-exposed mice, respectively (Fig. 5E). Similarly, in lung tissues from former smokers with or without chronic bronchitis, IC-IGF-1- and Bcl-2-positive cells were 7- and 5-fold increased, compared to subjects with no chronic bronchitis, respectively. As was observed for cells in culture, IC-IGF-1 and Bcl-2 were colocalized in airway epithelial cells (Fig. 5F).
Discussion
The present study identifies IL-1β and IGF-1 as the principal inflammatory mediators that induce Bcl-2 expression in airway epithelial cells. Induction in Bcl-2 expression involved both EGFR and IGF-1R pathways, and required the expression of ‘intracellular’ IGF-1. Several observations suggest that not the ‘secreted IGF-1’ but the intracellular(IC)-IGF-1 mediates induction in Bcl-2 expression. First, IGF-1 treatment of airway epithelial cells increased IC-IGF-1 and Bcl-2 levels both in-vitro and by intratracheal instillation of rats in-vivo. Second, immunofluorescent analysis of IL-1β-treated cells showed that IC-IGF-1 and Bcl-2 are co-localized in the same cells, while cells without IGF-1-positivity were Bcl-2-negative. Third, IL-1β-treatment of airway epithelial cells resulted in robust IC-IGF-1 expression and blocking IC-IGF-1 expression using shRNA suppressed Bcl-2 expression. Collectively, these data show that IL-1β and IGF-1 lead to induced Bcl-2 expression via upregulating IC-IGF-1. The mechanism by which IL-1β induces IC-IGF-1 is not clear. However, similar results were observed in prostate epithelial cells and in cardiac cells, where IL-1β induces anti-apoptotic and proliferative phenotype by upregulating IGF-1 transcription using the Janus activated kinase-signal transducer and activator of transcription (JAK-STAT) pathway (27, 30). The molecular mechanisms by which IL-1β induces IC-IGF-1 in airway epithelial cells are yet to be defined
In airway epithelial cells, EGFR pathway has been identified as a common denominator for proinflammatory responses that aide in epithelial cell hyperplasia and mucous cell metaplasia (19, 31–33). Tyner et al. (19) proposed that the EGFR pathway is associated with survival of airway epithelial cells in response to a viral challenge. However, the mechanism of EGFR-induced survival airway epithelial cells was not identified. Our data show that EGFR activation by inflammatory mediators result in induced Bcl-2 expression and present a new paradigm that EGFR activation sustains airway epithelial cells by inducing the antiapoptotic protein, Bcl-2. These findings are consistent with other studies which report TGFα-induced activation of EGFR mediates Bcl-2 induction in NCI-H292 cells (34) and in gastric mucosal cells (35). Moreover, the external stimuli such as exposure to ozone, LPS, CS, or allergen sensitization that upregulate Bcl-2 expression (36) are also known to activate EGFR in airway epithelial cells (37, 38). EGFR and IGF-1R pathways and their ligands have been shown to cross-talk, but mostly in transformed cells, with IGF-1 induction resulting in secretion of the EGFR ligands, TGFα and amphiregulin, and blocking of EGFR activation abolished the IGF-1R stimulated intracellular pathways (39, 40). In addition, continuous EGF stimulation required a functional IGF-1R pathway that resulted in ERK activation in mouse fibroblasts (41, 42). The observed increase in Bcl-2 immunopositivity as early as an hour post treatment indicates rapid convergence of these pathways leading to Bcl-2 induction.
Besides epithelial cell proliferation and survival signaling, EGFR mediates MUC5AC synthesis following exposure to LPS-, CS-, or infection with Pseudomonas aeruginosa (31, 37, 38). Interestingly, EGFR activation causes MUC5AC and Bcl-2 expression, (33, 34) and Bcl-2 expression sustains epithelial cell hyperplasia (7). Future studies will investigate whether Bcl-2 expression directly mediates MUC5AC expression. Moreover, Bcl-2 binds and suppresses NALP1, an integral component of inflammasome, to reduce caspase-1, originally identified as IL-1β-converting enzyme activation and, thus, inhibits IL-1β production (43, 44). Together, the present data suggest that the inflammatory mediators such as IL-1β and IGF-1 stimulate EGFR and IGF-1R pathways that converge to induce IC-IGF-1 to help upregulate Bcl-2 expression in epithelial cells, and induced Bcl-2 expression could, in a negative feedback mechanism, suppress inflammation (Fig. 6).
FIGURE 6. A schematic representation of a proposed molecular pathway by which IL-1β and IGF-1 induce IC-IGF-1 and Bcl-2-expression.

Bcl-2 expression is regulated at the transcriptional and post-translational levels. The P1 region harbors the positive regulatory elements for Bcl-2 transcription (25, 45) and the P2-region suppresses the P1 activity (25). Transcriptional factors such as the CREB and SP-1 (46, 47) interact with the P1 region to upregulate its transcriptional activity while factors such as nuclear NF-κB (11), Brn3a (29), C/EBP (46), and p53 (25) interact with the P2 region to either suppress or enhance its inhibitory role. However, in our experimental setting IGF-1 did not affect the promoter activity of various luciferase-reporter constructs containing Bcl-2 promoter regions. Instead, IGF-1 treatment stabilized Bcl-2 mRNA resulting in increased Bcl-2 mRNA levels. While the repressor that binds to the P2 region plays an important role in regulating Bcl-2 transcription as shown in our previous study (25), the lack of IGF-1 affecting the Bcl-2 promoter region suggests that IGF-1-induced Bcl-2 mRNA stability is not mediated by this repressor. However, cells that were transfected with a plasmid containing the P2 region consistently showed an additional increase in Bcl-2 mRNA when treated with IGF-1. In the absence of IGF-1, the transfection of P2 construct alone had no affect on Bcl-2 mRNA suggesting that the P2 region, when transfected as part of a plasmid, did not affect the repressor function of the P2 region. Taken together, these data suggest that the repressor-binding potential of the P2 region is not playing a role in the context of IGF-1-mediated induction of Bcl-2 mRNA. Therefore, we conclude that IGF-1 increased Bcl-2 mRNA stability by P2 serving as a sink for the putative destabilizing factor(s). Previous studies have shown that the P2 repressor activity is mediated by the region 412–607 that includes a 195 bp p53-dependent negative regulatory element. The present study showed that deleting the region 106–126 or mutating the core Brn3a binding site (28), 93–99, is sufficient to disrupt the IGF-1-mediated enhancement of Bcl-2 mRNA when transfecting the P2 region. These findings suggest that the regions interacting with the putative destabilizing factor and repressor are non-overlapping. Whether the repressor binding to the P2 region is competed out by the destabilizing factor causing steric hindrances or whether other factors are involved to exclude repressor binding to the transfected P2 region will require additional studies. Nonetheless, both IL-1β and IGF-1 induced the Bcl-2 mRNA in cells transfected with P2 promoter region implicating a common mechanism that likely involves IC-IGF-1. Stabilization of Bcl-2 mRNA has been recently reported as one of the mechanism by which Bcl-2 is upregulated in cancer cells (48, 49). Further studies will be carried out in identifying the mechanism(s) that mediates IGF-1 induced Bcl-2 mRNA stabilization but are beyond the scope of the present study.
In mouse models of acute or chronic lung injury using LPS or CS, respectively, we observed increased number of IC-IGF-1- and Bcl-2-positive airway epithelial cells. Similar findings in autopsied lung airways from patients with cystic fibrosis and chronic bronchitis suggest that this pathway is of clinical relevance and should be exploited to help regulate chronic airway epithelial inflammation. However, the numbers of IC-IGF-1-positive cells were consistently higher than the Bcl-2-positive cells, suggesting that IC-IGF-1 expression is upstream of Bcl-2 induction, and also that IC-IGF-1 may be involved in other prosurvival mechanisms besides Bcl-2 induction. It is also possible that as yet unknown factors or pathways affect IC-IGF-1-induced Bcl-2 expression in airway epithelial cells. Of an important note, Bcl-2 as a proto-oncogene is implicated in several cancers, including melanoma, breast, prostate, and lung carcinomas (3, 11, 50, 51), and IGF-1 and IGF-1R levels have been positively correlated with tumor progression (52, 53). In addition, EGFR, when constitutively active due to mutations, promotes carcinogenesis (54) and when heterodimerized with IGF-1R monomers (55) may enhance Bcl-2 expression. Therefore, the IC-IGF-1-mediated Bcl-2 induction could be one of the early molecular changes that sustain inflammation-induced hyperplasia and could be the germinal centers for preneoplastic lesions.
Supplementary Material
Acknowledgements
We thank Nick Johnson, Xin Zhang, Edward Roberts, Karen Cardon and Zekarias Woldegiorgis for their help in selected experiments. This study used biological specimens and data provided by the Lung Tissue Research Consortium (LTRC) supported by the National Heart, Lung, and Blood Institute (NHLBI).
Abbreviations
- CF
cystic fibrosis
- CS
cigarette smoke
- Bcl-2
B cell lymphoma factor-2
- IGF-1
Insulin-like growth factor-I
- IC-IGF-1
Intracellular-insulin-like growth factor-I
- IGF-1R
Insulin-like growth factor-I receptor
- EGFR
Epidermal growth factor receptor
- AG-1478
anilinoquinazoline, a tyrosine kinase inhibitor specific for EGFR
- PPP
Picropodophyllin
Footnotes
These studies were supported by grants from the National Institutes of Health (HL68111 and ES015482).
All datasets have been depostitied at the National Center for Biotechnology Information/Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36174) under accession number GSE36174).
References
- 1.Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 2.Tsujimoto Y, Croce CM. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci U S A. 1986;83:5214–5218. doi: 10.1073/pnas.83.14.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin B, Paesmans M, Berghmans T, Branle F, Ghisdal L, Mascaux C, Meert AP, Steels E, Vallot F, Verdebout JM, Lafitte JJ, Sculier JP. Role of Bcl-2 as a prognostic factor for survival in lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2003;89:55–64. doi: 10.1038/sj.bjc.6601095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–885. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- 5.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 6.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris JF, Fischer MJ, Hotchkiss JA, Monia BP, Randell SH, Harkema JR, Tesfaigzi Y. Bcl-2 sustains increased mucous and epithelial cell numbers in metaplastic airway epithelium. Am J Respir Crit Care Med. 2005;171:764–772. doi: 10.1164/rccm.200408-1108OC. [DOI] [PubMed] [Google Scholar]
- 8.Tesfaigzi Y, Fischer MJ, Martin AJ, Seagrave J. Bcl-2 in LPS- and allergen-induced hyperplastic mucous cells in airway epithelia of Brown Norway rats. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1210–L1217. doi: 10.1152/ajplung.2000.279.6.L1210. [DOI] [PubMed] [Google Scholar]
- 9.Tesfaigzi Y, Harris JF, Hotchkiss JA, Harkema JR. DNA synthesis and Bcl-2 expression during the development of mucous cell metaplasia in airway epithelium of rats exposed to LPS. Am J Physiol Lung Cell Mol Physiol. 2004;286:L268–L274. doi: 10.1152/ajplung.00172.2003. [DOI] [PubMed] [Google Scholar]
- 10.Vignola AM, Chiappara G, Siena L, Bruno A, Gagliardo R, Merendino AM, Polla BS, Arrigo AP, Bonsignore G, Bousquet J, Chanez P. Proliferation and activation of bronchial epithelial cells in corticosteroid-dependent asthma. J Allergy Clin Immunol. 2001;108:738–746. doi: 10.1067/mai.2001.119160. [DOI] [PubMed] [Google Scholar]
- 11.Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20:7342–7351. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 12.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 13.Kurland JF, Kodym R, Story MD, Spurgers KB, McDonnell TJ, Meyn RE. NF-kappaB1 (p50) homodimers contribute to transcription of the bcl-2 oncogene. J Biol Chem. 2001;276:45380–45386. doi: 10.1074/jbc.M108294200. [DOI] [PubMed] [Google Scholar]
- 14.Sepulveda P, Encabo A, Carbonell-Uberos F, Minana MD. BCL-2 expression is mainly regulated by JAK/STAT3 pathway in human CD34+ hematopoietic cells. Cell Death Differ. 2007;14:378–380. doi: 10.1038/sj.cdd.4402007. [DOI] [PubMed] [Google Scholar]
- 15.Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med. 2004;200:659–669. doi: 10.1084/jem.20040789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minshall C, Arkins S, Dantzer R, Freund GG, Kelley KW. Phosphatidylinositol 3'-kinase, but not S6-kinase, is required for insulin-like growth factor-I and IL-4 to maintain expression of Bcl-2 and promote survival of myeloid progenitors. J Immunol. 1999;162:4542–4549. [PubMed] [Google Scholar]
- 17.Li XC, Demirci G, Ferrari-Lacraz S, Groves C, Coyle A, Malek TR, Strom TB. IL-15 and IL-2: a matter of life and death for T cells in vivo. Nat Med. 2001;7:114–118. doi: 10.1038/83253. [DOI] [PubMed] [Google Scholar]
- 18.Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–144. doi: 10.1016/j.immuni.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyner JW, Kim EY, Ide K, Pelletier MR, Roswit WT, Morton JD, Battaile JT, Patel AC, Patterson GA, Castro M, Spoor MS, You Y, Brody SL, Holtzman MJ. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J Clin Invest. 2006;116:309–321. doi: 10.1172/JCI25167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doherty MM, Liu J, Randell SH, Carter CA, Davis CW, Nettesheim P, Ferriola PC. Phenotype and differentiation potential of a novel rat tracheal epithelial cell line. Am J Respir Cell Mol Biol. 1995;12:385–395. doi: 10.1165/ajrcmb.12.4.7535063. [DOI] [PubMed] [Google Scholar]
- 21.Randell SH, Liu JY, Ferriola PC, Kaartinen L, Doherty MM, Davis CW, Nettesheim P. Mucin production by SPOC1 cells--an immortalized rat tracheal epithelial cell line. Am J Respir Cell Mol Biol. 1996;14:146–154. doi: 10.1165/ajrcmb.14.2.8630264. [DOI] [PubMed] [Google Scholar]
- 22.Stout B, Melendez K, Seagrave J, Holtzman MJ, Wilson B, Xiang J, Tesfaigzi Y. STAT1 activation causes translocation of Bax to the endoplasmic reticulum during the resolution of airway mucous cell hyperplasia by IFNγ. J Immunol. 2007;178:8107–8116. doi: 10.4049/jimmunol.178.12.8107. [DOI] [PubMed] [Google Scholar]
- 23.Schwalm K, Stevens JF, Jiang Z, Schuyler MR, Schrader R, Randell SH, Green FH, Tesfaigzi Y. Expression of the pro-apoptotic protein bax is reduced in bronchial mucous cells of asthmatics. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1102–L1109. doi: 10.1152/ajplung.00424.2007. [DOI] [PubMed] [Google Scholar]
- 24.Greenberg JR. High stability of messenger RNA in growing cultured cells. Nature. 1972;240:102–104. doi: 10.1038/240102a0. [DOI] [PubMed] [Google Scholar]
- 25.Bredow S, Juri DE, Cardon K, Tesfaigzi Y. Identification of a novel Bcl-2 promoter region that counteracts in a p53-dependent manner the inhibitory P2 region. Gene. 2007;404:110–116. doi: 10.1016/j.gene.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.March TH, Bowen LE, Finch GL, Nikula KJ, Wayne BJ, Hobbs CH. Effects of strain and treatment with inhaled aII-trans-retinoic acid on cigarette smoke-induced pulmonary emphysema in mice. COPD. 2005;2:289–302. [PubMed] [Google Scholar]
- 27.Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, Kishita E, Matsui A, Katsume A, Yamaguchi S, Nishikawa K, Isoda K, Riches DW, Matoba S, Okigaki M, Matsubara H. Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ Res. 2009;105:1149–1158. doi: 10.1161/CIRCRESAHA.109.208199. [DOI] [PubMed] [Google Scholar]
- 28.Xiang M, Zhou L, Macke JP, Yoshioka T, Hendry SH, Eddy RL, Shows TB, Nathans J. The Brn-3 family of POU-domain factors: primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. J Neurosci. 1995;15:4762–4785. doi: 10.1523/JNEUROSCI.15-07-04762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Budhram-Mahadeo V, Morris PJ, Smith MD, Midgley CA, Boxer LM, Latchman DS. p53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J Biol Chem. 1999;274:15237–15244. doi: 10.1074/jbc.274.21.15237. [DOI] [PubMed] [Google Scholar]
- 30.Jerde TJ, Bushman W. IL-1 induces IGF-dependent epithelial proliferation in prostate development and reactive hyperplasia. Sci Signal. 2009;2:ra49. doi: 10.1126/scisignal.2000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao MX, Ueki IF, Nadel JA. Tumor necrosis factor alpha-converting enzyme mediates MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11618–11623. doi: 10.1073/pnas.1534804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeyama K, Dabbagh K, Jeong Shim J, Dao-Pick T, Ueki IF, Nadel JA. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: role of neutrophils. J Immunol. 2000;164:1546–1552. doi: 10.4049/jimmunol.164.3.1546. [DOI] [PubMed] [Google Scholar]
- 33.Casalino-Matsuda SM, Monzon ME, Forteza RM. EGFR Activation by EGF Mediates Oxidant-induced Goblet Cell Metaplasia in Human Airway Epithelium. Am J Respir Cell Mol Biol. 2006 doi: 10.1165/rcmb.2005-0386OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeyama K, Tamaoki J, Kondo M, Isono K, Nagai A. Role of epidermal growth factor receptor in maintaining airway goblet cell hyperplasia in rats sensitized to allergen. Clin Exp Allergy. 2008;38:857–865. doi: 10.1111/j.1365-2222.2008.02951.x. [DOI] [PubMed] [Google Scholar]
- 35.Kanai M, Konda Y, Nakajima T, Izumi Y, Takeuchi T, Chiba T. TGF-alpha inhibits apoptosis of murine gastric pit cells through an NF-kappaB-dependent pathway. Gastroenterology. 2001;121:56–67. doi: 10.1053/gast.2001.25544. [DOI] [PubMed] [Google Scholar]
- 36.Tesfaigzi Y. Roles of Apoptosis in Airway Epithelia. Am J Respir Cell Mol Biol. 2006;34:537–547. doi: 10.1165/rcmb.2006-0014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shao MX, Nakanaga T, Nadel JA. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L420–L427. doi: 10.1152/ajplung.00019.2004. [DOI] [PubMed] [Google Scholar]
- 38.Kohri K, Ueki IF, Shim JJ, Burgel PR, Oh YM, Tam DC, Dao-Pick T, Nadel JA. Pseudomonas aeruginosa induces MUC5AC production via epidermal growth factor receptor. Eur Respir J. 2002;20:1263–1270. doi: 10.1183/09031936.02.00001402. [DOI] [PubMed] [Google Scholar]
- 39.Baserga R, Porcu P, Rubini M, Sell C. Cell cycle control by the IGF-1 receptor and its ligands. Adv Exp Med Biol. 1993;343:105–112. doi: 10.1007/978-1-4615-2988-0_11. [DOI] [PubMed] [Google Scholar]
- 40.Doepfner KT, Spertini O, Arcaro A. Autocrine insulin-like growth factor-I signaling promotes growth and survival of human acute myeloid leukemia cells via the phosphoinositide 3-kinase/Akt pathway. Leukemia. 2007;21:1921–1930. doi: 10.1038/sj.leu.2404813. [DOI] [PubMed] [Google Scholar]
- 41.Putz T, Culig Z, Eder IE, Nessler-Menardi C, Bartsch G, Grunicke H, Uberall F, Klocker H. Epidermal growth factor (EGF) receptor blockade inhibits the action of EGF, insulin-like growth factor I, and a protein kinase A activator on the mitogen-activated protein kinase pathway in prostate cancer cell lines. Cancer Res. 1999;59:227–233. [PubMed] [Google Scholar]
- 42.Roudabush FL, Pierce KL, Maudsley S, Khan KD, Luttrell LM. Transactivation of the EGF receptor mediates IGF-1-stimulated shc phosphorylation and ERK1/2 activation in COS-7 cells. J Biol Chem. 2000;275:22583–22589. doi: 10.1074/jbc.M002915200. [DOI] [PubMed] [Google Scholar]
- 43.Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S, Terskikh AV, Faustin B, Reed JC. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 44.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Seto M, Jaeger U, Hockett RD, Graninger W, Bennett S, Goldman P, Korsmeyer SJ. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. Embo J. 1988;7:123–131. doi: 10.1002/j.1460-2075.1988.tb02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heckman CA, Wheeler MA, Boxer LM. Regulation of Bcl-2 expression by C/EBP in t(14;18) lymphoma cells. Oncogene. 2003;22:7891–7899. doi: 10.1038/sj.onc.1206639. [DOI] [PubMed] [Google Scholar]
- 47.Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–10766. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- 48.Otake Y, Soundararajan S, Sengupta TK, Kio EA, Smith JC, Pineda-Roman M, Stuart RK, Spicer EK, Fernandes DJ. Overexpression of nucleolin in chronic lymphocytic leukemia cells induces stabilization of bcl2 mRNA. Blood. 2007;109:3069–3075. doi: 10.1182/blood-2006-08-043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sengupta TK, Bandyopadhyay S, Fernandes DJ, Spicer EK. Identification of nucleolin as an AU-rich element binding protein involved in bcl-2 mRNA stabilization. J Biol Chem. 2004;279:10855–10863. doi: 10.1074/jbc.M309111200. [DOI] [PubMed] [Google Scholar]
- 50.Fahy BN, Schlieman MG, Mortenson MM, Virudachalam S, Bold RJ. Targeting BCL-2 overexpression in various human malignancies through NF-kappaB inhibition by the proteasome inhibitor bortezomib. Cancer Chemother Pharmacol. 2005;56:46–54. doi: 10.1007/s00280-004-0944-5. [DOI] [PubMed] [Google Scholar]
- 51.Benimetskaya L, Ayyanar K, Kornblum N, Castanotto D, Rossi J, Wu S, Lai J, Brown BD, Popova N, Miller P, McMicken H, Chen Y, Stein CA. Bcl-2 protein in 518A2 melanoma cells in vivo and in vitro. Clin Cancer Res. 2006;12:4940–4948. doi: 10.1158/1078-0432.CCR-06-1002. [DOI] [PubMed] [Google Scholar]
- 52.Jerome L, Shiry L, Leyland-Jones B. Deregulation of the IGF axis in cancer: epidemiological evidence and potential therapeutic interventions. Endocr Relat Cancer. 2003;10:561–578. doi: 10.1677/erc.0.0100561. [DOI] [PubMed] [Google Scholar]
- 53.Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- 54.Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer--is it becoming a reality? Nat Rev Clin Oncol. 2010;7:401–414. doi: 10.1038/nrclinonc.2010.64. [DOI] [PubMed] [Google Scholar]
- 55.Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006;66:10100–10111. doi: 10.1158/0008-5472.CAN-06-1684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.