

Abstract
Please cite this paper as: Bateman et al. (2012) Infectivity phenotypes of H3N2 influenza A viruses in primary swine respiratory epithelial cells are controlled by sialic acid binding. Influenza and Other Respiratory Viruses 6(6), 424–433.
Background In the late 1990s, triple reassortant H3N2 influenza A viruses emerged and spread widely in the US swine population. We have shown previously that an isolate representative of this virus‐lineage, A/Swine/Minnesota/593/99 (Sw/MN), exhibits phenotypic differences compared to a wholly human‐lineage H3N2 virus isolated during the same time period, A/Swine/Ontario/00130/97 (Sw/ONT). Specifically, Sw/MN was more infectious for pigs and infected a significantly higher proportion of cultured primary swine respiratory epithelial cells (SRECs). In addition, reverse genetics‐generated Sw/MN × Sw/ONT reassortant and point mutant viruses demonstrated that the infectivity phenotypes in SRECs were strongly dependent on three amino acids within the hemagglutinin (HA) gene.
Objectives To determine the mechanism by which Sw/MN attains higher infectivity than Sw/ONT in SRECs.
Methods A/Swine/Minnesota/593/99, Sw/ONT, and mutant (reverse genetics‐generated HA reassortant and point mutant) viruses were compared at various HA‐mediated stages of infection: initial sialic acid binding, virus entry, and the pH of virus–endosome fusion.
Results/Conclusions Sialic acid binding was the sole stage where virus differences directly paralleled infectivity phenotypes in SRECs, indicating that binding is the primary mechanism responsible for differences in the infectivity levels of Sw/MN and Sw/ONT.
Keywords: Infectivity, influenza, sialic acid, swine, virus
Introduction
H1N1 influenza viruses were introduced into North American pigs early in the twentieth century, near the time of the emergence of the related 1918 H1N1 “Spanish influenza” human pandemic virus. 1 , 2 For many decades, the predominant virus circulating in North American pigs was this “classical” H1N1 (cH1N1) swine virus. 2 However, influenza viruses infecting North American pigs have evolved extensively in recent years. In the late 1990s “triple reassortant” H3N2 (rH3N2) viruses containing genes from classical swine‐, avian‐, and human‐lineage viruses emerged and spread widely throughout the US swine population. 3 , 4 , 5 Further reassortment resulted in rH1N1, rH1N2, rH3N1, and rH2N3 viruses in pigs. 6 , 7 , 8 , 9 Many of these reassortant viruses have caused extensive outbreaks of disease in pigs, and these viruses also contributed six nucleic acid segments to the 2009 H1N1 human pandemic virus. 10 , 11 Thus, the emergence of these reassortant swine viruses has had major health implications for both pigs and humans.
To determine what allowed the initial rH3N2 viruses to successfully emerge, spread, and be maintained in the swine population, we previously compared a representative rH3N2 virus [A/Swine/Minnesota/593/99 (Sw/MN)] with a wholly human‐lineage virus isolated from a single pig during the same time period [A/Swine/Ontario/00130/97 (Sw/ONT)]. Sw/ONT has not been subsequently isolated from pigs, while the rH3N2 viruses spread and have established stable lineages in the North American swine population. To determine whether the differences in epidemiologic outcomes between Sw/MN and Sw/ONT could have been due to fundamental differences between the viruses, in vivo pig experiments were performed. In these experiments, Sw/MN was infectious at lower doses, exhibited earlier and more extensive nasal shedding, and caused more severe lung lesions than Sw/ONT. 12 Using reverse genetics (rg) to generate viruses in which the hemagglutinin (HA) and neuraminidase (NA) genes of Sw/MN and Sw/ONT were exchanged, we further showed that the HA and/or NA genes control the infectivity and replication phenotypes of rgMN and rgONT in vivo. 13 To examine detailed molecular mechanisms of virus infection, primary swine respiratory epithelial cells (SRECs), an in vitro model for virus infection of swine cells, were utilized. These cells are derived from the target cells of influenza viruses in vivo and express both α2‐3‐ and α2‐6‐linked sialic acids, 14 , 15 as seen in pigs. 16 , 17 , 18 (Influenza viruses initiate infection by binding to cell surface sialic acids). 19 Reverse genetics‐generated reassortant and point mutant viruses demonstrated that mutation of only three residues in the HA protein could modify rgMN and rgONT infectivity levels in SRECs. 14 Specifically, three mutations (G124D/A138S/G142E) were necessary to increase rgONT infectivity to the level of rgMN, whereas only a single mutation from the rgMN to the rgONT residue at amino acid 138 (S138A) decreased rgMN infectivity to the level of rgONT.
The mutant viruses generated in our previous study 14 serve as useful tools to examine the cellular basis of the infectivity phenotypes of rgMN and rgONT. The aim of the present study was to determine the step(s) in virus infection of SRECs where rgMN and rgONT differ and to determine whether differences paralleled the previously defined infectivity phenotypes 14 of mutant viruses in SRECs. The parental (rgMN and rgONT) and mutant (HA reassortant and point mutant) viruses were compared at different steps in the infection process that are mediated by the HA protein: initial virus binding to sialic acids, virus entry into cells, and the pH at which virus–endosome fusion occurs. The only step where viral differences paralleled infectivity levels was sialic acid binding, so it appears that this step is responsible for virus infectivity differences in SRECs and possibly in pigs in vivo as well.
Materials and methods
Cells and viruses
Madin Darby canine kidney (MDCK) cells were maintained in minimal essential medium (MEM; GIBCO/BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA, USA) and 1% penicillin/streptomycin/amphotericin (GIBCO/BRL) at 37°C in a 5% CO2 atmosphere. The SRECs were isolated and grown as described previously. 14 Briefly, distal tracheal specimens from pigs were placed in a tissue‐dissociation solution for 72 hours at 4°C. After incubation, cells were dislodged by gentle agitation, collected by centrifugation, and resuspended. Cells were incubated at 37°C and 5% CO2 in uncoated tissue culture dishes for 2–6 hours, and non‐adherent (epithelial) cells were collected and seeded into type VI collagen (Sigma Chemical Co., St. Louis, MO, USA)‐coated tissue culture flasks. SRECs were grown in bronchial epithelial growth media (BEGM; Lonza, Walkersville, MD, USA) at 37°C in a 5% CO2 atmosphere and passaged up to five times prior to infection. The collection and use of SRECs were approved by the Animal Care and Use Committee of the School of Veterinary Medicine. All viruses were generated using reverse genetics as previously described, 14 and virus titers were determined by 50% tissue culture infectious dose (TCID50) titrations in MDCK cells using the method of Reed and Muench. 20 To ensure highly accurate measurements of infectious particles, viruses were titered in three independent experiments using one‐fifth log dilutions in quadruplicate, and infected cells were identified by immunocytochemical staining for viral nucleoprotein (NP) expression as described previously. 12
Infection of swine respiratory epithelial cells
Cells were seeded into type VI collagen‐coated 24‐well cell culture plates at 50 000 cells per well, incubated at 37°C and 5% CO2, and grown to >95% confluency (approximately 48 hours post‐seeding). Prior to infection, cells were washed once with BEGM. Viruses were diluted in MEM media with 0·2% bovine serum albumin (BSA), 0·01% FBS and antimicrobials, and cells were inoculated at a ratio of three TCID50 per cell. Cells were incubated with virus for 1 hour at 37°C and washed twice with BEGM to remove the inoculum. At 12 hours post‐inoculation, the supernatant was removed and the cells were harvested for flow cytometric analysis.
Quantification of infected cells by flow cytometry
Infected cells were identified by viral NP expression using the anti‐influenza A NP antibody 68D2 (kindly provided by Y. Kawaoka, University of Wisconsin‐Madison), as described previously. 14 Briefly, cells were detached with 0·25% EDTA‐trypsin (GIBCO/BRL), fixed with formalin, permeabilized with 0·1% saponin (Sigma Chemical Co.), blocked with normal horse serum, and stained with a 1:6400 dilution of 68D2 followed by a 1:50 dilution of FITC‐labeled anti‐mouse antibody (Zymed Laboratories Inc., South San Francisco, CA, USA). Following resuspension of cells in 2% phosphate‐buffered saline (PBS)‐buffered paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA, USA), fluorescence intensity was measured with a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA) and analyses were completed using FLOWJO 8.5.3 (Treestar, Ashland, OR, USA).
Sialidase treatment of swine respiratory epithelial cells
Confluent SRECs in 24‐well cell culture plates were washed twice with BEGM and incubated in BEGM with various amounts of type III Vibrio cholerae sialidase (Sigma) for 3 hours at 37°C. Following incubation, cells were washed twice with BEGM and infected as described above.
Treatment of swine respiratory epithelial cells with N‐glycolylmannosamine
Cells were plated at 50 000 cells/well in 24‐well cell culture plates and grown in the presence of either 0·3 or 1·0 mm N‐glycolylmannosamine (ManNGc). ManNGc was solubilized in dimethyl sulfoxide (DMSO) and added to a final concentration of 0·05% DMSO. After 48 hours, confluent cells were washed twice with BEGM and infected as described above.
Hemagglutination of turkey and swine red blood cells
Twofold dilutions of each virus were added to round‐bottom plates, and an equal volume of turkey red blood cells (tRBCs) in PBS was added to a final concentration of 0·125% erythrocytes (Lampire, Pipersville, PA, USA). After 1 hour incubation at room temperature, the hemagglutination titer was determined as described previously. 21 Viruses were equilibrated based on tRBC hemagglutination units (HAU) and then tested for the ability to hemagglutinate swine red blood cells (swRBCs).
Solid‐phase glycopolymer binding assay
The sialic acid binding assay was conducted based on previously described methods. 22 Viruses were concentrated by ultracentrifugation [20 000 rpm for 1 hour, Beckman L8‐70M ultracentrifuge with SW‐28 rotor (Beckman Coulter Inc., Brea, CA, USA)] and added to 96‐well plates. To equilibrate the amount of each virus used in each assay, real‐time RT‐PCR 23 was used to quantify influenza matrix gene RNA. Virus containing approximately 25 000 copies of matrix gene RNA was bound per well in sodium bicarbonate (pH 9·3). After overnight incubation at 4°C, plates were rinsed with washing buffer (PBS + 0·01% Tween‐20) and serial twofold dilutions (beginning with 1·6 nm sialic acid) of biotinylated synthetic glycopolymers in the working buffer [PBS + 0·01% Tween‐20 + 0·1% BSA + 0·1 μm oseltamivir carboxylate (NA inhibitor, kindly provided by Dr. Alexander Klimov, Centers for Disease Control and Prevention)] were added to each well. Following a second overnight incubation, plates were rinsed with washing buffer and incubated with streptavidin‐peroxidase in the working buffer at 4°C for 1 hour. Finally, plates were rinsed, o‐phenylenediamine was added, and the absorbance (490 nm) of each sample was determined using a Model 680 microplate reader (Bio‐Rad, Hercules, CA, USA). Scatchard analyses of A490 (x‐axis) plotted against A490/concentration of sialic acid (y‐axis) were conducted, and the slopes from the Scatchard plots were used to determine the dissociation constants (K diss) of virus–glycopolymer complexes, as described previously. 22 The binding of glycopolymers to a virus with many HA molecules is multivalent, meaning that the values are not true equilibrium dissociation constants. However, they provide a reproducible measure of the relative affinities of each virus for each polymer. 22
AlexaFluor 488 virus labeling and glycan array binding
Viruses were labeled with AlexaFluor 488 (Invitrogen, Carlsbad, CA, USA) based on previously described methods. 15 , 24 , 25 , 26 Briefly, virus stocks grown in MDCK cells were pelleted by ultracentrifugation (20 000 rpm for 1 hour), resuspended in Tris‐buffered saline (TBS), and layered over a discontinuous 20–60% sucrose gradient in TBS. After centrifugation (20 000 rpm for 1 hour), the virus band at the 20–60% interface was extracted, diluted in TBS, and pelleted by centrifugation. Virus pellets were resuspended in 0·15 m NaCl + 0·25 mm CaCl2 + 0·8 mm MgCl2 pH 7·2, and HAU were determined using turkey erythrocytes as described above. Viruses were diluted to 50 000 HAU/ml. To 100 μl (5000 HAU) of each virus, 10 μl of 1·0 m sodium bicarbonate pH 9·0 was added, followed by 0·0005 μg/HAU of AlexaFluor solubilized in DMSO. Following a 1 hour incubation at room temperature, labeled viruses were dialyzed into TBS + 0·25 mm CaCl2 + 0·8 mm MgCl2 with Slide‐A‐Lyzer MINI Dialysis Units (7000 MWCO; Thermo Scientific, Waltham, MA, USA) overnight at 4°C. Following overnight incubation, HAU on tRBCs and infectious titer in MDCK cells were re‐determined to ensure that labeling with AlexaFluor did not affect virus binding or infectivity (data not shown). Viruses were frozen at −80°C until further analysis. Samples were then thawed, BSA and Tween‐20 were added to final concentrations of 1% and 0·05%, respectively, and virus binding was examined on a printed microarray (version 4.1) containing synthetic and natural glycans 27 (Consortium for Functional Glycomics, http://www.functionalglycomics.org).
Statistical analysis
Comparisons of infectivity levels of each virus in untreated, sialidase treated, and ManNGc‐treated cells were analyzed using anova‐protected Student’s t‐tests. Analyses were performed using the r statistical software (http://www.R‐project.org).
Results
Virus infectivity phenotypes in swine respiratory epithelial cells
We have previously found that rgMN infects a significantly higher percentage of SRECs than rgONT. 14 Generation of rgMN and rgONT reassortant and point mutant viruses via reverse genetics indicated that the infectivity levels of these viruses are dependent on amino acid differences in the HA protein. Specifically, using a series of reassortant viruses where either the HA and NA genes, the HA gene alone, or the NA gene alone was exchanged, we showed that the origin of the HA gene determined virus infectivity levels in SRECs. Further, mutating one to three amino acids of the HA protein to those of the other virus exchanged the virus infectivity levels (Table 1). In previously published results, the high infectivity phenotype viruses infected 70–95% of cells, whereas low infectivity viruses infected 25–40% of cells. 14 The parental, HA reassortant, and point mutant viruses were chosen for further analysis here.
Table 1.
Virus infectivity in swine respiratory epithelial cells (SRECs)*
| High infectivity phenotype | Low infectivity phenotype |
|---|---|
| rgMN | rgONT |
| rgONT + MN HA | rgMN + ONT HA |
| rgONT G124D/A138S/G142E | rgMN S138A |
HA, hemagglutinin.
*After cells were inoculated with three TCID50/cell of each virus for 1 hour, viruses with a high infectivity phenotype infected 70–95% of SRECs, whereas viruses with a low infectivity phenotype infected 25–40% of cells. 14
Virus susceptibility to cell surface sialidase treatment
To initially determine whether the infectivity differences between rgMN and rgONT viruses are due to a sialic acid‐mediated step, virus susceptibility to sialidase treatment of SRECs was examined. Prior to infection, cells were treated with V. cholerae sialidase, which removes α2‐3‐ and α2‐6‐linked sialic acids. 28 As expected, because influenza viruses initiate infection by binding to cell surface sialic acids, 19 the infectivity levels of each virus decreased significantly (P < 0·01) with cell surface sialidase treatment (Figure 1A). However, the viruses were differentially susceptible to sialidase treatment, and these differences paralleled the virus infectivity levels in SRECs. Specifically, the infectivity of rgONT and the other low infectivity viruses dropped to mock‐infection level after only 0·001 U sialidase treatment, whereas rgMN and the other high infectivity viruses retained approximately 50% of their initial infectivity after this treatment (Figure 1A). Treatment with 10× more sialidase (0·01 U) still did not completely abolish infectivity of rgMN and the other high infectivity viruses.
Figure 1.
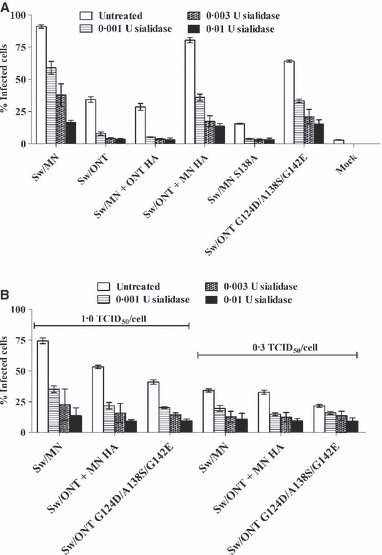
Virus infectivity in swine respiratory epithelial cells (SRECs) following cell treatment with Vibrio cholerae sialidase. Cells were treated with sialidase for 3 hours, washed, and infected with three TCID50/cell (A), one TCID50/cell (B), or 0·3 TCID50/cell (B). Results shown are mean ± SEM of three experiments performed in triplicate. Compared to untreated cells, each virus demonstrated significantly lower (P < 0·01) infectivity levels in SRECs treated with 0·001, 0·003, or 0·01 U of sialidase.
To ensure that the apparent relative resistance of the high infectivity phenotype viruses to sialidase treatment was not simply due to higher initial infectivity, rgMN and the high infectivity viruses were examined in the same assay at lower doses. After sialidase treatment, cells were infected with either 1·0 or 0·3 TCID50/cell of each virus (Figure 1B). At lower doses, the high infectivity phenotype viruses remained relatively resistant to sialidase, in that they retained infectivity even after SRECs were treated with 0·01 U of sialidase. This finding demonstrates that rgMN and the other high infectivity viruses interact with cell surface sialic acids in a fundamentally different way than rgONT and the low infectivity viruses. We hypothesize that the high infectivity viruses utilize residual sialic acids more efficiently than the low infectivity viruses. In summary, virus susceptibility to sialidase treatment of SRECs associated with virus infectivity levels, suggesting that the infectivity differences between rgMN and rgONT are due to a cell surface, sialic acid‐mediated mechanism.
Virus susceptibility to the modulation of cell surface sialic acid species
The two major sialic acid species in the swine respiratory tract are N‐acetylneuraminic acid (NeuAc) and N‐glycolylneuraminic acid (NeuGc). 29 To assess virus utilization of these different sialic acid forms, prior to infection, a sialic acid precursor was utilized to modulate the sialic acid forms on the SREC surface. This precursor, ManNGc, is an analog of sialic acid, and when added to cell culture media, it enters cells, is converted to its corresponding sialic acid (NeuGc), and is incorporated into glycolipids and glycoproteins on the cell surface. 30 Treatment with ManNGc did not alter cell morphology or viability, and a DMSO control showed no change in virus infectivity in the presence of this diluent (data not shown). We have previously shown by mass spectrometry that ManNGc treatment induces the expected changes in sialic acid species in SRECs, and that rgMN and rgONT preferentially utilize NeuAc over NeuGc to infect SRECs. 15 The mutant viruses examined here exhibited the same NeuAc preference, as the infectivity levels of all viruses decreased after SRECs were grown in the presence of ManNGc. However, as with virus susceptibility to sialidase treatment of SRECs, the viruses differed in their susceptibility to ManNGc treatment. Specifically, the infectivity levels of rgONT and the other low infectivity viruses decreased with as little as 0·03 mm ManNGc, while the infectivity of rgMN and the other high infectivity viruses did not change at this concentration of ManNGc (Figure 2). Further, 0·1 mm ManNGc completely abolished infectivity of rgONT and the other low infectivity viruses, whereas rgMN and the other high infectivity viruses retained at least 50% of their original infectivity (Figure 2). Thus, the high infectivity viruses were more resistant to changes in sialic acid species on the SREC cell surface, further suggesting that the difference in SREC infectivity levels involves a sialic acid‐mediated event.
Figure 2.
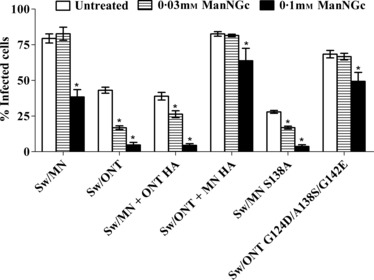
Virus infectivity in swine respiratory epithelial cells cultured in the presence of N‐glycolylmannosamine (ManNGc). Cells were cultured with ManNGc for 48 hours, washed, and infected with three TCID50/cell. Results shown are mean ± SEM of three experiments performed in triplicate. *P < 0·01 compared to untreated cells.
Hemagglutination of turkey red blood cells and swine red blood cells
In an initial assessment of virus binding, hemagglutination assays were performed. Hemagglutination has been extensively utilized to examine influenza virus binding. 31 , 32 , 33 In this assay, viruses bind to sialic acids on the RBC surface, agglutinating the cells. 34 Turkey RBCs are commonly used as a standard to examine hemagglutination activity, 35 , 36 , 37 and RBCs from different species have been used previously by others to investigate influenza virus binding. 38 , 39 Here, viruses were equilibrated by hemagglutination on tRBCs and then further examined for their ability to hemagglutinate swRBCs, which express both NeuAc and NeuGc. 40 The rgMN virus and other high infectivity viruses agglutinated swRBCs, while rgONT and the other low infectivity viruses did not (Figure 3). These results indicate that tRBCs contain sialic acids that all six viruses can bind. In contrast, the results suggest that swRBCs, compared to tRBCs, express (and/or express at an optimal density) specific sialic acids that only the high infectivity viruses can recognize and use to agglutinate the cells. The difference in swRBC agglutination between rgMN and rgONT, and the association of swRBC agglutination with infectivity in SRECs, provides additional evidence that virus infectivity levels are due to differences in virus binding to sialic acids.
Figure 3.
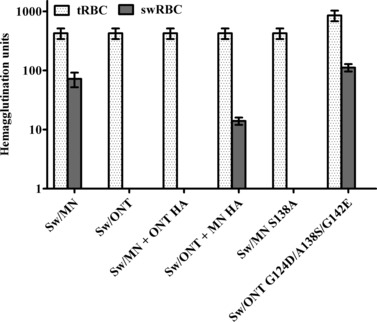
Hemagglutination of turkey and swine red blood cells (tRBCs and swRBCs). Results shown are mean ± SEM of four independent experiments. Similar hemagglutination patterns were seen with swRBCs collected from different donor pigs.
Virus binding to synthetic glycopolymers
A direct binding assay was used to examine virus binding to synthetic sialylated glycopolymers. Binding of rgMN, rgONT, rgMN S138A, and rgONT G124D/A138S/G142E to the following polymers was examined: NeuAcα2‐6Galβ1‐4GlcNAcβ (6′SLN), NeuAcα2‐6Galβ1‐4Glcβ (6′SL), NeuAcα2‐3Galβ1‐4GlcNAcβ (3′SLN), NeuAcα2‐3Galβ1‐4Glcβ (3′SL), and NeuGcα2‐6Galβ1‐4GlcNAcβ (6′Gc‐SLN). The human‐ and swine‐like receptor analog are 6′SLN, while the avian‐like receptor analog is 3′SL. 41 , 42 , 43 All viruses preferentially bound to α2‐6 sialylated glycans over α2‐3 glycans, as virus–polymer dissociation constants were lower (i.e., higher affinity binding) for 6′SLN and 6′SL than they were for 3′SLN and 3′SL [Figure 4; similar results were seen with HA reassortant viruses (data not shown)]. With regard to sialic acid species, all viruses preferentially bound to 6′SLN over 6′Gc‐SLN, demonstrating a binding preference for NeuAc over NeuGc. Taken together, the data indicate that the viruses bind α2‐6‐ over α2‐3‐linked sialic acids and NeuAc over NeuGc sialic acids. However, no substantial differences between the viruses were seen, as binding affinities to this set of sialylated glycopolymers were similar regardless of virus infectivity levels in SRECs. Thus, while this assay indicated which sialic acid linkages and species the viruses preferentially bind, it was not able to discern differences in virus binding that could account for differences in virus infectivity in SRECs.
Figure 4.
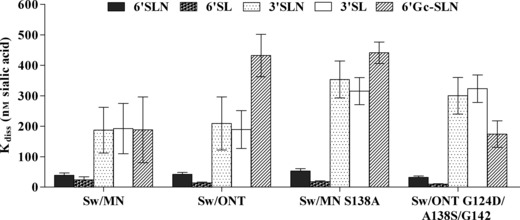
Binding affinity of viruses to synthetic glycopolymers. Linear regression analysis was conducted using Scatchard plots, and values are presented as the constant of dissociation (K diss). Lower values indicate higher affinity binding. Results shown are mean ± SEM of six independent experiments.
Virus binding to glycan microarray
To examine virus binding to a wider range of structurally different sialic acid‐containing glycans, viruses were labeled with AlexaFluor 488 as described previously 24 , 25 , 26 and binding was examined in a glycan microarray that contains synthetic and natural glycans. 27 We have previously published microarray binding of the parental rgMN and rgONT viruses, showing that while both viruses preferentially bound α2‐6‐ over α2‐3‐linked and NeuAc over NeuGc sialylated glycans, subtle differences in binding were seen between these two viruses. 15 Shown here are data examining microarray binding of the HA reassortant and point mutant viruses. Results from the glycan array confirmed those of the synthetic glycopolymer binding assay, in that all viruses preferred α2‐6‐ over α2‐3‐linked and NeuAc over NeuGc sialylated glycans. Specifically, viruses bound many glycans that contained α2‐6‐linked sialic acid, but no virus showed appreciable binding to asialo, α2‐3‐, or α2‐8‐linked sialylated glycans (Figure 5). The viruses also only bound to glycans containing the NeuAc form of sialic acid. Two NeuGc‐containing glycans are present on the array, and none of the viruses bound to either NeuGc glycan (data not shown). In addition, one glycan (6′SLN) is present on the microarray in both NeuAc and NeuGc forms, and all viruses bound NeuAc much more strongly [between 14× to 600× more strongly, depending on the virus (data not shown)], demonstrating preferential binding for NeuAc over NeuGc sialic acids.
Figure 5.
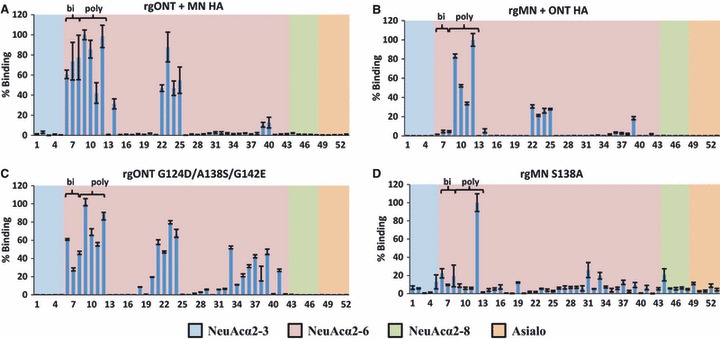
Glycan microarray analysis of virus binding. (A) rgONT + MN HA, (B) rgMN + ONT HA, (C) rgONT G124D/A138S/G142E, and (D) rgMN S138A binding to glycans was performed on microarray version 4.1 from the Consortium for Functional Glycomics. Results shown are the average of four replicate spots ±SEM after the highest and lowest readings of six were excluded, with the highest value set to 100. As the binding of all α2‐3‐linked, α2‐8‐linked, and asialo glycans was low, the structures of only five of each glycan are plotted on the graph for the clarity of presentation. Glycan structures can be found in Table S1. Bi = biantennary α2‐6‐containing sialylated glycans (the first three NeuAcα2‐6 bars). Poly = polylactosamine α2‐6‐containing sialylated glycans (the second four NeuAcα2‐6 bars). Binding data for parental rgMN and rgONT have been published previously. 15 HA, hemagglutinin; NeuAc, N‐acetylneuraminic acid.
When binding to specific NeuAcα2‐6 sialylated glycans available on the microarray is examined in detail, more subtle differences in virus binding profiles become evident. Two major types of glycans that viruses bound to were biantennary and polylactosamine glycans. Lactose, which consists of one galactose and one glucose moiety, is integral to both of these glycan types. Biantennary glycans terminate in two antennae (each of which contain one lactose moiety capped with sialic acid), whereas polylactosamine glycans terminate in two or more repeat units of lactose capped with sialic acid. Our results demonstrate that among NeuAcα2‐6 sialylated glycans, rgMN bound both biantennary and polylactosamine approximately equally, while rgONT preferentially bound polylactosamine glycans 15 (and data not shown). As expected, the rgONT + MN HA virus had a binding profile similar to rgMN, as it bound biantennary and polylactosamine glycans to approximately equal degrees (Figure 5A). Conversely, rgMN + ONT HA exhibited similar binding to rgONT, by preferentially binding polylactosamine glycans (Figure 5B). Thus, exchanging the HA gene reverses both SREC infectivities and the binding profiles of the viruses. Furthermore, with respect to biantennary and polylactosamine glycan binding, rgONT G124D/A138S/G142E bound similarly to rgMN, and rgMN S138A bound similarly to rgONT (Figure 5C–D). In summary, high infectivity viruses bind to both biantennary and polylactosamine glycans, whereas low infectivity viruses bind polylactosamine glycans but have little to no affinity for biantennary glycans. The association between the binding profiles and SREC infectivity levels further indicates that the binding differences are likely responsible for virus infectivity phenotypes in SRECs.
Virus entry kinetics
Although the results of the above experiments indicate that differences in sialic acid‐mediated events are likely responsible for the infectivity phenotypes of rgMN and rgONT in SRECs, we considered the possibility that more than one mechanism could be involved. As such, virus entry kinetics and the pH of virus–endosome fusion were also examined. To examine virus entry kinetics, the time to virus–endosome fusion was examined using inhibitors of endosomal acidification. Influenza viruses that have undergone fusion with endosomes are resistant to endosomal acidification inhibition, so time to resistance is a measure for virus entry kinetics up to the fusion event. 44 , 45 , 46 SRECs were infected with each virus, and inhibitors were added every 40 minutes after infection to determine the time to which viruses attained resistance to the inhibitors. All viruses became resistant to the inhibitors at approximately the same time (200–240 minutes), and the resistance kinetics of all viruses were comparable (Figure S1). Thus, it is unlikely that rgMN and rgONT infectivity levels in SRECs are owing to differences in virus entry kinetics.
pH stability of viruses
In the final step of influenza virus entry through endosomes, endosomal acidification triggers the viral HA protein to undergo irreversible conformational changes to induce virus–endosome fusion. Influenza viruses typically fuse between pH 5·0 and pH 6·0, but individual viruses fuse at different specific pH levels. 19 To determine whether the high infectivity viruses fuse with endosomes at a different pH than the low infectivity viruses, the fusion pH of each virus was examined with an inactivation assay. If the pH triggers a conformational change in the HA protein, the HA is not in the correct formation to bind sialic acids and the virus is not infectious. 47 , 48 The HA of rgMN underwent its conformational change at higher pH than the HA of rgONT, as rgMN lost infectious titer at a higher pH (pH 5·4), whereas rgONT retained infectivity down to pH 5·2 (Figure S2). Physiologically, as the viruses enter cells, this could mean that rgMN fuses with endosomes sooner than rgONT. However, when mutant viruses were examined, this difference did not parallel SREC infectivity levels, so it is unlikely that fusion pH explains virus infectivity levels in SRECs.
Discussion
Elucidating the mechanism responsible for the infectivity levels of rgMN and rgONT in SRECs further informs our understanding of host range specificity of influenza viruses and may provide insight at a cellular level as to why the triple reassortant H3N2 viruses emerged and spread widely throughout the North American swine population. Results of SREC infectivity experiments described here demonstrated that the only HA‐mediated stage of infection where differences paralleled infectivity levels was sialic acid binding.
Sialic acid binding has previously been found to be a major determinant of influenza virus species specificity, as avian viruses preferentially bind α2‐3‐linked sialic acids, while human and swine viruses preferentially bind α2‐6 sialylated glycans. 32 , 33 , 41 , 49 Our studies further support the importance of sialic acid binding specificity in determining influenza virus infectivity, but previous studies have not dissected fine details of sialic acid binding by swine influenza viruses in parallel with studies of infectivity in swine cells. The rgMN and rgONT viruses both preferentially bound to α2‐6‐linked sialic acids (4, 5). This is not unexpected, as both viruses contain human‐lineage HA genes. Our experiments went beyond this initial characterization to examine more subtle binding differences.
All viruses examined in the microarray bound only to α2‐6 sialylated glycans, as no binding was seen to asialo or α2‐3 sialylated glycans (Figure 5). This confirms the results of the polymer binding assay (Figure 4) and complements functional data demonstrating that both rgMN and rgONT utilize α2‐6 sialylated glycans to infect SRECs. 15 These results are also consistent with recent glycan microarray binding analyses of human and swine viruses, where selectivity for α2‐6 and very limited binding to α2‐3 glycans were consistently observed. 50 , 51 The viruses examined in the current study did not bind to every α2‐6 sialylated glycan on the array, indicating that the overall α2‐6 structure is necessary, but not sufficient, for binding. Two major forms of glycans that viruses bound were biantennary and polylactosamine (Figure 5); binding to these glycans was also reported with other swine and human viruses. 50 , 51 Others have reported that human‐origin viruses preferentially bind to α2‐6 polylactosamine glycans. 52 , 53 Our results are in accordance with these reports, as rgONT, a wholly human virus isolated from a single pig, preferentially bound polylactosamine glycans. 15 Conversely, rgMN, representative of the rH3N2 viruses that spread widely throughout the swine population, bound both biantennary and polylactosamine glycans. It is interesting to speculate that rgMN’s ability to bind both biantennary and polylactosamine glycans might be an adaptation to increase infectivity and/or transmission among pigs.
A possible alternative explanation for the infectivity differences described here is that high infectivity viruses use a non‐sialic acid alternative receptor to infect SRECs. However, we believe that the data strongly support the importance of sialic acid‐mediated events in SREC infectivity of these viruses. First, the amino acids important in rgMN and rgONT infectivity levels in SRECs are located in or near the sialic acid binding site of the HA protein. 14 In particular, residue 138 is directly across the binding site from residue 190 and at a right angle to residue 226; all three of these positions are associated with sialic acid binding. 54 (For a further discussion of the topology of these residues, please refer to Busch et al. 14 ) Second, modulation of cell surface sialic acids, which likely affects the sialic acid density, differentially affects high versus low infectivity viruses (1, 2). The relationship between sialic acid density and influenza virus binding has been reported previously. 54 Third, high infectivity viruses are able to agglutinate swRBCs while low infectivity viruses are not (Figure 3). Lastly, the glycan array binding profiles of the parental, HA reassortant, and point mutant viruses consistently paralleled virus infectivity in SRECs (Figure 5).
Our previous mass spectrometry analysis of the N‐ and O‐linked glycans expressed by SRECs showed that SRECs express quantitatively more α2‐6‐ than α2‐3‐linked sialic acid, and that both polylactosamine and biantennary glycans are present. 15 In particular, biantennary glycans on the glycan microarray are almost identical to SREC glycans defined by mass spectrometry (the only difference is that the SREC glycans contain one fucose moiety on the reducing end GlcNAc). Polylactosamine glycans on the microarray are also expressed on SRECs (again, SREC glycans differ only in that they contain one fucose moiety on the reducing end GlcNAc). Thus, the differences seen in glycan array binding are likely to be biologically relevant and directly related to SREC infection.
Previous studies reported that some influenza viruses bind to α2‐8‐linked sialic acids. 55 However, none of the viruses examined here bound to α2‐8 sialylated glycans, so it appears that rgMN and rgONT do not bind this moiety and thus are unlikely to use it to infect SRECs. The lack of virus binding to α2‐8 sialylated glycans complements our prior analysis of SREC glycans by mass spectrometry, where this moiety was not found on SRECs. 15
In summary, sialic acid binding is the most likely determinant of the infectivity phenotypes of rgMN and rgONT in SRECs. The data presented herein expand our understanding of the factors that control infectivity of influenza viruses across species barriers; in this case, viruses with human virus‐lineage HA that either did (Sw/MN) or did not (Sw/ONT) adapt to readily infect pigs. Both rgMN and rgONT preferentially bound NeuAc over NeuGc and α2‐6 over α2‐3 sialylated glycans. However, glycan microarray binding data exposed subtle differences in binding, wherein rgMN and other high infectivity viruses bound equally to biantennary and polylactosamine glycans, while rgONT and other low infectivity viruses preferentially bound polylactosamine glycans. Virus acquisition of binding to these α2‐6 sialylated glycans may be important in the emergence and persistence of novel influenza viruses in the swine population and serve as a potential marker for swine adaptation of influenza viruses.
Supporting information
Figure S1. Virus entry kinetics, measured as time to virus acquisition of resistance to ammonium chloride (A) or monensin (B).
Figure S2. Virus titer after incubation at various pH.
Table S1. Structure of microarray glycans included in Figure 5, as well as virus binding to each glycan, expressed as a percentage of maximum binding.
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
We thank the Consortium for Functional Glycomics, and Drs David Smith and Jamie Heimburg‐Molinaro in particular, for helpful comments regarding labeling viruses with AlexaFluor 488 and for performing the glycan array binding. Thanks also to Dr. Ronald Schnaar of Johns Hopkins University for providing the ManNGc sialic acid precursor, to Dr. M. Suresh of the University of Wisconsin‐Madison for flow cytometry assistance, and to Dr. Jim Gern and Rebecca Brockman‐Schneider of the University of Wisconsin‐Madison for assistance with primary cells. This work was supported by the NIH NIAID (grant R01AI060646).
References
- 1. Tumpey TM, Basler CF, Aguilar PV et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 2005; 310:77–80. [DOI] [PubMed] [Google Scholar]
- 2. Olsen CW, Brown IH, Easterday BC, van Reeth K. Swine influenza; in Straw BE, Zimmerman JJ, D’Allaire SD, Taylor DJ. (eds): Diseases of Swine, 9th edn Ames, IA: Iowa State Press, 2006; 469. [Google Scholar]
- 3. Zhou NN, Senne DA, Landgraf JS et al. Genetic reassortment of avian, swine, and human influenza A viruses in American pigs. J Virol 1999; 73:8851–8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karasin AI, Schutten MM, Cooper LA et al. Genetic characterization of H3N2 influenza viruses isolated from pigs in North America, 1977–1999: evidence for wholly human and reassortant virus genotypes. Virus Res 2000; 68:71–85. [DOI] [PubMed] [Google Scholar]
- 5. Webby RJ, Swenson SL, Krauss SL, Gerrish PJ, Goyal SM, Webster RG. Evolution of swine H3N2 influenza viruses in the United States. J Virol 2000; 74:8243–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karasin AI, Landgraf J, Swenson S et al. Genetic characterization of H1N2 influenza A viruses isolated from pigs throughout the United States. J Clin Microbiol 2002; 40:1073–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma W, Gramer M, Rossow K, Yoon KJ. Isolation and genetic characterization of new reassortant H3N1 swine influenza virus from pigs in the midwestern United States. J Virol 2006; 80:5092–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vincent AL, Ma W, Lager KM, Gramer MR, Richt JA, Janke BH. Characterization of a newly emerged genetic cluster of H1N1 and H1N2 swine influenza virus in the United States. Virus Genes 2009; 39:176–185. [DOI] [PubMed] [Google Scholar]
- 9. Ma W, Vincent AL, Gramer MR et al. Identification of H2N3 influenza A viruses from swine in the United States. Proc Natl Acad Sci U S A 2007; 104:20949–20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garten RJ, Davis CT, Russell CA et al. Antigenic and genetic characteristics of swine‐origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009; 325:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith GJ, Vijaykrishna D, Bahl J et al. Origins and evolutionary genomics of the 2009 swine‐origin H1N1 influenza A epidemic. Nature 2009; 459:1122–1125. [DOI] [PubMed] [Google Scholar]
- 12. Landolt GA, Karasin AI, Phillips L, Olsen CW. Comparison of the pathogenesis of two genetically different H3N2 influenza A viruses in pigs. J Clin Microbiol 2003; 41:1936–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Landolt GA, Karasin AI, Schutten MM, Olsen CW. Restricted infectivity of a human‐Lineage H3N2 influenza A virus in pigs is hemagglutinin and neuraminidase gene dependent. J Clin Microbiol 2006; 44:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Busch MG, Bateman AC, Landolt GA et al. Identification of amino acids in the HA of H3 influenza viruses that determine infectivity levels in primary swine respiratory epithelial cells. Virus Res 2008; 133:269–279. [DOI] [PubMed] [Google Scholar]
- 15. Bateman AC, Karamanska R, Busch MG, Dell A, Olsen CW, Haslam SM. Glycan analysis and influenza A virus infection of primary swine respiratory epithelial cells: the importance of NeuAc{alpha}2‐6 glycans. J Biol Chem 2010; 285:34016–34026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ito T, Couceiro JN, Kelm S et al. Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J Virol 1998; 72:7367–7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nelli RK, Kuchipudi SV, White GA, Perez BB, Dunham SP, Chang KC. Comparative distribution of human and avian type sialic acid influenza receptors in the pig. BMC Vet Res 2010; 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trebbien R, Larsen LE, Viuff BM. Distribution of sialic acid receptors and influenza A virus of avian and swine origin in experimentally infected pigs. Virol J 2011; 8:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 2000; 69:531–569. [DOI] [PubMed] [Google Scholar]
- 20. Reed L, Muench H. A simple method of estimating 50 percent endpoints. Am J Hyg 1938; 27:493–497. [Google Scholar]
- 21. Kendal AP, Pereira MS. Concepts and procedures for laboratory‐based influenza surveillance. U.S. Department of Health and Human Services 1982.
- 22. Matrosovich M, Tuzikov A, Bovin N et al. Early alterations of the receptor‐binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J Virol 2000; 74:8502–8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landolt GA, Karasin AI, Hofer C, Mahaney J, Svaren J, Olsen CW. Use of real‐time reverse transcriptase polymerase chain reaction assay and cell culture methods for detection of swine influenza A viruses. Am J Vet Res 2005; 66:119–124. [DOI] [PubMed] [Google Scholar]
- 24. Kumari K, Gulati S, Smith DF, Gulati U, Cummings RD, Air GM. Receptor binding specificity of recent human H3N2 influenza viruses. Virol J 2007; 4:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lugovtsev VY, Smith DF, Weir JP. Changes of the receptor‐binding properties of influenza B virus B/Victoria/504/2000 during adaptation in chicken eggs. Virology 2009; 394:218–226. [DOI] [PubMed] [Google Scholar]
- 26. Gulati S, Smith DF, Air GM. Deletions of neuraminidase and resistance to oseltamivir may be a consequence of restricted receptor specificity in recent H3N2 influenza viruses. Virol J 2009; 6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blixt O, Head S, Mondala T et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci U S A 2004; 101:17033–17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shukla AK, Schauer R. Analysis of sialidase and N‐acetylneuraminate pyruvate‐lyase substrate specificity by high‐performance liquid chromatography. Anal Biochem 1986; 158:158–164. [DOI] [PubMed] [Google Scholar]
- 29. Suzuki T, Horiike G, Yamazaki Y et al. Swine influenza virus strains recognize sialylsugar chains containing the molecular species of sialic acid predominantly present in the swine tracheal epithelium. FEBS Lett 1997; 404:192–196. [DOI] [PubMed] [Google Scholar]
- 30. Collins BE, Fralich TJ, Itonori S, Ichikawa Y, Schnaar RL. Conversion of cellular sialic acid expression from N‐acetyl‐ to N‐glycolylneuraminic acid using a synthetic precursor, N‐glycolylmannosamine pentaacetate: inhibition of myelin‐associated glycoprotein binding to neural cells. Glycobiology 2000; 10:11–20. [DOI] [PubMed] [Google Scholar]
- 31. Wang SP, Lin TY. Laboratory studies on some biological properties of the Far East influenza virus. J Infect Dis 1958; 103:178–182. [DOI] [PubMed] [Google Scholar]
- 32. Rogers GN, Paulson JC, Daniels RS, Skehel JJ, Wilson IA, Wiley DC. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 1983; 304:76–78. [DOI] [PubMed] [Google Scholar]
- 33. Rogers GN, D’Souza BL. Receptor binding properties of human and animal H1 influenza virus isolates. Virology 1989; 173:317–322. [DOI] [PubMed] [Google Scholar]
- 34. Killian ML. Hemagglutinatibon assay for the avian influenza virus; in Spackman E. (ed.): Methods in Molecular Biology: Avian Influenza Virus. Totown, NJ: Humana Press, 2008; 47–52. [DOI] [PubMed] [Google Scholar]
- 35. Leuwerke B, Kitikoon P, Evans R, Thacker E. Comparison of three serological assays to determine the cross‐reactivity of antibodies from eight genetically diverse U.S. swine influenza viruses. J Vet Diagn Invest 2008; 20:426–432. [DOI] [PubMed] [Google Scholar]
- 36. Stephenson I, Wood JM, Nicholson KG, Charlett A, Zambon MC. Detection of anti‐H5 responses in human sera by HI using horse erythrocytes following MF59‐adjuvanted influenza A/Duck/Singapore/97 vaccine. Virus Res 2004; 103:91–95. [DOI] [PubMed] [Google Scholar]
- 37. Dong J, Matsuoka Y, Maines TR et al. Development of a new candidate H5N1 avian influenza virus for pre‐pandemic vaccine production. Influenza Other Respi Viruses 2009; 3:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ito T, Suzuki Y, Mitnaul L, Vines A, Kida H, Kawaoka Y. Receptor specificity of influenza A viruses correlates with the agglutination of erythrocytes from different animal species. Virology 1997; 227:493–499. [DOI] [PubMed] [Google Scholar]
- 39. Ito T, Kawaoka Y, Nomura A, Otsuki K. Receptor specificity of influenza A viruses from sea mammals correlates with lung sialyloligosaccharides in these animals. J Vet Med Sci 1999; 61:955–958. [DOI] [PubMed] [Google Scholar]
- 40. Corfield AP, Veh RW, Wember M, Michalski JC, Schauer R. The release of N‐acetyl‐ and N‐glycolloyl‐neuraminic acid from soluble complex carbohydrates and erythrocytes by bacterial, viral and mammalian sialidases. Biochem J 1981; 197:293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gambaryan AS, Karasin AI, Tuzikov AB et al. Receptor‐binding properties of swine influenza viruses isolated and propagated in MDCK cells. Virus Res 2005; 114:15–22. [DOI] [PubMed] [Google Scholar]
- 42. Gambaryan AS, Tuzikov AB, Piskarev VE et al. Specification of receptor‐binding phenotypes of influenza virus isolates from different hosts using synthetic sialylglycopolymers: non‐egg‐adapted human H1 and H3 influenza A and influenza B viruses share a common high binding affinity for 6′‐sialyl(N‐acetyllactosamine). Virology 1997; 232:345–350. [DOI] [PubMed] [Google Scholar]
- 43. Owen RE, Yamada E, Thompson CI et al. Alterations in receptor binding properties of recent human influenza H3N2 viruses are associated with reduced natural killer cell lysis of infected cells. J Virol 2007; 81:11170–11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoshimura A, Ohnishi S. Uncoating of influenza virus in endosomes. J Virol 1984; 51:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gujuluva CN, Kundu A, Murti KG, Nayak DP. Abortive replication of influenza virus A/WSN/33 in HeLa229 cells: defective viral entry and budding processes. Virology 1994; 204:491–505. [DOI] [PubMed] [Google Scholar]
- 46. Misinzo G, Delputte PL, Nauwynck HJ. Inhibition of endosome‐lysosome system acidification enhances porcine circovirus 2 infection of porcine epithelial cells. J Virol 2008; 82:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wagner R, Heuer D, Wolff T, Herwig A, Klenk HD. N‐Glycans attached to the stem domain of haemagglutinin efficiently regulate influenza A virus replication. J Gen Virol 2002;83(Pt 3): 601–609. [DOI] [PubMed] [Google Scholar]
- 48. Korte T, Ludwig K, Booy FP, Blumenthal R, Herrmann A. Conformational intermediates and fusion activity of influenza virus hemagglutinin. J Virol 1999; 73:4567–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matrosovich MN, Gambaryan AS, Teneberg S et al. Avian influenza A viruses differ from human viruses by recognition of sialyloligosaccharides and gangliosides and by a higher conservation of the HA receptor‐binding site. Virology 1997; 233:224–234. [DOI] [PubMed] [Google Scholar]
- 50. Chen LM, Rivailler P, Hossain J et al. Receptor specificity of subtype H1 influenza A viruses isolated from swine and humans in the United States. Virology 2011; 412:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bradley KC, Jones CA, Tompkins SM et al. Comparison of the receptor binding properties of contemporary swine isolates and early human pandemic H1N1 isolates (Novel 2009 H1N1). Virology 2011; 413:169–182. [DOI] [PubMed] [Google Scholar]
- 52. Chandrasekaran A, Srinivasan A, Raman R et al. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol 2008; 26:107–113. [DOI] [PubMed] [Google Scholar]
- 53. Srinivasan A, Viswanathan K, Raman R et al. Quantitative biochemical rationale for differences in transmissibility of 1918 pandemic influenza A viruses. Proc Natl Acad Sci U S A 2008; 105:2800–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martin J, Wharton SA, Lin YP et al. Studies of the binding properties of influenza hemagglutinin receptor‐site mutants. Virology 1998; 241:101–111. [DOI] [PubMed] [Google Scholar]
- 55. Wu W, Air GM. Binding of influenza viruses to sialic acids: reassortant viruses with A/NWS/33 hemagglutinin bind to alpha2,8‐linked sialic acid. Virology 2004; 325:340–350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Virus entry kinetics, measured as time to virus acquisition of resistance to ammonium chloride (A) or monensin (B).
Figure S2. Virus titer after incubation at various pH.
Table S1. Structure of microarray glycans included in Figure 5, as well as virus binding to each glycan, expressed as a percentage of maximum binding.
Supporting info item
Supporting info item
Supporting info item