

Abstract
After separating from a primary tumor, metastasizing cells enter the circulatory system and interact with host cells before lodging in secondary organs. Previous studies have implicated the surface glycoproteins CD44 and carcinoembryonic antigen (CEA) in adhesion, migration, and invasion, suggesting that they may influence metastatic progression. To elucidate the role of these multifunctional molecules while avoiding the potential drawbacks of ectopic expression or monoclonal antibody treatments, we silenced the expression of CD44 and/or CEA in LS174T colon carcinoma cells and analyzed their ability to metastasize in 2 independent mouse models. Quantitative PCR revealed that CD44 knockdown increased lung and liver metastasis >10-fold, while metastasis was decreased by >50% following CEA knockdown. These findings were corroborated by in vitro experiments assessing the metastatic potential of LS174T cells. Cell migration was decreased as a result of silencing CEA but was enhanced in CD44-knockdown cells. In addition, CD44 silencing promoted homotypic aggregation of LS147T cells, a phenotype that was reversed by additional CEA knockdown. Finally, CD44-knockdown cells exhibited greater mechanical compliance than control cells, a property that correlates with increased metastatic potential. Collectively, these data indicate that CEA, but not CD44, is a viable target for therapeutics aimed at curbing colon carcinoma metastasis.—Dallas, M. R., Liu, G., Chen, W.-C., Thomas, S. N., Wirtz, D., Huso, D. L., Konstantopoulos, K. Divergent roles of CD44 and carcinoembryonic antigen in colon cancer metastasis.
Keywords: CEA, cytoplasmic compliance, migration
Blood-borne metastasis is a multifaceted process that begins when cancerous cells detach from a primary tumor; a subset of these cells infiltrate the vasculature and travel to distant sites where they can extravasate and colonize secondary organs. The progression of a primary tumor to metastasis is dictated by a number of factors, including but not limited to insensitivity to growth suppressors, unlimited potential to proliferate, migratory and invasive potential, and apoptosis resistance (1). We have recently identified sialofucosylated CD44 and carcinoembryonic antigen (CEA) as functional selectin ligands expressed by metastatic colon carcinoma cells (2, 3). Interestingly, sialofucosylated CD44 and CEA cooperate to mediate colon carcinoma cell binding to E- and L-selectin in shear flow (3). The interactions between these ligands displayed by circulating tumor cells and selectins expressed by host cells play a critical role in the establishment of distant metastases by facilitating the tethering of tumor cells to the blood vessel wall and ultimately their extravasation from the vasculature (4).
In addition to their functional role as selectin ligands, both CD44 and CEA have been implicated in other steps of the metastatic cascade. Specifically, CD44 is a receptor for hyaluronic acid (HA), an extracellular matrix component; CD44-HA binding is thought to play a key role in the regulation of tumor cell migration (5). CD44 has also been reported to bind to fibrin; this interaction is believed to protect circulating tumor cells from immunological and physiological stresses in the bloodstream (5, 6). In addition, CD44 has been implicated in various signaling cascades and angiogenesis (5).
Despite this knowledge, the ultimate role of CD44 in metastasis is a point of considerable debate. While some clinical studies in colon cancer have shown that the expression of CD44 on tumor cells correlates with metastasis and overall tumor progression (7), others have shown that CD44 expression is inversely correlated with tumor progression or is reduced in metastatic tumors (8). Contradictory observations regarding the role of CD44 in metastasis have also been made in breast and prostate cancers (9, 10). Similarly, Gao et al. (11) demonstrated that transfection of prostate cancer cells with CD44 cDNA reduces their ability to metastasize, while Harada et al. (12) showed that transfection with CD44 antisense oligonucleotides attenuates colon cancer metastasis to the liver. The debate is nicely framed by a number of review articles (13, 14).
Due to its high expression in many tumor types and low expression in normal adult tissue, CEA has become one of the most extensively used clinical tumor markers (15). CEA has been linked to a number of processes relevant to cancer progression. These include mediation of both hetero- and homotypic cell-cell interactions (3, 16), apoptosis resistance (17), and immunomodulation (18). Unlike CD44, the body of literature pertaining to the role of CEA in metastasis is not controversial. The expression of CEA is consistently associated with a number of human cancers (19–21), while treatment with anti-CEA antibodies limits the ability of tumor cells to interact with endothelial cells, migrate, and invade in vitro (22).
Much of the literature lending mechanistic insight into the roles of both CD44 and CEA in metastasis relies on interventions such as ectopic expression or monoclonal antibodies (mAbs). The post-translational modifications of ectopically expressed glycoproteins may not reflect those of the naturally expressed molecule. Moreover, mAbs may unintentionally modulate cellular signaling pathways. In light of these shortcomings, in this work we used an RNA interference (RNAi)-based approach to stably silence the endogenous expression of CD44 and CEA in LS71T colon carcinoma cells to test the roles of these molecules in colon cancer metastasis. In doing so, we used 2 independent experimental metastasis models. In the first model, resulting primarily in the formation of lung metastases, wild-type and knockdown LS174T cells are injected into nonobese diabetic severe combined immunodeficient interleukin (IL)-2 receptor-γ null (NSG) mice via the tail vein. The second model, in which tumor cells are injected into the spleens of NSG mice, allows for simultaneous growth of primary tumor in the spleen and metastatic development, largely in the liver.
Utilizing quantitative polymerase chain reaction (qPCR) as a highly sensitive measure of tumor burden (23), we have determined that CD44 and CEA expressed by LS174T colon carcinoma cells have divergent effects on the ability of these cells to metastasize. In each of our models, CD44-knockdown (CD44-KD) LS174T cells exhibited a large increase in metastatic potential when compared with the parental line. Conversely, CEA-knockdown (CEA-KD) cells displayed a reduced ability to metastasize to major organs. These in vivo model results were consistent with in vitro wound healing, aggregation, and microrheology assays. Taken together, our findings provide evidence that CEA, but not CD44, is a potential target for the treatment and prevention of colon carcinoma metastasis.
MATERIALS AND METHODS
Mice
All experimental procedures were in compliance with guidelines provided by the Office of Laboratory Animal Welfare at the National Institutes of Health, and protocols were approved by the Johns Hopkins University Animal Care and Use Committee. Adult male and female NSG mice were used in all in vivo studies.
Cell culture
The human colorectal carcinoma cell line LS174T was obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in recommended medium. CD44-KD, CEA-KD, and CD44/CEA-double knockdown cells were generated in the LS174T cell line via short-hairpin RNA (shRNA; refs. 2, 3). Before use, cells were harvested via mild trypsinization (0.25% trypsin plus EDTA·4Na for 5 min at 37°C) and incubated at 37°C for 2 h to regenerate surface glycoproteins (24, 25).
Flow cytometry
Surface expression levels of CD44 and CEA were measured via flow cytometry. Briefly, cells were resuspended in phosphate-buffered saline with 0.1% bovine serum albumin (wt/vol) at 1 × 106/ml. Cell suspensions were incubated with fluorescein isothiocyanate-conjugated anti-CEA (C365D3; AbD Serotec, Raleigh, NC, USA) or phycoerythrin-conjugated anti-CD44 mAbs (515; BD Pharmingen, San Diego, CA, USA) for 30 min at room temperature. Cell suspensions were diluted to 2 × 105/ml before flow cytometry.
SDS-PAGE and immunoblotting
Protein from cell lysate or supernatant was diluted with reducing sample buffer and separated via 4-20% SDS-PAGE Tris-HCl gels (Bio-Rad Laboratories, Hercules, CA, USA). Resolved proteins were transferred to Immun-blot polyvinylidene difluoride and blocked with StartingBlock (Pierce Biotechnology, Rockford, IL, USA) for 15 min. Immunoblots were stained with anti-CD44 (2C5; R&D Systems, Minneapolis, MN, USA), anti-CEA (COL-1; BD Pharmingen), or anti-β-actin (C4/actin; BD Transduction Laboratories) mAbs, rinsed with TBS/0.1% Tween 20, and incubated with appropriate alkaline phosphatase (AP)- or horseradish peroxidase-conjugated anti-mouse IgG. Western blue AP substrate (Promega, Madison, WI, USA) or SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology) was used to develop immunoblots. Staining intensity was quantified using ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA).
Detection of secreted CEA
Cells were allowed to grow to 80% confluence, at which point growth medium was replaced with serum-free medium. After 48 h, supernatant was collected, filtered through 0.2 μm sterile filters, and submitted directly for immunoblot analysis.
Experimental metastasis assays
Cells were suspended in serum-free medium and stored on ice before injection. Two experimental metastasis models were established in NSG mice.
Tail-vein injection
Mice (12–15 mice/experimental group) were injected with 1 × 105 cells in a volume of 100 μl (1×106 cells/ml) via the tail vein. Animals were killed or died 33-35 d postinjection, and metastasis was detected and quantified in the liver and lungs by qPCR amplification of human long interspersed nuclear element-1 (hLINE-1; ref. 23).
Splenic injection
An incision was made in the left, ventral abdomen, the spleen was exposed, and 1 × 105 cells in 50 μl were injected directly into the spleen of 3-5 mice/experimental group. Animals were killed 35 d postinjection, and tumor burden was detected and quantified in the liver and spleen by qPCR amplification of hLINE-1. Primary tumor burden in the spleen was also assessed by direct measurement of tumor mass.
DNA extraction from mouse tissue
DNA was extracted from mouse tissue using the DNeasy blood and tissue kit (Qiagen, Valenica, CA, USA) as recommended. Extraction was performed in a sterile biological safety cabinet to minimize the risk of human DNA contamination. Lysis was performed in 200 μl vol per 25 mg of fixed (10% buffered formalin) or unfixed liver or lung tissue; 200 μl vol was used for 10 mg of splenic tissue; and 200 μl lysed tissue was used in all subsequent purification steps. Elutions were performed in 200 μl vol and subsequently analyzed via qPCR.
hLINE-1 quantification
qPCR was performed as reported previously (23) with minor modifications. All reactions were prepared in a biological safety cabinet to minimize the risk of human DNA contamination. Briefly, qPCR was performed in 15 μl vol with the following components: 7.5 μl 2× iQ SYBR Green Supermix (Bio-Rad), 1.5 μl each of 10 μM forward (5′-TCACTCAAAGCCGCTCAACTAC-3′) and reverse (5′-TCTGCCTTCATTTCGTTATGTACC-3′) primers (desalted, 25 nmol; Invitrogen, Carlsbad, CA, USA), and 4.5 μl purified DNA. The reaction was monitored on an iCycler/iQ5 (Bio-Rad) with the following cycling: (94°C, 2 min) × 1, (94°C, 10 s; 67°C, 15 s; 70°C, 15 s) × 3, (94°C, 10 s; 64°C, 15 s; 70°C, 15 s) × 3, (94°C, 10 s; 61°C, 15 s; 70°C, 15 s) × 3, and (94°C, 10 s, 59°C, 15 s; 70°C, 15 s) × 35. Threshold cycle number was calculated using Bio-Rad iQ5 software. Dilutions of human DNA purified from LS174T colon carcinoma cells were included in each plate to serve as standards.
Histopathology of lung tissue
Lung samples for pathology were fixed in 10% buffered formalin. Samples to be histologically examined were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin using standard techniques. Other fixed samples were examined visually for gross metastatic foci.
Wound healing assay
Wound healing assays were performed as reported previously (22), with some alterations. Cells were plated at 100% confluence and allowed to attach to tissue culture-treated polystyrene for 6 h. Thin wounds were created with a 25-cm cell scraper (Sarstedt, Numbrecht, Germany) and images (t=0) were taken with an inverted microscope, ×10 objective (Nikon, Tokyo, Japan). Subsequent images were taken every 24 h for a total of 4 d, with the medium being changed every 48 h. Wound width was quantified with ImageJ.
Growth rate assessment
Cells were plated at 50% confluence and allowed to grow for 48 h, at which point the cells were trypsinized, resuspended as singlets, and counted. Half of the trypsinized cells were replated at 50% confluence and allowed to grow for 48 h, after which the cell counting process was repeated. Doubling time was calculated assuming logarithmic growth using initial and final cell counts.
Homotypic cell-cell aggregation assay
Following mild trypsinization, cells were allowed to regenerate surface glycoproteins and aggregate for 2 h under mild agitation. To encourage aggregation, the cells were not vortexed. Samples were immediately imaged using a Countess cell counter (Invitrogen). Images were subsequently analyzed for number of singlets, doublets, triplets, and aggregates with ≥4 cells using ImageJ.
Ballistic particle injection and nanoparticle tracking microrheology
Cells were plated on 35-mm dishes before ballistic injection. When cells reached ∼90% confluence, 100-nm-diameter fluorescent nanoparticles (Invitrogen) were used for injection with a Biolistic PDS-1000/HE particle-delivery system (Bio-Rad). Nanoparticles were coated on microcarriers (Bio-Rad) and allowed to completely dry for 6 h. The 1100-psi rupture disks (Bio-Rad) were used in conjunction with a hepta adapter. After ballistic bombardment, cells were immediately and repeatedly washed with Hanks' balanced salt solution to remove excess particles. Cells were allowed to recover in fresh medium overnight before nanoparticles in cytosol were tracked with a high-magnification objective (×60 Plan Apo lens, N.A. 1.4; Nikon). Movies of the brownian motion of the fluorescent nanoparticles were taken at 30 frames/s for 20 s with an electron-multiplying charge-coupled device (EMCCD) camera (Andor Technology, Belfast, Ireland) mounted on a Nikon TE2000 microscope controlled by Nikon NIS-Element software. Tracking of particle trajectories was performed with customized MATLAB codes (MathWorks, Natick, MA, USA).
Statistical analysis
Data are presented as means ± se. qPCR samples outside the 99.9% confidence interval were identified as outliers and not included in subsequent analyses. One-way analysis of variance (ANOVA) or 2-tailed Student's unpaired t test was used to determine significance. The statistical significance minimum was set at P < 0.05.
RESULTS
CD44 knockdown facilitates, while CEA knockdown limits, metastasis
To study the effect of silencing endogenously expressed CD44 and CEA on the metastatic spread of colon carcinoma cells, we employed an experimental tail-vein metastasis model in NSG mice, injecting parental or stable CD44-KD, CEA-KD, or CD44/CEA-KD cells. Before injection, cells were examined for CD44 and CEA expression via flow cytometry and immunoblot to confirm selective knockdown (Supplemental Fig. S1). Of note, total sLex/a and β1 integrin levels were unaltered by shRNA-mediated silencing of CD44 or CEA, as previously reported (2, 3). In addition to depleting surface-expression levels, knockdown of CEA was also shown to limit CEA secretion (Supplemental Fig. S2). At 5 wk postinjection, animals were killed and lung tissue was isolated to assess tumor burden. Tissue from mice not injected with tumor cells was used as negative control; hLINE quantification in negative control samples was consistently >3 orders of magnitude below metastatic samples (Fig. 1A, B).
Figure 1.

CD44 knockdown facilitates, while CEA knockdown diminishes, metastasis in NSG mice. A, B) Tail-vein (A) or splenic (B) injection metastasis models were initiated with LS174T or CD44-KD, CEA-KD, or CD44/CEA-KD cells. Tumor burden in the lungs resulting from tail-vein injection (A; n≥12) or liver resulting from splenic injection (B; n≥3) was probed via qPCR. Organs from animals not injected with tumor cells were used as negative controls. C) Lungs were isolated 35 d following tail-vein injection of LS174T, CD44-KD, CEA-KD, or CD44/CEA-KD cells. Metastatic foci visible on the surface of a single lung lobe were quantified. D) Representative images of lung surface foci. Scale bar = 3 mm. Bar graphs represent means ± se; n ≥ 3 mice/group. *P < 0.05 vs. parental LS174T cells; §P < 0.05 vs. LS CD44/CEA-KD cells.
Knockdown of CD44 resulted in a 20-fold increase in the concentration of hLINE DNA in the lungs of tail-vein-injected mice. In contrast, CEA knockdown reduced the metastatic potential of LS174T cells by >50% (Fig. 1A). These trends are also observed when tumor burden is evaluated through quantification of lung surface foci; the lungs of mice injected with CD44-KD cells display over twice as many visible metastases, while lungs injected with CEA-KD cells show <50% compared with those of mice injected with the parental cell line (Fig. 1C, D). Histological examination of lung samples is also in agreement with these results, as micrometastases are prevalent in CD44-KD lungs but are difficult to locate in lungs injected with CEA-KD cells (Fig. 2). Similar results were obtained with a second CD44-KD clone (data not shown).
Figure 2.
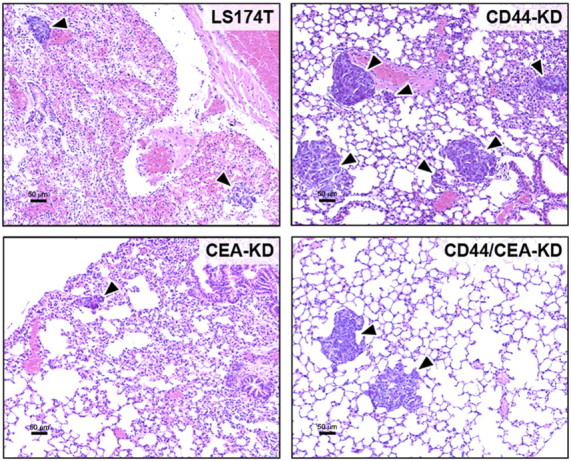
Representative histology of lungs following tail-vein injection. Lungs were isolated 35 d following tail-vein injection of LS174T, CD44-KD, CEA-KD, or CD44/CEA-KD cells. A single lobe from each animal was fixed, H&E stained, and examined for signs of lung metastasis (indicated by arrowheads). Scale bars = 50 μm.
The effect of CD44 or CEA knockdown on metastatic potential was also examined in a splenic injection model, in which parental or knockdown cells were injected directly into the spleens of NSG mice. hLINE analysis of the liver, the most likely site for metastasis from the spleen, showed a large increase in the metastasis of CD44-KD cells but a decrease in metastasis of CEA-KD cells when compared with their wild-type counterparts (Fig. 1B).
The opposing outcomes due to CD44 or CEA knockdown combined to result in CD44/CEA-KD cells whose metastatic potential falls between those in which either molecule has been individually silenced. These effects can be seen in lung hLINE DNA levels, surface metastatic foci, and qualitatively in histological sections (Figs. 1 and 2). The double-knockdown cells also allow us to make alternative, independent assessments of the result of CD44 or CEA knockdown in LS174T cells. The increase in metastasis to the lung from parental to CD44-KD cells is similar to the increase from CEA-KD to CD44/CEA-KD cells (Fig. 1). Similarly, the decrease in metastasis to the lung from parental to CEA-KD cells is comparable to the decrease from CD44-KD to CD44/CEA-KD cells. These data taken together illustrate the divergent effect of CD44 and CEA on the metastatic potential of colon carcinoma cells in 2 distinct metastasis models.
Knockdown of CD44, but not CEA, increases the tumorigenicity of LS174T cells in vivo
To ensure that the metastatic potentials observed in both models were not simply a function of tumorigenicity, we sought to assess the effect of CD44 and/or CEA knockdown on the ability for LS174T cells to form primary tumors in vivo. As such, spleens injected with LS174T parental, CD44-KD, CEA-KD, or CD44/CEA-KD cells were examined for tumor growth. CD44 silencing increased the ability for LS174T cells to form splenic tumors ∼2-fold. This increase was noted using 2 distinct assays: weighing of the spleen and associated tumor (Fig. 3A) and quantification of hLINE DNA in splenic tissue (Fig. 3B). This growth rate disparity was not observed in vitro (Supplemental Fig. S3) and cannot fully account for the dramatic (≥10-fold) increases in metastatic potential of CD44-KD cells. In contrast, tumor burden in the spleens of mice injected with CEA-depleted cells was statistically indistinguishable from their parental counterpart in vivo (Fig. 3). Growth rates were also similar in vitro (Supplemental Fig. S3).
Figure 3.

CD44-KD increases tumor development in vivo. LS174T, CD44-KD, CEA-KD, or CD44/CEA-KD cells (1×105) suspended in serum-free MEM were injected into the spleens of NSG mice. Mice were killed at 35 d postinjection, and primary tumor growth at the site of injection was quantified by direct measurement of tumor mass (A) or via qPCR of hLINE DNA of splenic/tumor tissue (B). Data represent means ± se; n ≥ 4 mice/group. *P < 0.05 vs. parental LS174T cells.
CD44 and CEA differentially affect migration, aggregation, and compliance in vitro
In light of the opposing consequences of CD44 and CEA silencing on LS174T metastatic potential, we sought to test the effect of these knockdowns on LS174T behavior in vitro. Cell migration was first investigated through a wound-healing assay because migration represents a pivotal aspect of cancer metastasis; tumor cells depend on motility to navigate through interstitial tissues and seed new metastatic foci (4). LS174T parental or CD44-KD, CEA-KD, or CD44/CEA-KD cells were plated at confluence and allowed to attach to tissue culture-treated polystyrene. Thin wounds were created, and their rate of closure was tracked over the course of 4 d. Important trends became apparent 2 d after wounding: CD44-KD cells spread faster than parental LS174T cells, while CEA-KD cells filled the same wound more slowly (Fig. 4). Notably, CD44/CEA-KD cells exhibited behavior that combined the consequences of the individual knockdown of either CD44 or CEA in parental cells. In comparing the wound healing capacity of CEA-KD and CD44/CEA-KD cells, which differ only in their expression of CD44, CD44/CEA-KD cells filled the wounds more quickly. This is in agreement with the results of wild-type vs. CD44-KD cells. When the same comparisons were made in CD44-KD and CD44/CEA-KD cells, the decrease in healing rate of double-knockdown cells was similar to that between wild-type and CEA-KD cells. Wound healing results cannot be accounted for by intrinsic growth rate differences, because no significant changes in growth rate were associated with CD44 or CEA knockdown (Supplemental Fig. S3).
Figure 4.
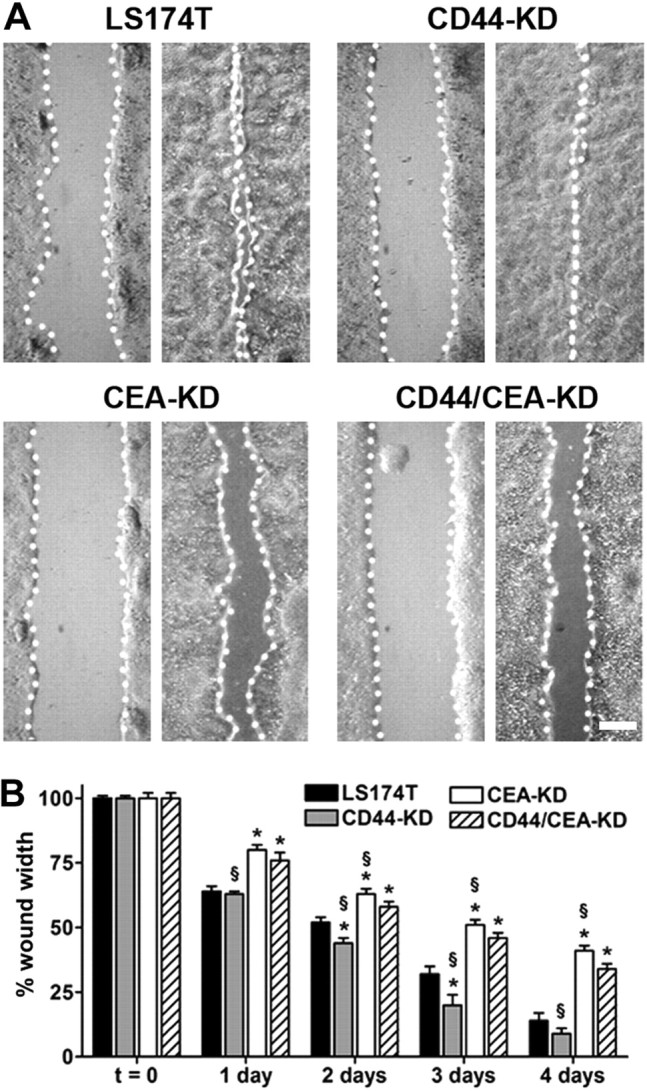
CD44 knockdown augments, while CEA knockdown limits, wound healing. LS174T parental and CD44-KD, CEA-KD, and CD44/CEA-KD cells were detached and plated at 100% confluence; cells were allowed to attach to tissue culture-treated polystyrene for 6 h. Thin wounds were created and imaged (t=0), with subsequent images tracking wound closure taken at 24 h intervals up to 4 d. Initial wound widths for LS174T and CD44-KD, CEA-KD, and CD44/CEA-KD cells were 550 ± 60, 600 ± 6, 620 ± 10, and 570 ± 9 μm, respectively. A) Representative micrographs demonstrate initial (left panels) and final (right panels) wound widths. Scale bar = 200 μm. B) Bars indicate mean ± se wound widths for LS174T (solid bars), CD44-KD (shaded bars), CEA-KD (open bars), and CD44/CEA-KD (hatched bars). *P < 0.05 vs. LS174T cells; §P < 0.05 vs. CD44/CEA-KD cells.
Tumor cell aggregation and metastatic potential are linked due to the propensity for large cell aggregates to arrest in the microvasculature (26, 27). For this reason, we studied the tendency for cells to aggregate in vitro. We observed that CD44-KD cells were 50% more likely to form large clusters, which we defined as aggregates that contained ≥4 cells. This enhanced homotypic aggregation was eliminated by additional knockdown of CEA in CD44/CEA-KD cells (Fig. 5). Of note, E-cadherin levels remain unaltered following knockdown of CD44 and CEA (Supplemental Fig. S1C).
Figure 5.

Homotypic aggregation is increased following CD44 knockdown; additional CEA knockdown eliminates aggregation. A) LS174T parental and CD44-KD, CEA-KD, and CD44/CEA-KD cells were trypsinized and allowed to regenerate cell surface glycoproteins and aggregate under mild agitation for 2 h. Scale bar = 150 μm. B) Clusters made up of ≥4 cells were counted and compared with total number of cell singlets and clusters observed. *P < 0.05 vs. LS174T cells; §P < 0.05 vs. CD44/CEA-KD cells.
Finally, mechanical compliance was assessed following CD44 and CEA knockdown; while the underlying mechanism behind the correlation is currently unknown (4), increased cytoplasmic compliance is an earmark of metastatic tumor cells (28–30). Cells were bombarded with fluorescent nanoparticles, the movement of which was tracked to assess the overall stiffness of LS174T parental and knockdown cells (Fig. 6A and ref. 31). Nanoparticle diffusion through the cytosol of CD44-KD cells was significantly enhanced compared with diffusion through parental cells (Fig. 6B, C), strongly suggesting that CD44-KD cells are more compliant than parental LS174T cells. Although we observed a reduction in nanoparticle diffusion through CEA knockdown compared with parental cell cytoplasm, this difference was not statistically significant.
Figure 6.

CD44 knockdown increases compliance of LS174T cells. LS174T and CD44-KD, CEA-KD, and CD44/CEA-KD cells were ballistically injected with fluorescent nanoparticles whose brownian motion was tracked to assess cytoplasmic compliance. A) Representative phase-contrast/fluorescent overlay of LS174T cell following ballistic injection. Scale bar = 20 μm. Inset: ×3 view of indicated area. B) Particles were tracked for 20 s at a rate of 30 frames/s. Images show representative particle tracks where passage of time is indicated by color change. Scale bar = 5 μm. C) Bar graph represents means ± se of mean squared displacement (MSD) of ≥65 particles. *P < 0.05 vs. LS174T WT cells; §P < 0.05 vs. LS CD44/CEA-KD cells.
DISCUSSION
Although numerous studies have examined the roles of CD44 in cancer metastasis, they provide conflicting data as to the role of CD44 expression in cancer progression. Where several reports show that CD44 is strongly expressed in aggressive tumors (32, 33), others suggest that the presence of CD44, in fact, acts to inhibit tumor progression (34, 35). There are also many contradictory studies discussing the associations of CD44 variant (CD44v) isoforms with an increase in metastatic potential. Some of these indicate that CD44v can be correlated with an increase in metastasis or with progression of colon (36), pancreatic (37), and liver (38) cancers. However, there are a number of reports revealing that CD44v is down-regulated in or has no relevance to the progression of colon (39) and prostate (40) cancers. To date, much of what is known regarding the function of CD44 in cancer metastasis is based on experiments utilizing either ectopic expression of CD44 or anti-CD44 mAb treatments. These interventions may complicate data interpretation, as ectopic expression of glycoproteins may not recapitulate naturally expressed glycans. In addition, mAb treatments may inadvertently activate signaling cascades by cross-linking surface antigens (41). In this work, we have exploited shRNA-mediated gene silencing to demonstrate that the expression of CD44 in LS174T colon carcinoma cells is inversely proportional with their metastatic potential.
It is noteworthy that our results are in direct contrast with those reported by Harada et al. (12), where it was reported that antisense-mediated down-regulation of CD44 leads to a decrease in the metastatic potential of LS174T cells. While we observed an increase in tumorigenicity in the spleen following CD44 knockdown (Fig. 3), Harada et al. observed no increase in in vivo growth potential in subcutaneous tumors using LS174T cells transfected with antisense CD44. In addition, our in vivo observations for increased metastatic potential of CD44-depleted colon carcinoma cells are supported by several independent in vitro assays showing increased migratory potential, cytoplasmic compliance, and propensity to form aggregates. These in vitro measurements directly correlate with an enhanced metastatic potential that we observe in vivo.
CD44 is often considered an adhesive molecule, as its principal ligand, HA, is a common component of the extracellular matrix. Interestingly, the interaction between CD44 and HA has been implicated in invasion of tumor cells in vitro (42). It is unlikely, however, that our wound-healing results can be attributed to CD44-HA binding, as wound-healing experiments performed with exogenously added HA showed the same trends as the HA-free assays (data not shown).
It has been shown in a number of studies that metastatic tumor cells are significantly more compliant than cells that cannot metastasize (28–30). Although the mechanism behind this observation has not yet been elucidated (4), the phenomenon has been made in a variety of tumor types and confirmed with assays, including atomic force microscopy (28) and optical cell stretching (29). The results we report here further this association. Using particle tracking microrheology (31), a technique that allows for direct measurement of cytoplasmic stiffness throughout the cytosol, we show that nanoparticle diffusion through the cytoplasm of the highly metastatic CD44-KD cells is significantly increased (Fig. 6), strongly suggesting that these cells are more compliant than the parental LS174T cells. The cytoplasmic tail of CD44 interacts with ankyrin, merlin, and members of the ezrin, radixin, moesin (ERM) family (5), which act as crosslinkers of integral membrane proteins and the actin cytoskeleton. The increased compliance and migratory potential of CD44-KD cells may be attributed to an altered physical connection between the actin network and plasma membrane. Interestingly, CD44 is also a target of the Wnt signaling cascade, which regulates cell migration (43, 44).
CEA has been shown to be overexpressed by many cancers, including those derived from colon, rectal, and pancreatic tissue (19, 21). The role of CEA as an adhesion molecule has been well documented in both homotypic and heterotypic interactions. We previously demonstrated that CEA possesses E- and L-selectin ligand activity, which facilitates tumor cell-host cell interactions in the vasculature (3). Homotypic cell-cell interactions may be largely mediated by CEA alone, as demonstrated by a reduction of cell clumping in vitro following treatment with anti-CEA antibody fragment antigen binding (Fab′) fragments (27). In this work, we show that CD44 knockdown in LS174T cells results in an increased propensity for these cells to form large clumps (Fig. 5). The increase in aggregation is reversed when CEA is additionally knocked down, suggesting that the clumping behavior is predominantly CEA dependent. This inhibition of clumping would be expected to limit arrest in the microvasculature due to size exclusion and, as a result, the metastatic potential of these cells. Indeed, prior studies have verified that pretreatment of CEA-expressing cells with anti-CEA antibodies leads to a reduction in metastasis to the liver, perhaps due to the prevention of cell clumping (22, 45).
Blumenthal et al. (22) reported that CEA may play an integral role in cell migration. Their treatment of LS174T cells with antibodies that bind to the A1B1 or N domains found in CEA led to a reduction in the in vitro migration and invasion (22). These results compare well with our observation that CEA-KD cells show reduced potential for wound healing. Less migratory cells would be expected to exhibit a reduced capacity to metastasize, which is precisely what we observe in CEA-KD cells in vivo (Figs. 1 and 2).
In addition to supporting cell-cell adhesion and promoting cell migration, secreted CEA can facilitate metastasis through modulation of the host immune response, mainly through Kupffer cells. These liver-specific macrophages clear secreted CEA from the circulation and in doing so initiate a signaling cascade resulting in production of proinflammatory cytokines, such as IL-1, IL-6, and tumor necrosis factor-α (TNF-α) (46, 47). Release of these cytokines increases expression of endothelial-cell adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin, all of which promote tumor cell adhesion to, and ultimately extravasation through, vascular endothelium (48). In addition, activated Kupffer cells release IL-10, which enhances tumor cell survival by inhibiting inducible nitric oxide synthetase, nitric oxide, and reactive oxygen species (18, 49). Interactions between CEA and transforming growth factor-β (TGF-β) have also been reported to inhibit TGF signaling and promote tumor cell proliferation, although the mechanism is unknown (15). Taken together, the large quantities of CEA secreted by LS174T parental and CD44-KD cells may aid these cells in adhering to vascular endothelium and surviving microenvironmental stresses, thereby increasing their metastatic potential. Similarly, the decrease in CEA secretion in CEA- and CD44/CEA-KD cells may be partially responsible for the observed decrease in metastatic potential of these cells compared with parental and CD44-KD cells, respectively.
Recently, the expression of CEA has been associated with cancer stem cells (CSCs; refs. 50, 51). These cells share characteristics with embryonic stem cells, namely in that they have the ability to indefinitely self-renew, differentiate into diverse populations, and resist apoptosis (50, 51). Wirth et al. (17) demonstrated that cells in which endogenous CEA levels are reduced via tetracycline controlled ribozyme targeting exhibit increased apoptosis in the presence of 5-fluorouracil and γ-interferon when compared with cells with baseline CEA levels. These baseline CEA cells also showed higher colony forming ability in vitro and in vivo (17). A separate report shows that long-term propagation of CD133+ colon cancer cells results in a population of CSC-like cells that are enriched in CEA (51). If CEA is in fact a marker of CSCs, reducing its expression may divert the cells in question away from CSC-like behavior. Cells of this nature would exhibit a reduced ability to form metastatic colonies in vivo, as we have observed in CEA-KD cells.
We have demonstrated that expression of CD44 and CEA have contrasting effects on the metastatic potential of LS174T cells. We observe CD44, as expressed by LS174T colon carcinoma cells, functions as a tumor suppressor. When the expression of CD44 is silenced, we see a dramatic increase in metastatic potential. This trend is observed in multiple metastatic models and cannot simply be attributed to an increased in vivo growth rate. Conversely, CEA acts to promote the metastatic dissemination of tumor cells. On depletion of CEA, the metastatic potential of LS174T cells is dramatically reduced. As was true for CD44, the effect of CEA silencing is independent of model, organ system, and growth rate in vivo. Altogether, our data provide evidence that therapeutic interventions against CD44-expressing cells may not control tumor progression or metastasis. Alternatively, we have shown that CEA may be a practical target molecule for the eradication of metastatic colon carcinoma cells.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
Acknowledgments
This work was supported by U.S. National Institutes of Health/National Cancer Institute grants R01-CA-101135 and U54-CA-143868.
The authors thank Philip Yang for critical input to this work.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- CD44-KD
- CD44-knockdown
- CD44v
- CD44 variant
- CEA
- carcinoembryonic antigen
- CEA-KD
- carcinoembryonic antigen-knockdown
- CSC
- cancer stem cell
- HA
- hyaluronic acid
- hLINE
- human long interspersed nuclear element-1
- IL
- interleukin
- mAb
- monoclonal antibody
- NSG
- nonobese diabetic severe combined immunodeficient interleukin-2 receptor-γ null
- qPCR
- quantitative polymerase chain reaction
- shRNA
- short-hairpin RNA
REFERENCES
- 1.Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell , 646–674 [DOI] [PubMed] [Google Scholar]
- 2.Napier S. L., Healy Z. R., Schnaar R. L., Konstantopoulos K. (2007) Selectin ligand expression regulates the initial vascular interactions of colon carcinoma cells: the roles of CD44v and alternative sialofucosylated selectin ligands. J. Biol. Chem. , 3433–3441 [DOI] [PubMed] [Google Scholar]
- 3.Thomas S. N., Zhu F., Schnaar R. L., Alves C. S., Konstantopoulos K. (2008) Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E-and L-selectin in shear flow. J. Biol. Chem. , 15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wirtz D., Konstantopoulos K., Searson P. C. (2011) The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer , 512–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ponta H., Sherman L., Herrlich P. a. (2003) CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. , 33–45 [DOI] [PubMed] [Google Scholar]
- 6.Alves C. S., Yakovlev S., Medved L., Konstantopoulos K. (2009) Biomolecular characterization of CD44-fibrin(ogen) binding. J. Biol. Chem. , 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wielenga V. J. M., Heider K. H., Johan G., Offerhaus A., Adolf G. R., van den Berg F. M., Ponta H., Herrlich P., Pals S. T. (1993) Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res. , 4754. [PubMed] [Google Scholar]
- 8.Weg-Remers S., Anders M., von Lampe B., Riecken E. O., Schuder G., Feifel G., Zeitz M., Stallmach A. (1998) Decreased expression of CD44 splicing variants in advanced colorectal carcinomas. Eur. J. Cancer , 1607–1611 [DOI] [PubMed] [Google Scholar]
- 9.Kaufmann M., von Minckwitz G., Heider K., Ponta H., Herrlich P., Sinn H. (1995) CD44 variant exon epitopes in primary breast cancer and length of survival. Lancet , 615–619 [DOI] [PubMed] [Google Scholar]
- 10.Noordzij M. A., van Steenbrugge G. J., Schröder F. H., van der Kwast T. H. (1999) Decreased expression of CD44 in metastatic prostate cancer. Int. J. Cancer , 478–483 [DOI] [PubMed] [Google Scholar]
- 11.Gao A. C., Lou W., Dong J. T., Isaacs J. T. (1997) CD44 is a metastasis suppressor gene for prostatic cancer located on human chromosome 11p13. Cancer Res. , 846–849 [PubMed] [Google Scholar]
- 12.Harada N., Mizoi T., Kinouchi M., Hoshi K., Ishii S., Shiiba K., Sasaki I., Matsuno S. (2001) Introduction of antisense CD44s CDNA down-regulates expression of overall CD44 isoforms and inhibits tumor growth and metastasis in highly metastatic colon carcinoma cells. Int. J. Cancer , 67–75 [DOI] [PubMed] [Google Scholar]
- 13.Jothy S. (2003) CD44 and its partners in metastasis. Clin. Exp. Metastasis , 195–201 [DOI] [PubMed] [Google Scholar]
- 14.Louderbough J. M. V., Schroeder J. A. (2011) Understanding the dual nature of CD44 in breast cancer progression. Mol. Cancer Res. , 1573–1586 [DOI] [PubMed] [Google Scholar]
- 15.Li Y., Cao H., Jiao Z., Pakala S. B., Sirigiri D. N. R., Li W., Kumar R., Mishra L. (2010) Carcinoembryonic antigen interacts with TGF-β receptor and inhibits TGF-β signaling in colorectal cancers. Cancer Res. , 8159–8168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitsuki H., Katano M., Morisaki T., Torisu M. (1995) CEA-mediated homotypic aggregation of human colorectal carcinoma cells in a malignant effusion. Cancer Lett. , 7–13 [DOI] [PubMed] [Google Scholar]
- 17.Wirth T., Soeth E., Czubayko F., Juhl H. (2002) Inhibition of endogenous carcinoembryonic antigen (CEA) increases the apoptotic rate of colon cancer cells and inhibits metastatic tumor growth. Clin. Exp. Metastasis , 155–160 [DOI] [PubMed] [Google Scholar]
- 18.Jessup J. M., Laguinge L., Lin S., Samara R., Aufman K., Battle P., Frantz M., Edmiston K. H., Thomas P. (2004) Carcinoembryonic antigen induction of IL-10 and IL-6 inhibits hepatic ischemic/reperfusion injury to colorectal carcinoma cells. Int. J. Cancer , 332–337 [DOI] [PubMed] [Google Scholar]
- 19.Hammarström S. (1999) The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Sem. Cancer Biol. , 67–81 [DOI] [PubMed] [Google Scholar]
- 20.Shively J. E., Beatty J. D. (1985) CEA-related antigens: molecular biology and clinical significance. Crit. Rev. Oncol. Hematol. , 355–399 [DOI] [PubMed] [Google Scholar]
- 21.Goldenberg D., Sharkey R., Primus F. (1976) Carcinoembryonic antigen in histopathology: immunoperoxidase staining of conventional tissue sections. J. Natl. Cancer Inst. , 11–22 [DOI] [PubMed] [Google Scholar]
- 22.Blumenthal R. D., Hansen H. J., Goldenberg D. M. (2005) Inhibition of adhesion, invasion, and metastasis by antibodies targeting CEACAM6 (NCA-90) and CEACAM5 (carcinoembryonic antigen). Cancer Res. , 8809–8817 [DOI] [PubMed] [Google Scholar]
- 23.Rago C., Huso D. L., Diehl F., Karim B., Liu G., Papadopoulos N., Samuels Y., Velculescu V. E., Vogelstein B., Kinzler K. W., Diaz L. a. (2007) Serial assessment of human tumor burdens in mice by the analysis of circulating DNA. Cancer Res. , 9364–9370 [DOI] [PubMed] [Google Scholar]
- 24.McCarty O. J. T., Mousa S. A., Bray P. F., Konstantopoulos K. (2000) Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood , 1789. [PubMed] [Google Scholar]
- 25.Jadhav S., Bochner B. S., Konstantopoulos K. (2001) Hydrodynamic shear regulates the kinetics and receptor specificity of polymorphonuclear leukocyte-colon carcinoma cell adhesive interactions. J. Immunol. , 5986–5993 [DOI] [PubMed] [Google Scholar]
- 26.Updyke T. V., Nicolson G. L. (1986) Malignant melanoma cell lines selected in vitro for increased homotypic adhesion properties have increased experimental metastatic potential. Clin. Exp. Metastasis , 273–284 [DOI] [PubMed] [Google Scholar]
- 27.Hashino J., Fukuda Y., Oikawa S., Nakazato H., Nakanishi T. (1994) Metastatic potential of human colorectal carcinoma SW1222 cells transfected with cDNA encoding carcinoembryonic antigen. Clin. Exp. Metastasis , 324–328 [DOI] [PubMed] [Google Scholar]
- 28.Cross S. E., Jin Y.-S., Rao J., Gimzewski J. K. (2007) Nanomechanical analysis of cells from cancer patients. Nat. Nanotech. , 780–783 [DOI] [PubMed] [Google Scholar]
- 29.Guck J., Schinkinger S., Lincoln B., Wottawah F., Ebert S., Romeyke M., Lenz D., Erickson H. M., Ananthakrishnan R., Mitchell D., Käs J., Ulvick S., Bilby C. (2005) Optical deformability as an inherent cell marker for testing malignant transformation and metastatic competence. Biophys. J. , 3689–3698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swaminathan V., Mythreye K., O'Brien E. T., Berchuck A., Blobe G. C., Superfine R. (2011) Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. , 5075–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wirtz D. (2009) Particle-tracking microrheology of living cells: principles and applications. Annu. Rev. Biophys. , 301–326 [DOI] [PubMed] [Google Scholar]
- 32.Terpe H. J., Störkel S., Zimmer U., Anquez V., Fischer C., Pantel K., Günthert U. (1996) Expression of CD44 isoforms in renal cell tumors: Positive correlation to tumor differentiation. Am. J. Pathol. , 453–463 [PMC free article] [PubMed] [Google Scholar]
- 33.Yüce I., Bayram A., Cağlı S., Canöz O., Bayram S., Güney E. (2011) The role of CD44 and matrix metalloproteinase-9 expression in predicting neck metastasis of supraglottic laryngeal carcinoma. Am. J. Otolaryngol. , 141–146 [DOI] [PubMed] [Google Scholar]
- 34.Lopez J. I., Camenisch T. D., Stevens M. V., Sands B. J., McDonald J., Schroeder J. A. (2005) CD44 attenuates metastatic invasion during breast cancer progression. Cancer Res. , 6755–6763 [DOI] [PubMed] [Google Scholar]
- 35.Choi S. H., Takahashi K., Eto H., Yoon S. S., Tanabe K. K. (2000) CD44s expression in human colon carcinomas influences growth of liver metastases. Int. J. Cancer , 523–526 [DOI] [PubMed] [Google Scholar]
- 36.Mulder J. W. R., Sewnath M., Offerhaus G., Pals S. (1994) Colorectal cancer prognosis and expression of exon-v6-containing CD44 proteins. Lancet , 1470–1472 [DOI] [PubMed] [Google Scholar]
- 37.Klingbeil P., Marhaba R., Jung T., Kirmse R., Ludwig T., Zöller M. (2009) CD44 variant isoforms promote metastasis formation by a tumor cell-matrix cross-talk that supports adhesion and apoptosis resistance. Mol. Cancer Res. , 168–179 [DOI] [PubMed] [Google Scholar]
- 38.Yang X.-R., Xu Y., Yu B., Zhou J., Qiu S.-J., Shi G.-M., Zhang B.-H., Wu W.-Z., Shi Y.-H., Wu B., Yang G.-H., Ji Y., Fan J. (2010) High expression levels of putative hepatic stem/progenitor cell biomarkers related to tumour angiogenesis and poor prognosis of hepatocellular carcinoma. Gut , 953–962 [DOI] [PubMed] [Google Scholar]
- 39.Morrin M., Delaney P. (2002) CD44v6 is not relevant in colorectal tumour progression. Int. J. Colorectal Dis. , 30–36 [DOI] [PubMed] [Google Scholar]
- 40.De Marzo A. M., Bradshaw C., Sauvageot J., Epstein J. I., Miller G. J. (1998) CD44 and CD44v6 downregulation in clinical prostatic carcinoma: relation to Gleason grade and cytoarchitecture. Prostate , 162–168 [DOI] [PubMed] [Google Scholar]
- 41.Murray E. W., Robbins S. M. (1998) Antibody cross-linking of the glycosylphosphatidylinositol-linked protein CD59 on hematopoietic cells induces signaling pathways resembling activation by complement. J. Biol. Chem. , 25279–25284 [DOI] [PubMed] [Google Scholar]
- 42.Kim H.-R., Wheeler M. A., Wilson C. M., Iida J., Eng D., Simpson M. A., McCarthy J. B., Bullard K. M. (2004) Hyaluronan facilitates invasion of colon carcinoma cells in vitro via interaction with CD44. Cancer Res. , 4569–4576 [DOI] [PubMed] [Google Scholar]
- 43.Zeilstra J., Joosten S. P. J., Dokter M., Verwiel E., Spaargaren M., Pals S. T. (2008) Deletion of the WNT target and cancer stem cell marker CD44 in Apc(Min/+) mice attenuates intestinal tumorigenesis. Cancer Res. , 3655–3661 [DOI] [PubMed] [Google Scholar]
- 44.Nelson W. J., Nusse R. (2004) Convergence of Wnt, beta-catenin, and cadherin pathways. Science , 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshioka T., Masuko T., Kotanagi H., Aizawa O., Saito Y., Nakazato H., Koyama K., Hashimoto Y. (1998) Homotypic adhesion through carcinoembryonic antigen plays a role in hepatic metastasis development. Jpn. J. Cancer Res. , 177–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gangopadhyay A., Bajenova O., Kelly T. M., Thomas P. (1996) Carcinoembryonic antigen induces cytokine expression in Kupffer cells: implications for hepatic metastasis from colorectal cancer. Cancer Res. , 4805. [PubMed] [Google Scholar]
- 47.Edmiston K. H., Gangopadhyay A., Shoji Y., Jessup J. M., Nachman A. P., Thomas P. (1997) In vivo induction of murine cytokine production by carcinoembryonic antigen. Cancer Res. 4432–4436 [PubMed] [Google Scholar]
- 48.Gangopadhyay A., Lazure D. A., Thomas P. (1998) Adhesion of colorectal carcinoma cells to the endothelium is mediated by cytokines from CEA stimulated Kupffer cells. Clin. Exp. Metastasis , 703–712 [DOI] [PubMed] [Google Scholar]
- 49.Jessup J. M., Samara R., Battle P., Laguinge L. M. (2005) Carcinoembryonic antigen promotes tumor cell survival in liver through an IL-10-dependent pathway. Clin. Exp. Metastasis , 709–717 [DOI] [PubMed] [Google Scholar]
- 50.O'Brien C. A., Pollett A., Gallinger S., Dick J. E. (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature , 106–110 [DOI] [PubMed] [Google Scholar]
- 51.Fang D. D., Kim Y. J., Lee C. N., Aggarwal S., Mckinnon K., Mesmer D., Norton J., Birse C. E., He T., Ruben S. M., Moore P. A. (2010) Expansion of CD133+ colon cancer cultures retaining stem cell properties to enable cancer stem cell target discovery. Br. J. Cancer , 1265–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.