

Abstract
Salt-sensitive hypertension and chronic kidney disease (CKD) following recovery from acute kidney injury (AKI) may occur secondary to incomplete repair, or by activation of circulating factors stimulated by injury. We created two types of renal injury induced by unilateral ischemia-reperfusion (I/R); in a direct/ipsilateral AKI group, rats were subjected to unilateral I/R and the untouched contralateral kidney was removed by unilateral nephrectomy after 5 wk to isolate effects on the injured kidney. In the remote/contralateral AKI group, the injured kidney was removed after 5 wk to isolate effects on the untouched kidney. When these animals were subsequently challenged with elevated dietary sodium for an additional 4 wk (0.4 to 4%), both remote/contralateral and direct/ipsilateral AKI rats manifested a significant increase in blood pressure relative to sham-operated controls. Similarly, in acute studies, both ipsilateral and contralateral kidneys had impaired pressure natriuresis and hemodynamic responses. Reductions in vascular density were observed following direct/ipsilateral injury, but were not observed in the remote/contralateral kidney. However, both remote/contralateral and direct/ipsilateral kidneys contained interstitial cells, some of which were identified as activated (low CD62L/CD4+) T lymphocytes. In contrast, only the direct/ipsilateral AKI group demonstrated significant CKD following exposure to elevated salt. This was characterized by a significant reduction in creatinine clearance, an increase in albuminuria, and a dramatic expansion of interstitial inflammation. Taken together, these data suggest that the salt-sensitive features of AKI on hypertension and CKD are segregable such that effects on hemodynamics and hypertension occur independent of direct renal damage. However, prior direct injury to the kidney is required to elicit the full manifestation of CKD induced by elevated sodium intake.
Keywords: hemodynamics, blood pressure, T lymphocytes
acute kidney injury (AKI) due to renal ischemia or sepsis represents a major clinical problem associated with significant mortality. In addition, AKI is becoming widely recognized as a risk factor for the development of chronic kidney disease (CKD) leading to end-stage renal disease (13, 16, 28). Studies using rodent models have demonstrated that AKI induced by ischemia or nephrotoxins predisposes CKD characterized by interstitial fibrosis and proteinuria and may lead to a secondary reduction in glomerular filtration rate (GFR) (4, 29). Several factors have been proposed to contribute to CKD following AKI, including hypoxia secondary to vascular dropout and persistent activation of inflammatory and profibrotic cytokines; the contribution of these factors is enhanced in the setting of reduced renal mass (3, 12, 29, 30). Alterations in renal function that are present chronically following recovery from AKI include impaired renal hemodynamic and natriuretic responses (22), which may contribute to hypertension and renal scarring observed when post-AKI rats are exposed to elevated dietary salt (25). Interestingly, chronic alterations in function are also present at remote, nonrenal locations, as demonstrated by the increased pressor responses to ANG II in skeletal muscle arterioles following recovery from AKI (23).
The basis for impaired hemodynamic and natriuretic responses that underlie salt sensitivity post-AKI is not clear. A role for capillary loss was suggested by a study in which treatment of rats with vascular endothelial growth factor (VEGF)-121 during the early post-ischemia-reperfusion (I/R) period attenuated the loss of renal microvessels and significantly reduced the degree of secondary damage resulting from subsequent exposure to elevated dietary salt (5). However, Pechman et al. (21) demonstrated that administration of the lymphocyte inhibitor mycophenolate mofetil (MMF) beginning at 5 wk postinjury mitigated the development of hypertension, albuminuria, and fibrosis in response to a subsequent increase in dietary sodium intake. Taken together, these data suggest that aspects of both microvascular loss combined with activation of immune function are required for the full manifestation of salt sensitivity following recovery from I/R-induced AKI.
It is well-known that injury to the kidney results in alterations in function at remote sites and that this is due to the activation of proinflammatory cytokines and systemic immune function (14). In a study by Burne-Taney et al. (8) splenocytes from post-AKI mice were adoptively transferred into naive mice resulting in the late development of albumunuria, suggesting that AKI-activated immune cells may chronically affect renal function. Based on these observations, we hypothesized that renal I/R alters renal function, hemodynamic responses, and salt sensitivity via circulating factors independent of direct renal injury, per se. To investigate this, we developed an experimental paradigm to study the effects of remote kidney injury that develops in the contralateral untouched kidney following unilateral renal I/R. Alterations in renal function were evaluated by determining the response to elevated sodium intake and by studying hemodynamic and natriuretic responses in anesthetized animals. The results indicate there are discrete aspects of salt sensitivity post-AKI that result secondary to direct injury to the renal parenchyma as well as effects due to circulating factors, independent of direct parenchymal injury, per se.
METHODS
Animals.
Male Sprague-Dawley rats (250–300 g) were purchased from Harlan (Indianapolis, IN) and used for all studies. Care of the rats before and during the experimental procedures was conducted in accordance with the policies of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All protocols had received prior approval by the Institutional Animal Care and Use Committees at Indiana University.
Study protocols.
Study I was designed to determine the effect of either direct/ipsilateral or remote/contralateral kidney injury on long-term renal function and salt-sensitive hypertension. The time line and study groups are outlined in Fig. 1A. In these studies, rats were acclimated to a standard lab diet (AIN 76A, Dyets) containing standard 0.4% NaCl before surgery. Rats were subjected to unilateral I/R injury or sham surgery and allowed to recover for ∼5 wk. To distinguish between the chronic effects of direct or remote injury, rats were subjected to unilateral nephrectomy (UNx) at 33 days postsurgery, and subsequently (day 35) exposed to elevated NaCl diet (AIN76A, plus 4% NaCl). The group that is defined as the “direct/ipsilateral” AKI group is created when the untouched kidney was removed by UNx leaving only the kidney that had been subjected to I/R (Fig. 1, group C). In contrast, the group defined as the “remote/contralateral” group is created when the post-I/R kidney was removed leaving the untouched kidney (see Fig. 1, group B). At day 57 (just following the 8-wk urine collection), telemetric blood pressure transducers were implanted to measure blood pressure before termination of the study.
Fig. 1.

Experimental schema of studies carried out to distinguish the effects of unilateral injury on chronic function in both direct/ipsilateral and remote/contralateral injured kidneys. A: illustrates groups used in study I to distinguish between direct and remote effects of ischemia-reperfusion (I/R) injury on salt sensitivity. The time line is similar to that used in previous studies (25). All rats are maintained on standard 0.4% NaCl diet during recovery for 5 wk of unilateral I/R (red circle) or sham I/R (black circle). The “direct/contralateral” injury group is studied by subjecting rats to unilateral nephrectomy (UNx; depicted by X) of the uninjured, contralateral kidney at 33 days, such that only the injured kidney remains. The “remote/contralateral” injury group is prepared by removal of the injured kidney at 33 days, such that contralateral kidney remains. UNx is also carried out in the sham group as a control for both direct and remote injury groups. At 35 days, rats are exposed to an elevated NaCl diet (4.0%) for the remaining 4 wk of the study. At 8 wk, telemetric transducers are implanted to evaluate the effects of treatments on mean arterial blood pressure, which was evaluated on the final 3 consecutive days of the study. B: illustrates the time line for study II, in which the effects of “direct/ipsilateral” and “remote/contralateral” injury to the kidney are subjected to acute renal function studies. In these studies, animals recover for 1 wk following UNx (from week 5–6) and are maintained for the entire study period on standard 0.4% NaCl diet. At 6 wk of recovery, the effects of these injuries on renal autoregulation and pressure natriuresis were studied.
The purpose of study II was to determine the effects of direct/ipsilateral or remote/contralateral injury on acute renal hemodynamic and Na-handling responses. As shown in Fig. 1B, this study was similar to study I with the following exceptions: 1) rats were maintained on the standard 0.4% NaCl-containing diet for the entire study and 2) at 6 wk, animals were prepared for studies to examine acute renal function, hemodynamics, and Na-handling responses. The injuries and unilateral nephrectomies were carried out such that acute renal function studies were performed on the left kidney.
Surgeries.
AKI was induced by unilateral I/R injury to the kidneys by clamping the renal pedicle for 35 min using a surgical approach that has been described previously under anesthesia-induced ketamine (100 mg/kg) and pentobarbital sodium (25–50 mg/kg) (5, 21, 22). However, the current study used only unilateral renal ischemia, while the contralateral kidney was untouched (see Fig. 1). For sham surgeries, the kidneys were examined and not touched.
At 33 days, UNx was carried out under anesthesia induced by a cocktail of ketamine (60 mg/kg), xylazine (3 mg/kg), and acepromazine (1 mg/kg). Using a midline incision, the kidney to be removed was isolated by blunt dissection. A 1–0 sterile silk was used to ligate the renal pedicle, and the kidney was excised with microscissors distal to the ligature.
For study I, rats were subjected to a third survival surgery at 8 wk following I/R or sham surgery. This surgery was used to implant the telemetric blood pressure transducers. This surgery was carried out under anesthesia induced by ketamine (60 mg/kg), xylazine (3 mg/kg), and acepromazine (1 mg/kg). The catheter tip of was implanted into the right femoral artery and the body of the pressure transducer (TA11PA-C40, Data Science) was implanted subcutaneously as described previously (25).
Measurement of renal function in chronic studies.
For measurement of serum creatinine, tail blood samples (0.2 ml) were collected into heparinized tubes and plasma was obtained by centrifugation. Serum and urine creatinine were determined with a Beckman Creatinine Analyzer II (Beckman Coulter). Urine volume from metabolic cages was determined gravimetrically at times indicated in results. Urine albumin content was measured using the albumin blue 580 fluorescence method (Fluka), as previously described (4, 5).
Measurement of blood pressure for study I was similar to that previously described (21, 25). Blood pressure was measured from 0900 to 1200 by using DSI Dataquest ART acquisition software for the final 3 consecutive days of the study period before death (Days 60–62), similar to sampling protocols carried out in previous studies (21, 25).
Measurement of renal function in acute studies.
In study II, acute evaluation of renal function followed protocols described previously (22). After 5 wk of recovery from unilateral I/R injury, and one additional week of recovery from UNx, rats were anesthetized with ketamine HCl (60 mg/kg), followed by Inactin (50–100 mg/kg ip). Rats were placed on a heated surgical board to maintain body temperature at 37°C. The trachea was cannulated to facilitate respiration, and catheters were placed in the carotid and femoral artery to measure renal perfusion pressure (RPP). The femoral vein was cannulated for intravenous infusions. A midline abdominal incision was made, and a flow probe was placed around the renal artery for measurement of renal blood flow (RBF) via an ultrasonic Doppler flowmeter (model T206, Transonic Systems, Ithaca, NY). An inflatable cuff was placed around the aorta above the renal arteries, and a loose ligature was placed around the celiac and mesenteric arteries so RPP could be adjusted during the experiment. The left ureter was cannulated for urine collection. The left kidney was placed in a stainless steel cup to secure the kidney in place and optical fibers for laser Doppler flowmetry were implanted to a depth of ∼4.0 mm beneath the surface for measurements in the renal medulla.
Animals were volume expanded with an intravenous infusion of 2% bovine serum albumin in 0.9% NaCl at a rate of 2 ml·h−1·100 g body wt−1. The volume status of the anesthetized rats provides urine flow and sodium excretion rates in the range of those observed in rats fed a 4.0% NaCl diet. The kidney was denervated by stripping the renal artery of all visible nerves and coating the artery with 5% phenol in ethanol. Circulating levels of extrarenal sodium and water-retaining hormones were set at maximal physiological levels by intravenous infusion of aldosterone (6.67 ng·min−1·100 g body wt−1; Sigma, St. Louis, MO), vasopressin (16.67 pg·min−1·100 g body wt−1; Sigma), norepinephrine (33.3 ng·min−1·100 g body wt−1; Sigma), and corticosterone (3.33 μg·min−1·100 g body wt−1; Sigma) as previously described (20, 24). Using these approaches, neural and hormonal influences on the kidney were fixed by denervation and hormone infusion since manipulation of RPP with aortic cuffs may affect renal sympathetic activity and circulating levels of aldosterone and vasopressin (20, 24).
Following the surgical preparation, the aortic cuff superior to the kidney was constricted to establish the lower RPP at ∼95 mmHg. After an equilibration period, two consecutive urine collection periods were made for 30 min each. RPP was then permitted to increase to ∼120 mmHg by releasing the aortic cuff, occluding the superior mesenteric and celiac arteries, and, if necessary, clamping the aorta below the kidney. The renal excretory parameters and hemodynamic parameters were measured again at this elevated level of RPP.
To measure glomerular filtration rate, inulin clearance measurements were carried out as described previously (27). A priming dose of inulin (10% in saline, 0.2 ml/100 g body wt; Sigma) was provided, followed by an infusion of 0.1% inulin in the saline/BSA used to provide volume expansion described above (i.e., 2 ml·h−1·100 g body wt−1). Plasma and urine samples were collected at the indicated times and the inulin concentrations were determined using the anthrone method. GFR was calculated using the standard clearance formula Uinulin·V/Pinulin, where V represents the urine flow rate over each collection period in ml·min−1·100 g body wt−1. Urine sodium concentration was measured by flame photometry (Buck Scientific).
Renal histology and immunohistochemistry.
Following the conclusion of the study, kidneys were removed from deeply anesthetized rats, bisected, and fixed by immersion into either 10% buffered formalin and stored at room temperature. Formalin-fixed tissues were embedded in paraffin and stained using perodic acid Schiff for standard histological assessment. ED-1-positive macrophage staining was carried out as described previously (21). Staining for renal capillaries and quantitative assessment of renal peritubular capillary density were carried out using cablin immunohistochemistry on methanol-fixed tissues and confocal microscopy exactly as described previously (6).
FACS analysis.
To determine the effects of renal injury on chronic T cell deposition, kidneys were harvested from rats identical to those described in Fig. 1, but were isolated at 33 days postinjury, without UNx or exposure to elevated salt diet. Harvested kidneys were minced in 20 mM HEPES in PBS and digested in collagenase (Type II, 0.85 mg/ml; Sigma) for 1 h at 37°C with gentle agitation and bubbling with 100% O2. The digested tissue was gently forced through a 100-μm filter mesh and washed by gentle centrifugation three times. The cell pellet was then separated in Ficoll (Histopaque 1083, Sigma). The total cells after Ficoll separation were counted by hemocytometer. To evaluate T lymphocytes, the cells were stained with antibodies against rat CD4 (PE-Cy7), CD8a (Alexa 647), CD25 (FITC), and CD62L (PE). All antibodies were obtained from BD Biosciences. Cells were scanned using flow cytometry (FACSCalibur, BD Biosciences) and scans were analyzed using Flowjo software (Tree Star, Ashland, OR). The exact numbers of the different T cell populations in the harvested kidney were calculated using the pecentage of each cell type and the total cell numbers estimated for each kidney.
Statistical analysis.
All data are expressed as means ± SE. Differences in means were established by Student's t-test or one-way ANOVA with Student-Newman-Keuls post hoc test.
RESULTS
The effects of unilateral renal I/R on renal function are shown in Fig. 2, A and B. Serum creatinine values rose slightly but significantly in response to unilateral I/R injury in both the direct/ipsilateral and remote/contralateral injury groups by 24 h relative to the sham-operated control. Recovery from unilateral I/R was evident as serum creatinine and creatinine clearance values were not different in either injury group relative to the sham-operated control group at 33 days following injury (Fig. 2, A and B).
Fig. 2.

Effect of direct and remote injury on serum creatinine and creatinine clearance. A: serum creatinine values are shown for sham, remote/contralateral, and direct/ipsilateral groups at 24 h, 7 days, 33 days, and 56 days post-I/R or sham. B: creatinine clearance values are shown for sham, remote/contralateral, and direct/ipsilateral groups based on 24-h urine collections at the indicated times; n = 7, 11, and 7 for sham, remote/contralateral, and direct/ipsilateral injury groups, respectively. Data in A and B are means ± SE. *P < 0.05 vs. sham-operated control by ANOVA and Student-Newman-Keuls post hoc test. NS, not significant.
Histological examination of kidneys following 33 days of recovery in ipsilateral or contralateral kidneys was facilitated by removal of these kidneys at the time of UNx. The ipsilateral kidney (i.e., derived from the remote/contralateral AKI group; Fig. 1) illustrated a largely intact renal epithelium with only modest evidence of persistent damage (Fig. 3, C vs. A). At 33 days post-I/R, the directly injured ipsilateral kidney showed a slight but statistically significant reduction in kidney/body weight ratio (Fig. 4A). In contrast, at 33 days in the contralateral kidney (the untouched kidney from the direct/ipsilateral group), the epithelium showed no obvious evidence of damage, but there was a modest increase the number of interstitial cells (black arrow; Fig. 3B). In contrast to the ipsilateral kidney, the contralateral kidney showed a slight but significant increase in kidney/body weight ratio at 33 days of recovery (Fig. 4A).
Fig. 3.
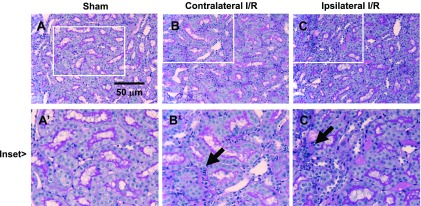
Renal histology of direct and remote I/R injury at 33 days of recovery. Representative renal histology of ipsilateral and contralateral kidneys following unilateral I/R, obtained at the time of UNx, before exposure to elevated dietary salt. Shown are representative periodic acid Schiff (PAS)-stained sections of a kidney from a sham-operated animal at day 33 (A), a contralateral injured kidney (B), and an ipsilateral injured kidney (C). Note renal tubular morphology is largely intact in both direct and remote injury. Evidence of increased interstitial cells is present in both the contralateral and ipsilateral kidneys (black arrows). Magnification is shown.
Fig. 4.

Effect of unilateral I/R on kidney weight of direct/ipsilatereal and remote contralateral kidneys. A: shown are kidney-to-body wt ratios at 33 days, representing weights at the time of UNx. At 33 days, the contralateral kidney is from direct injury group and is significantly hypertrophic relative to the sham. The kidney direct/ipsilateral kidney is from the remote group and is slightly hypotrophic. B: kidney-to-body wt ratios for sham, direct/ipsilateral, and remote/contralateral injury at 63 days. Data in A and B are means ± SE. * And #P < 0.05 vs. sham-operated control by Student's t-test.
All animals in study I were exposed to an elevated NaCl diet beginning at 35 days following I/R and monitored for the development of CKD and hypertension. Animals in the direct/ipsilateral injury group manifested a significant increase in albuminuria within 1 wk of exposure to elevated NaCl (42 days), which was markedly enhanced on day 49 and day 56 (Fig. 5). In contrast, remote/contralateral injury did not result in increased albuminura relative to sham. Both the sham and remote/contralateral group showed a moderate but statistically significant increase in albumin excretion following exposure to elevated sodium (Fig. 5).
Fig. 5.

Effect of elevated NaCl intake on albuminuria in sham, remote/contralateral acute kidney injury (AKI), and direct/ipsilateral AKI groups. Albumin excretion rates were derived from 24-h urine collections at the indicated times. Data for day 63 time points were not obtained due to the need to collect blood pressure measurements. Data are means ± SE. *P < 0.05 vs. sham-operated control by ANOVA and Student-Newman-Keuls post hoc test. (a, b)Significant increases in sham-operated and remote injury groups following exposure to elevated NaCl relative to the final collection under standard salt conditions (i.e., day 33).
Moreover, direct/ipsilateral injury in combination with elevated salt intake prominently reduced GFR, indicated by a significant increase in serum creatinine (Fig. 2A) and an ∼40% reduction in creatinine clearance by day 56 relative to the sham-operated control group (Fig. 2B). In contrast, creatinine clearance was not compromised by elevated salt intake in either the remote/contralateral group or sham-operated control groups (Fig. 2B).
Blood pressure was measured via telemetry between days 60–62. At this time point, following UNx and exposure to elevated dietary NaCl, both remote/contralateral injury and direct/ipsilateral injury groups had significantly elevated blood pressures relative to the sham (124 ± 5 and 125 ± 6 mmHg, respectively, vs. 109 ± 2 mmHg; Fig. 6). Of note, fewer animals contributed to the data in Fig. 6 than in Figs. 2, 4, and 5, since some animals died following implantation of transmitters on day 57 (see figure legends).
Fig. 6.

Effect of direct/ipsilateral injury and remote/contralateral injury on the salt-sensitive changes in blood pressure. Values shown are mean blood pressures derived from telemetry measurements averaged over the 3-day period from days 60–62 of study I. Due to loss of animals at the time of implantation of transducers, n = 5, 6, and 3 for the sham, remote, and direct injury groups. Data are means ± SE. *P < 0.05 vs. sham-operated control by Student's t-test.
The effects of direct injury to the ipsilateral kidney and remote injury to the contralateral kidney following UNx and elevated sodium intake on renal morphology is shown in Fig. 7. Proximal tubule structure was largely intact in the remote injury kidney (Fig. 7B, right) but several dilated tubules were occasionally observed suggesting evidence of new focal damage (Fig. 7B, inset). There remained a persistent elevation of tubular interstitial cells in the remotely injured kidney (black arrows) compared with the corresponding sham (Fig. 7, A vs. B). In contrast, ipsilateral kidneys following direct injury (with UNx and elevated NaCl) demonstrated prominent renal damage, with numerous dilated tubular structures and large areas of substantial interstitial infiltration (black arrows; Fig. 7C). Immunohistochemical analysis demonstrated both direct/ipsilateral injured kidneys and remote/contralateral injured kidneys contained ED-1+ macrophages, which were located primarily but not exclusively in the outer medulla (Fig. 8). However, the majority of interstitial cells were negative for ED-1.
Fig. 7.
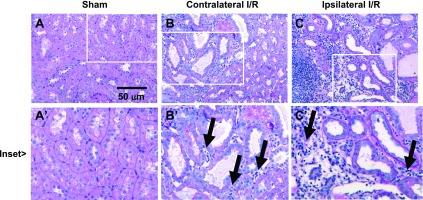
Representative renal histology from direct/ipsilateral and remote/contralateral injured kidneys following reduction in renal mass by UNx and exposure to elevated dietary salt. Shown are representative PAS-stained sections of a kidney from a sham-operated animal at day 63 (A), a remotely injured kidney (B), and a directly injured kidney (C). Magnification is shown in C. Insets for each micrograph reveal the presence of interstitial infiltrate in remotely injured kidneys and a more severe infiltration in directly injured kidneys.
Fig. 8.

Macrophage deposition following direct/ipsilateral AKI and remote/contralateral AKI. ED-1-positive cells were identified in both remote/contralateral (A) and direct/ipsilateral injured kidneys (B) and are indicated by the black arrows. Magnification is shown.
The development of salt-sensitive hypertension suggests that both remote/contralateral injury and direct/ipsilateral injury may alter intrinsic renal hemodynamic and salt-handling processes. To evaluate this, we studied hemodynamic and natriuretic responses in anesthetized rats following both types of AKI, without elevated dietary sodium (study II; Fig. 1B). In sham-operated controls, GFR values were well autoregulated when RPP were increased to higher values (Fig. 9A). As expected, sham-operated control rats responded to increased RPP by increasing urinary flow rate and renal sodium output (Fig. 9, B and C). In addition, renal medullary blood flow significantly increased in response to increased RPP as determined by laser Doppler flowmetry (Fig. 9D).
Fig. 9.
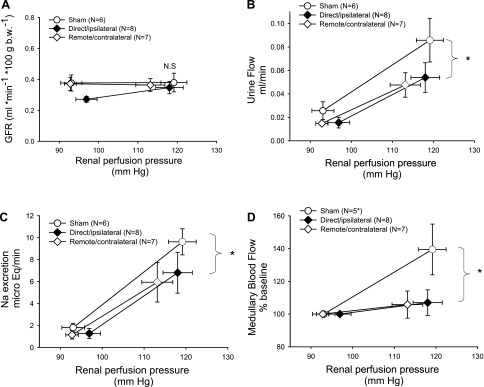
Effect of direct/ipsilateral and remote/contralateral injury on renal function, pressure natriuresis, and medullary blood flow. Rats from study II were analyzed at 6 wk following I/R and renal hemodynamics were measured in response to increases in renal perfusion pressure. A: glomerular filtration rate (GFR) measured by inulin clearance. B: urinary flow rate. C: sodium excretion. D: medullary blood flow by laser Doppler flow. The N for each group is shown. *P < 0.05 by ANOVA and Student-Newman-Keuls post hoc test measuring the high-pressure point in remote and direct groups relative to the sham-operated control group.
Relative to the sham-operated control, GFR was slightly but significantly diminished in direct/ipsilateral injured kidneys at the low RPP point (Fig. 9A), but GFR was not different from sham in either of the injury groups at the elevated RPP points (Fig. 9A). Interestingly, both direct/ipsilateral injury and remote/contralateral injury resulted in a significant compromise in the diuretic and natriuretic response to increased RPP (Fig. 9, B and C) and this was associated with a blunted medullary blood flow response in both injury groups, relative to shams (Fig. 9D).
The reason for impaired hemodynamics is not clear. Previous studies in bilateral I/R models showed a decrease in renal vascular density that has been proposed to participate in progression of injury and salt sensitivity (4, 6). To investigate differences in vascular density, we stained renal sections with cablin and carried out quantitative analysis of microvascular density. Consistent with previous studies, there was a significant reduction in vascular density in direct/ipsilateral injured kidneys relative to the sham-operated control. In contrast, despite the impaired hemodynamic responses, there was no effect of remote/contralateral injury on vascular density (Fig. 10).
Fig. 10.

Effect of unilateral I/R on capillary density in remote/contralateral and direct/ipsilateral kidneys. Quantitative analysis of vessel density scoring based on cablin immunohistochemistry is shown. Data are means ± SE and the N for each group is shown. *P < 0.05 in directly injured vs. sham-operated control by Student's t-test, while remote injury did not influence blood vessel density (N.S.).
Lymphocytes may alter renal function following AKI (10, 11). We sought to determine whether either remote or direct renal injury resulted in the persistent deposition of T lymphocytes. Following 33 days of recovery from injury, there was a significant increase in the CD4+ and CD8+ cells observed in remote/contralateral injured kidneys relative to sham-operated controls and this value was further enhanced in kidneys following recovery from direct/ipsilateral injury (Fig. 11, C and D). Moreover, compared with the population of cells derived from the spleen, CD4+ cells from kidney showed a distribution in the expression of the naive T cell marker CD62L and the activation marker CD25 (Fig. 11B). Specifically, CD4+ cells from direct/ipsilateral AKI kidneys had the lowest level of CD62L, CD4+ cells from sham kidneys had the highest expression, and CD4+ cells from the remote/contralateral AKI kidneys had intermediate expression of CD62L (Fig. 11E). A reciprocal profile for CD25 expression on CD4+ cells was also observed; such that sham-operated kidneys contained high low CD25, while direct/ipsilateral injury kidneys had high CD25+ cells (Fig. 11F).
Fig. 11.

FACS analysis of renal T cell deposition in sham kidneys, remote/contralateral kidneys, or direct/ipsilateral kidneys 33 days following unilateral I/R. A: forward and side scatter are shown with representative CD4+ and CD8+ characterization corresponding to the indicated population. B: representative histograms of CD62L and CD25 are indicated for the CD4-positive population in spleen, and kidney from sham, direct and remote AKI kidneys. The total numbers of CD4 and CD8 lymphocytes per kidney are shown in C and D, respectively; while the mean fluorescence intensities of CD62L and CD25 of the CD4-positive populations are shown in E and F, respectively. Data are means ± SE and are based on N = 5 per group. *P < 0.05 by Student's t-test relative to sham-operated control. Differences between remote and direct injury groups are indicated above each of these bars in the graphs.
DISCUSSION
I/R chronically alters renal function and predisposes CKD. Our laboratory demonstrated that renal I/R chronically reduces peritubular capillary density (4) and impairs renal hemodynamic responses (22). It has been proposed that this impairment promotes hypertension and accelerates CKD when post-AKI animals are exposed to elevated sodium intake (25). The fact that VEGF-121 preserved renal microvascular density following I/R and attenuated secondary features of CKD induced by dietary salt supports the suggestion that capillary loss predisposes these chronic effects of AKI (18).
Injury-induced alterations in vascular reactivity could also conceivably alter renal hemodynamic responses regulating sodium control independent of capillary loss. I/R injury to the kidney liberates cytokines, suggested to mediate remote effects on distant organs by altering barrier function in vascular beds outside of the kidney (e.g., lung, brain), which may contribute to overall mortality and morbidity (14, 15). We also demonstrated that there are chronic alterations in remote vascular function following AKI by showing that skeletal muscle arterioles are hypersensitive to ANG II stimulation at 5 wk of recovery from I/R, which occurs independent of changes in circulating renin or ANG II levels (2). This suggests that the post-AKI kidney liberates a factor(s) that alters systemic vascular responses.
The identities of such factors remain unknown but may be secondary to immune or inflammatory activity within the injured kidney. Persistent activation of immune and inflammatory activity has been suggested to promote progressive CKD following AKI. For example, Burne-Taney et al. (8) demonstrated that splenocytes derived from mice following I/R induced renal fibrosis and albuminuria in recipient, noninjured mice. In addition, Takada et al. (26) demonstrated that blockade of the B7 costimulatory pathway improved long-term function in rats following cold renal I/R. Finally, our group demonstrated that the late administration of the lymphocyte inhibitory agent MMF, beginning at 5 wk post-AKI, blocked the subsequent hypertension and CKD following exposure of rats to elevated dietary sodium (21). Because impaired renal medullary blood flow is typically associated with salt-sensitive hypertension (19), the beneficial effect of MMF after established capillary rarefaction suggests that capillary loss, per se, may not be the dominant feature that alters hemodynamic responses and imparts salt sensitivity following I/R in rats.
Therefore, the current studies were conceived to determine the potential that acute I/R could chronically influence renal hemodynamic function and salt sensitivity in the absence of direct kidney injury. We carried out UNx just before 5 wk of recovery so that we could isolate the effects of the postischemic milieu in the presence of the directly injured ipsilateral kidney, or the remotely injured contralateral kidney. The 5-wk time point is historically relevant to our previous studies as it is typically associated with complete repair; however, when challenged with elevated sodium, rats develop hypertension and CKD (25). It is important to note the sham-operated control group used in this study has some deficiencies as it does not control for the early loss of GFR that is measurable over the first week of recovery following unilateral I/R. In this regard, it is worth noting that the contralateral kidney following unilateral I/R has a greater mass relative to the sham group at day 33, which likely reflects compensatory activity secondary to the loss of GFR. Thus, in addition to being exposed to the ischemic milieu, the contralateral kidney undergoes compensatory growth activity for a longer duration than the sham-I/R group, which is not subjected to UNx until day 33. Conceivably, the prolonged compensatory activity in the I/R groups may have contributed to some of the functional responses reported here. In the future, the inclusion of a control group in which UNx is carried out at day 0 may allow us to address this issue.
To investigate the potential underlying mechanism of sodium-sensitive nature of the hypertension, previous studies from our group demonstrated that post-AKI rats at 5 wk of recovery on standard salt diet have impaired pressure natriuresis and renal medullary blood flow responses to increased RPP (22). Under these conditions, blood pressure of post-AKI rats is the same as shams. In addition, GFR, percent fractional Na excretion, and total RBF are largely recovered, while a persistent urinary concentrating defect is present (4, 22). The experimental approach uses denervated rats under hormone clamp conditions and is useful in investigating differences in renal function when extrarenal influences are held constant. As previous studies showed the importance of renal medullary blood flow in Na excretion and the development of hypertension (19), our prior results are consistent with the hypothesis that impaired renal medullary blood flow responses may impart salt sensitivity post-AKI. However, we cannot exclude that post-AKI rats have altered sensitivity post-AKI to the hormone clamp-denervated conditions used in these studies. Moreover, within the current investigation, it is unclear whether alterations in sodium handling and medullary hemodynamics could be affected in the setting of contralateral/remote AKI.
Taking into consideration these important caveats, the results of the current study do indicate that remote/contralateral injury chronically alters renal hemodynamic responses having a similar impairment in pressure natriuresis and medullary blood flow responses as direct injury. The reason for the impaired hemodynamic response in remote/contralateral AKI rat kidneys is not yet clear. Renal I/R is known to reduce the density of renal capillaries, but the effects of I/R on capillary density in contralateral kidneys have not been determined. Previous studies showed that hepatic I/R could result in renal damage and peritubular capillary endothelial cell apoptosis (17). Such a result suggests that remote injury could alter renal vascular structure. However, in the current study, capillary loss was associated with only direct injury in the ipsilateral kidney, but not remote injury to the contralateral kidney. Given that both direct and remote injury models are associated with impaired blood flow responses, the data indicate that factors independent of capillary loss can impair renal hemodynamic responses post-AKI.
In our previous studies, we demonstrated that renal I/R was associated with persistent increases in plasma malondialdehyde levels and demonstrated evidence of oxidant stress within peripheral skeletal muscle arteriole 5 wk following injury (23). Similarly, it is possible that local oxidant stress in the remote/contralateral kidney will mediate altered vascular responses. Unfortunately, we did not evaluate the effect of direct vs. remote injury on renal oxidant stress levels in the current study.
Additional potential mediators, which are perhaps related to or may contribute to oxidant stress, are infiltrating immune or inflammatory cells. Clearly, inflammatory cells represent a source of increased superoxide activity and De Miguel et al. (10, 11) showed that T lymphocytes derived from Dahl S rats express all of the necessary components of NADPH and local renin-angiotensin system. Thus, these cells may contribute to alterations in local hemodynamic control in a paracrine fashion. In the current study, interstitial cells were consistently observed in remotely injured kidneys at 5 wk of recovery (i.e., before the elevated sodium intake phase). Based on the results of FACS analysis, the increased interstitial cellularity may be due to the persistent elevations in T lymphocytes following AKI. Little information has been available regarding the chronic effects of AKI on persistent T cell deposition. In a previous study by Burne Taney et al. (9), evidence of persistent T cell deposition was found in mouse kidneys up to 6 wk following a severe episode of I/R injury and our data are consistent with this report. However, the current study also demonstrates that there is a significant population of T cells that reside in the remote contralateral kidney with low CD62L and high CD25 expression, suggesting that they are in an activated state. In light of our previous results demonstrating that MMF treatment attenuated salt-sensitive hypertension post-AKI, these observations suggest that T lymphocytes may modulate renal hemodynamic activity following AKI.
One of the most interesting aspects of the current study is the observation that salt-sensitive hypertension is segregable from the secondary damage and CKD induced by dietary salt post-AKI. Despite the fact that both remote/ipsilateral and direct/contralateral injury groups showed similar degrees of hypertension, only the direct injury animals manifested significant proteinuria and evidence of significant tissue damage. In previous studies, we showed that reduced renal mass and elevated sodium diet hasten proteinuria and secondary tissue damage (3, 25). In the current study, we applied both of these maneuvers, resulting in a more severe CKD than in previous experimental approaches, including an ∼40% reduction in creatinine clearance within 3 wk of elevated sodium intake. In addition, previous studies in rats demonstrate that post-AKI kidneys are hypertrophic with evidence of fibrosis and inflammation. However, in the current study, the combination of direct injury with reduced renal mass and elevated sodium intake was associated with lower renal mass, representing a more advanced or accelerated model of CKD.
The observation that significant CKD did not develop in the remote/contralateral injury group exposed to elevated salt was also noteworthy as it is not consistent with the observation brought forth by Burne-Taney et al. (8) in which adoptive transfer of splenocytes from AKI mice promoted albuminuria and damage in naive recipient mice. Presumably, these experimental approaches have some similarities as they both can interrogate I/R-induced immune responses on an untouched kidney, albeit using very different approaches. We can only speculate on why CKD developed in the study by Burne Taney et al., but not in the remote AKI group in our current study. Important factors may be due to differences in species responses, the amount of cells used for transfer, or the duration of time allowed for recovery and the manifestation of disease. In our remote/contralateral group, there was modest renal damage and it is possible that more significant damage may have developed with a longer duration recovery.
Based on the cumulative evidence of previous studies showing the protective effects of both lymphocyte inhibition by MMF, and vascular preservation by VEGF-121, and the data from the current studies, we suggest that aspects of direct tissue injury in combination with activated immune system synergize to promote the AKI to CKD transition following I/R injury. The nature of the interaction we cannot determine, but it has been proposed that renal ischemia may activate lymphocyte activity by the exposure of neo-antigens (1, 7). The persistent reduction in renal capillaries may be associated with a persistent expression of neo-antigens, due either directly to hypoxia or indirectly by affecting the complete resolution of renal parenchymal structure during repair. In this state, elevated dietary sodium may provide an opportunity to amplify inflammatory signals by reexposing antigens or by producing other local regulatory factors that amplify an existing immune response. Future studies will be required to determine the relative contribution of circulating cells and the nature of renal injury that activates immune function and promotes the responses associated with CKD following recovery from AKI.
GRANTS
This work was supported by National Institutes of Health Grant DK063114 (to D. Basile) and the Life Health Science Internship Program of Indiana University Purdue University Indianapolis (to D. Tonade).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.P.B. conception and design of research; D.P.B., E.C.L., J.L.F., and S.G. analyzed data; D.P.B. and S.G. interpreted results of experiments; D.P.B. and S.G. prepared figures; D.P.B. drafted manuscript; D.P.B. and S.G. edited and revised manuscript; D.P.B. approved final version of manuscript; E.C.L., D.T., and J.L.F. performed experiments.
REFERENCES
- 1. Anders HJ. Toll-like receptors and danger signaling in kidney injury. J Am Soc Nephrol 21: 1270–1274, 2010. [DOI] [PubMed] [Google Scholar]
- 2. Basile DP, Donohoe DL, Phillips SA, Frisbee JC. Enhanced skeletal muscle arteriolar reactivity to Ang II following recovery from ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol 289: R1770–R1776, 2005. [DOI] [PubMed] [Google Scholar]
- 3. Basile DP, Donohoe DL, Roethe K, Mattson DL. Chronic renal hypoxia following ischemia/reperfusion injury: effects of l-arginine on hypoxia and secondary damage. Am J Physiol Renal Physiol 284: F338–F348, 2003. [DOI] [PubMed] [Google Scholar]
- 4. Basile DP, Donohoe DL, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol 281: F887–F899, 2001. [DOI] [PubMed] [Google Scholar]
- 5. Basile DP, Fredrich K, Chelladurai B, Leonard EC, Parrish AR. Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol 294: F928–F936, 2008. [DOI] [PubMed] [Google Scholar]
- 6. Basile DP, Friedrich JL, Spahic J, Knipe NL, Mang HE, Leonard EC, Ashtiyani SC, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol 300: F721–F733, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int 66: 480–485, 2004. [DOI] [PubMed] [Google Scholar]
- 8. Burne-Taney MJ, Liu M, Ascon D, Molls RR, Racusen L, Rabb H. Transfer of lymphocytes from mice with renal ischemia can induce albuminuria in naive mice: a possible mechanism linking early injury and progressive renal disease? Am J Physiol Renal Physiol 291: F981–F986, 2006. [DOI] [PubMed] [Google Scholar]
- 9. Burne-Taney MJ, Yokota N, Rabb H. Persistent renal and extrarenal immune changes after severe ischemic injury. Kidney Int 67: 1002–1009, 2005. [DOI] [PubMed] [Google Scholar]
- 10. De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 298: R1136–R1142, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol 300: F734–F742, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geng H, Lan R, Wang G, Siddiqi AR, Naski MC, Brooks AI, Barnes JL, Saikumar P, Weinberg JM, Venkatachalam MA. Inhibition of autoregulated TGF-β signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am J Pathol 174: 1291–1308, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldstein S, Devarajan P. Acute kidney injury in childhood: should we be worried about progression to CKD? Pediatr Nephrol 26: 509–522, 2011. [DOI] [PubMed] [Google Scholar]
- 14. Grams ME, Rabb H. The distant organ effects of acute kidney injury. Kidney Int In press. [DOI] [PubMed] [Google Scholar]
- 15. Hassoun HT, Grigoryev DN, Lie ML, Liu M, Cheadle C, Tuder RM, Rabb H. Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol 293: F30–F40, 2007. [DOI] [PubMed] [Google Scholar]
- 16. Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 20: 223–228, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee HT, Park SW, Kim M, D'Agati VD. Acute kidney injury after hepatic ischemia and reperfusion injury in mice. Lab Invest 89: 196–208, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leonard EC, Friderich J, Basile DP. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am J Physiol Renal Physiol 295: F1648–F1657, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mattson DL. Importance of the renal medullary circulation in the control of sodium excretion and blood pressure. Am J Physiol Regul Integr Comp Physiol 284: R13–R27, 2003. [DOI] [PubMed] [Google Scholar]
- 20. Mattson DL, Raff H, Roman RJ. Influence of angiotensin II on pressure natriuresis and renal hemodynamics in volume-expanded rats. Am J Physiol Regul Integr Comp Physiol 260: R1200–R1209, 1991. [DOI] [PubMed] [Google Scholar]
- 21. Pechman K, Basile DP, Lund H, Mattson DL. Immune suppression blocks sodium sensitive hypertension following recovery from acute renal failure. Am J Physiol Regul Integr Comp Physiol 294: R1234–R1239, 2008. [DOI] [PubMed] [Google Scholar]
- 22. Pechman KR, De Miguel C, Lund H, Leonard EC, Basile DP, Mattson DL. Recovery from renal ischemia-reperfusion injury is associated with altered renal hemodynamics, blunted pressure natriuresis, and sodium-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol 297: R1358–R1363, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Phillips SA, Pechman KR, Leonard EC, Friedrich JL, Bian JT, Beal AG, Basile DP. Increased ANG II sensitivity following recovery from acute kidney injury: role of oxidant stress in skeletal muscle resistance arteries. Am J Physiol Regul Integr Comp Physiol 298: R1682–R1691, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roman R, Cowley A, Garcia-Estan J, Lombard J. Pressure-diuresis in volume-expanded rats. Cortical and medullary hemodynamics. Hypertension 12: 168–176, 1988. [DOI] [PubMed] [Google Scholar]
- 25. Spurgeon-Pechman KR, Donohoe DL, Mattson DL, Lund H, James L, Basile DP. Recovery from acute renal failure predisposes hypertension and secondary renal disease in response to elevated sodium. Am J Physiol Renal Physiol 293: F269–F278, 2007. [DOI] [PubMed] [Google Scholar]
- 26. Takada M, Chandraker A, Nadeau KC, Sayegh MH, Tilney NL. The role of the B7 costimulatory pathway in experimental cold ischemia/reperfusion injury. J Clin Invest 100: 1199–1203, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanner GA, Sloan KL, Sophasan S. Effects of renal artery occlusion on kidney function in the rat. Kidney Int 4: 377–389, 1973. [DOI] [PubMed] [Google Scholar]
- 28. Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298: F1078–F1094, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zager RA, Johnson ACM. Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. Am J Physiol Renal Physiol 296: F1032–F1041, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]