

Abstract
During viral infection, fusion of the viral envelope with endosomal membranes and nucleocapsid release were thought to be concomitant events. We show here that for the vesicular stomatitis virus, they occur sequentially, at two successive steps of the endocytic pathway. Fusion already occurs in transport intermediates between early and late endosomes, presumably releasing the nucleocapsid within the lumen of intra-endosomal vesicles, where it remains hidden. Transport to late endosomes is then required for the nucleocapsid to be delivered to the cytoplasm. The latter step, which initiates infection, depends on the late endosomal lipid lysobisphosphatidic acid (LBPA) and its putative effector Alix/AIP1 and is regulated by PI3P signaling via the PI3P-binding protein SNX16. We conclude that the nucleocapsid is exported into the cytoplasm after the back-fusion of internal vesicles with the limiting membrane of late endosomes, and that this process is controlled by the phospholipids LBPA and PI3P, and by their effectors.
Keywords: Animals; Biological Transport; physiology; Cattle; Cell Line; Cricetinae; Cytosol; metabolism; ultrastructure; Endosomal Sorting Complexes Required for Transport; Endosomes; metabolism; ultrastructure; Epithelial Cells; virology; Fibroblasts; virology; Hela Cells; Humans; Lysophospholipids; physiology; Membrane Fusion; drug effects; physiology; Microscopy, Electron; Microscopy, Fluorescence; Monoglycerides; Nucleocapsid; metabolism; Phosphatidylinositol Phosphates; physiology; Phosphoproteins; genetics; physiology; RNA, Viral; biosynthesis; metabolism; Signal Transduction; physiology; Sorting Nexins; Time Factors; Transport Vesicles; metabolism; ultrastructure; Vesicular Transport Proteins; genetics; physiology; Vesicular stomatitis Indiana virus; physiology; Virus Replication; genetics
INTRODUCTION
Endocytosed proteins and lipids first appear in early endosomes 1, from where some molecules are recycled to the cell surface or transported to the trans-Golgi network (TGN), while others are routed to late endosomes and lysosomes for degradation 2–4. Along the latter route, major progress has been made in understanding the mechanisms that control sorting of downregulated receptors. These are selectively incorporated into intra-luminal vesicles of the forming endosomal transport intermediates, which exhibit a typical multivesicular appearance. Both receptor sorting and the invagination process are regulated by PI3P signaling via Hrs, which contains the PI3P-binding motive FYVE, and by ESCRT-I, II and III complexes sequentially 5,6. Then, multivesicular endosomal intermediates transport their cargo of intraluminal vesicles towards late endosomes and lysosomes, where intraluminal vesicles will eventually be degraded 6.
The intra-luminal membranes of the multivesicular — or multilamellar — regions of late endosomes, however, also contain molecules that are not destined for degradation. These include tetraspanins 7, mannose-6-phosphate receptor in transit 8, and MHC class II molecules in antigen-presenting cells 9, as well as the poorly degradable phospholipid lysobisphosphatidic acid (LBPA). This lipid, which is only found in late endosomes, plays a direct role in protein and lipid transport through this compartment 10–14. Little is known about the mechanisms regulating membrane invagination within late endosomes, except that LBPA itself may control the process via its putative effector Alix 15, a protein that also interacts with ESCRTs during HIV budding at the plasma membrane 16,17.
It has long been known that vesicular stomatitis virus (VSV) and many other enveloped viruses must be endocytosed for infection to proceed, and that virions are then transported along the endocytic pathway leading to lysosomes. Beyond early endosomes 18,19, but before the virions reach lysosomes, the acidic pH triggers the fusion of the viral envelope with endosomal membranes, releasing the nucleocapsid into the cytosol, where replication of the viral genome occurs 20,21. We recently found that Alix down-expression with siRNAs inhibits VSV infection 15, perhaps suggesting that endosomes play an active role in the infection process. Here, we followed the VSV infection pathway in vivo (by monitoring viral envelope fusion, viral RNA replication and viral protein synthesis) and nucleocapsid release in vitro, to investigate whether the dynamics of endosomal membrane control viral fusion or nucleocapsid release.
RESULTS
Time-course of viral fusion
To detect fusion, VSV was labeled with self-quenching amounts of the fluorescent long-chain dialkylcarbocyanine dye Dil 22,23. Low, physiologically relevant amounts of VSV were then used in all our experiments (0.1–1.0 MOI, see supplementary materials). After VSV pre-binding to the cell surface at 4°C, cells were incubated at 37°C, so that endocytosis resumed, and analyzed by time-lapse microscopy. Little fluorescent signal was detected in cells kept at 4°C (Fig 1A, frame t = 0), even when incubated at pH 5.0 (Fig 1B), as observed in vitro 22, or in cells incubated at 37°C in the presence of the V-ATPase inhibitor bafilomycinA1 (Fig 1B), confirming the validity of the assay. By contrast, brightly fluorescent spots appeared after incubation at 37°C in the absence of the drug (Fig 1A). This sequence is shown both with DIC optics (DIC.avi) and Dil fluorescence (FLUO.avi) in the supplementary materials. After a short lag-time, the fluorescence emitted by individual spots increased rapidly, reaching a maximum after 20–25 min (Fig 1C illustrates the time-course of two characteristic fusion events that cover the time-frame observed in these experiments), much like influenza virus 23. Similarly, the number of cells containing a fluorescent signal increased rapidly at 37°C and then leveled off (Fig 1D), perhaps suggesting that viral fusion was triggered before most virions reached late endocytic compartments. Indeed, it takes 30–60min at 37°C for the bulk of an endocytosed tracer, including the glycoprotein G of VSV envelope 24, to reach late endosomes and lysosomes in the same (BHK) cells 25 (Fig 1E illustrates the colocalization of G-protein with a fluid phase tracer in late endosomes containing LBPA after 45 min).
Figure 1. VSV fusion.
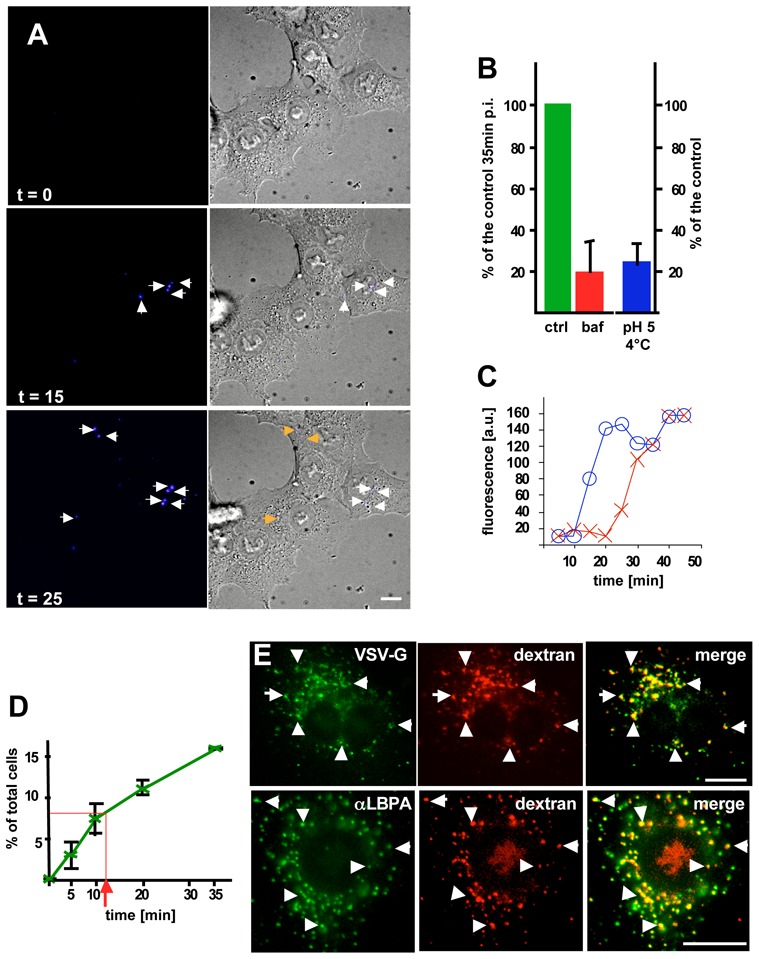
(A) BHK cells were incubated with low amounts (0.3 MOI) Dil-labeled VSV at 4°C. The temperature was raised to 37°C, and cells were imaged by time-lapse confocal microscopy. The figure shows frames captured at the indicated time, and arrows point at fluorescent spots that represent fusion events. (B) Cells were treated as in (A) in the presence (baf) or absence (ctrl) of 1μM bafilomycinA1. The number of cells containing fused viruses was counted after 35 min at 37°C (post-infection: p-i ). Alternatively, cells with bound virions were incubated at pH 5.0 at 4°C, and fusion events were immediately counted and expressed as a percentage of the control at 37°C. (C) The figure shows two representative examples of the endocytic time-course of viral fusion (as in A): the emitted fluorescence was quantified after tracking each spot in the sequence. (D) The number of cells treated as in (A) containing fused viruses was counted at the indicated times during the 37°C incubation, and is expressed as a percentage of the total cell number. (E) Excess virus (50μg/1.3x107 cells) were pre-bound to the cell surface (as in A), and then co-endocytosed with rhodamine-dextran for 5 min at 37°C, followed by a 40 min chase without dextran. Cells were processed for immnofluorescence and labeled with the indicated antibodies. VSV-G colocalized with endocytosed dextran (upper panels), which itself colocalized with LBPA (lower panels). Number of experiments: B, 3; D, 4. Bars, A: 2,5μm; E: 4μm.
Fusion occurs in endosomal transport intermediates
Transport from early to late endosomes is facilitated by microtubules 24,26,27 (see outline Fig S1, supplementary materials). Consistently, a pulse of endocytosed tracers failed to reach late endosomes containing LBPA after a 45 min incubation at 37°C in the presence of the microtubule depolymerizing drug nocodazole, and yet was no longer detected in early endosomes containing the Rab5 effector EEA1 (Fig S2A, supplementary materials). As expected 24, nocodazole also inhibited G-protein degradation, as did the V-ATPase inhibitor concanamycinB (Fig S2B). Microtubule depolymerization did not affect viral fusion to any significant extent (Fig 2A–B). Fluorescence ratio imaging showed that the pH of endosomes containing a pH-sensitive probe after microtubule depolymerization was approximately 5.5 (Fig 2C), as acidic as late endosomes, and significantly more acidic than early endosomes (pH=6.2) 28. These observations thus indicate that fusion occurs within acidic endosomal vesicles, but before late endosomes. These vesicles correspond to the operational definition of endosomal transport intermediates, or ECV/MVBs 6, which accumulate endocytosed VSV G-protein or other tracers in the absence of microtubules 24.
Figure 2. Microtubule-dependent transport.
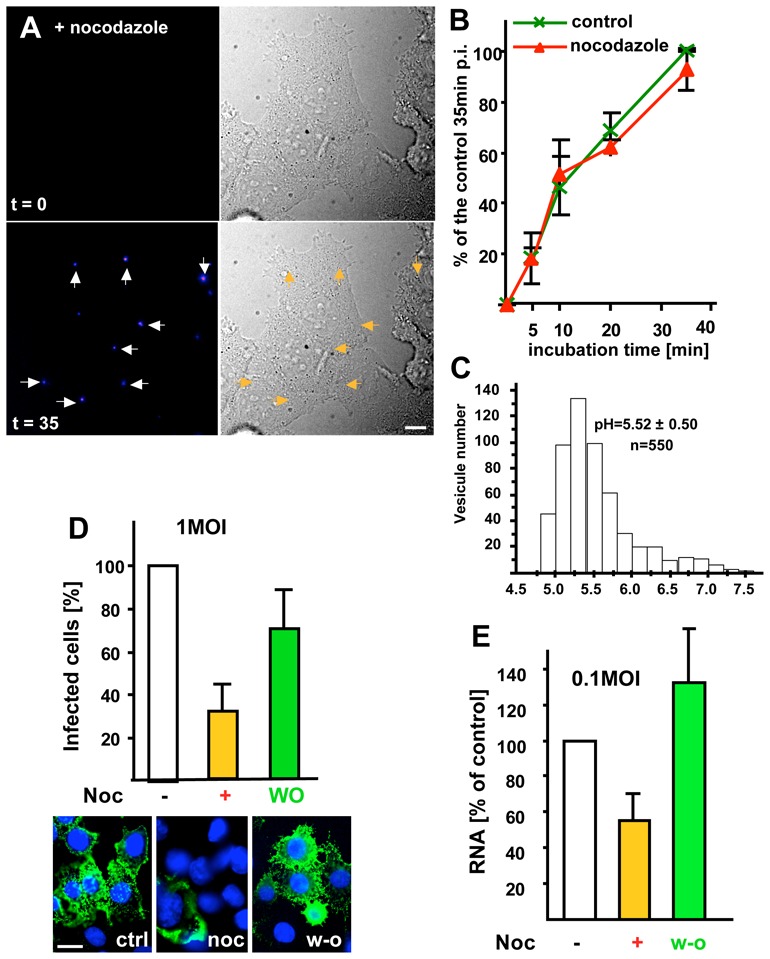
(A) Viral fusion was studied as in Fig 1A in cells pre-treated with 10μM nocodazole for 2h (the drug was present throughout the experiment), and frames were captured at the indicated time. (B) After treatment with or without nocodazole as in (A), the number of cells containing fused viruses was counted at the indicated times during the 37°C incubation. Values are expressed as a percentage of the untreated control after 35min, as in Fig 1B. (C) After microtubule depolymerization as in (A), BCECF-dextran was endocytosed for 10 min at 37°C and chased for 35min. The pH of individual endosomes was measured by BCECF-dextran fluorescence ratio imaging. The histogram shows the pH distribution of 550 endosomes with the means ± SD indicated on the figure. (D) VSV (1MOI) was bound at 4°C to the surface of BHK cells preincubated without (control, ctrl) or with nocodazole (noc), as in (A). Cells were incubated for 3h at 37°C to allow infection to proceed. When indicated (W-O, wash out), nocodazole was removed, and incubation continued without drugs for 2h. Cells were analyzed by immunofluorescence microscopy using antibodies against VSV-G after labeling nuclei with DAPI (blue). Typically, ≈70% of the cells were infected under control conditions, and the number of infected cells is expressed as a percentage of the untreated control. (E) Cells treated or not with nocodazole (as in D) were infected with 0.1 MOI VSV. Replication of VSV RNA minus strand was quantified by TaqMan-RT-PCR and results are expressed as a percentage of the untreated control. Number of experiments: B, 3; D, 6; E, 4. Bars, A: 2,5μm; D: 4μm.
Transport to late endosomes is required for efficient infection and RNA replication
Despite the lack of nocodazole effects on viral fusion (Fig 2A–B), infection, as monitored by G-protein synthesis, was markedly reduced by nocodazole — to the same extent as transport to late endosomes 24,27 — and restored to control levels upon drug wash-out (Fig 2D). This was not due to a role of the microtubules in the transport of free cytosolic capsids, since nocodazole had no effect on infection (Fig S3A supplementary materials) when VSV fusion was artificially induced at the plasma membrane 24,29, in agreement with previous observations 30. Then, however, infection was much less efficient (10% of controls), supporting the view that VSV highjacked the endocytic pathway to reach the perinuclear region more efficiently. It thus seems that, under physiological conditions, efficient infection, but not fusion, requires transport to late endosomes.
To interfere selectively with late endosome functions 11–14,31, we used our monoclonal antibody against LBPA. When internalized by fluid phase endocytosis, the antibody accumulates in late endosomes upon binding to its antigen 11. The endocytosed antibody, but not control antibodies, inhibited in a dose-dependent manner infection with VSV (Fig 3A), but not Sendaï virus (Fig 3B) — which undergoes fusion at the plasma membrane. This inhibition was not due to some indirect effects of the antibody on the virions. Indeed, VSV pre-treatment with anti-LBPA antibodies did not affect VSV binding (Fig S3B, supplementary materials) or infection (Fig 3C), while an antibody against an exposed viral epitope 32 efficiently blocked infection (Fig 3C) by preventing VSV binding to the cell surface (Fig S3B supplementary materials). Strikingly, however, endocytosed anti-LBPA antibodies had little effect on viral fusion (Fig 3D), even at high doses, further supporting the notion that most fusion events had already occurred in ECV/MVBs, prior to arrival in LBPA-containing late endosomes. When nocodazole was added to cells pre-treated with anti-LBPA antibodies, infection was further reduced when compared to the drug alone, and returned to the levels of the antibody alone upon drug wash-out (Fig 4A). Hence, the reduced infection rate detected with nocodazole (Fig 2D) might be caused by some virions that reached late endosomes even in the absence of microtubules. Indeed, microtubules facilitate transport to late endosomes, but transport may continue, albeit inefficiently, after depolymerization 27.
Figure 3. VSV infection requires transport to late endosome.
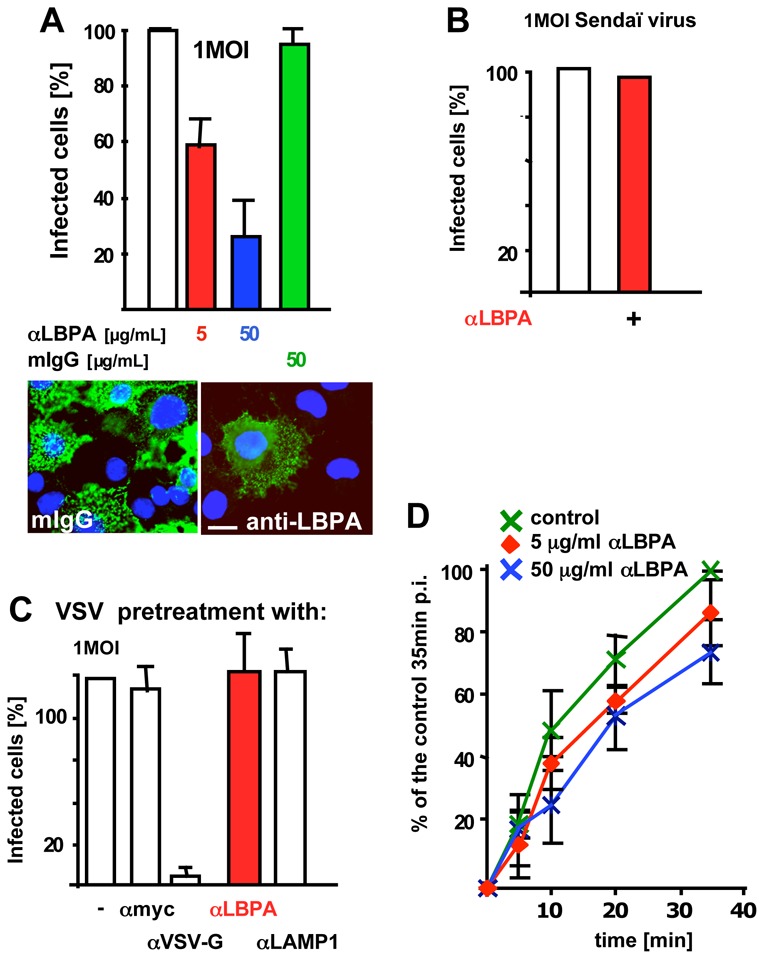
(A) Cells were preincubated with 5 or 50μg/ml anti-LBPA (αLBPA) antibody or 50μg/ml mouse IgG (mIgG) for 14h. Cells were then infected with 1MOI VSV for 3h, fixed, analyzed by fluorescence microscopy (example shown with 50μg/ml antibody). Infected cells were quantified as in Fig 2D. (B) Cells treated as in A with 50μg/ml anti-LBPA antibodies were infected with recombinant Sendaï virus expressing RedFP (1MOI) for 14h, and analyzed by fluorescence microscopy. Infected cells are expressed as a percentage of the untreated controls — typically, 25% of the cells were infected in controls. (C) VSV was preincubated for 1h at 4°C with antibodies against the indicated antigens, bound to the cell surface for 1h at 4°C and used for cell infection as in (A). Infected cells were quantified as in Fig 2D. (D) Cells were pre-treated with anti-LBPA antibodies, as in A. Then, viral fusion was studied as in Fig 1A, and quantified as in Fig 2B. Number of experiments: A, 4; B, 3; C, 3; D: 4. Bar, A: 4μm
Figure 4. Combination of treatments and RNA replication.
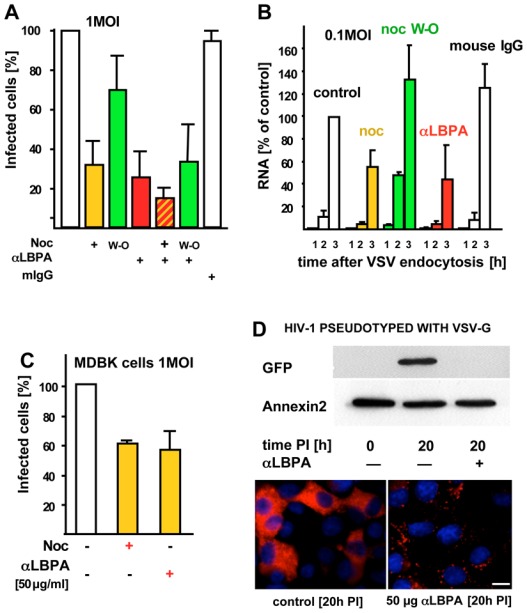
(A) Cells pre-treated with 50μg/ml anti-LBPA antibody or mouse IgG, as in Fig 3A, were then incubated with nocodazole as in Fig 2D with or without wash-out (W-O), and infected with 1MOI VSV. Then cells were analyzed by fluorescence microscopy, and infected cells were quantified as in Fig 2D. (B) Cells treated essentially as in (A) were infected with 0.1 MOI VSV. Replication of VSV RNA minus strand was quantified by RT-PCR, as in Fig 2E, at the indicated times after VSV endocytosis. Results are expressed as a percentage of RNA replication measured in the 3h control. It should be noted that all experiments in (A) or (B), but also in Fig 2D and E, and Fig 5C were performed in parallel on the same day. Data were split into separate figures for the sake of clarity. (C) Madin-Darby bovine kidney (MDBK) cells were infected with VSV without or with nocodazole or anti-LBPA antibodies, as in Fig 2A and 3A, respectively. Effects of anti-LBPA antibodies were less pronounced than in BHK or HeLa cells, presumably because MDBK cells did not take up antibodies as efficiently. (D) BHK cells, treated (αLBPA) or not with anti-LBPA antibodies as in Fig 3A, were infected with HIV-1 pseudotyped with VSV-G and expressing GFP. After 20h (20h PI), GFP expression was analyzed by SDS gel electrophoresis and Western blotting using anti-GFP antibodies, or anti-annexin2 antibodies as a loading marker (upper panel). Alternatively cells were analyzed by immunofluorescence using a monoclonal anti-GFP antibody, followed by rhodamine-labeled anti-mouse antibodies. Treated cells show a punctate staining pattern, corresponding to the endocytosed anti-LBPA (revealed by the secondary anti-mouse antibody), but not the diffuse GFP staining pattern visible in the controls. Number of experiments: A–B, 4; C, 3. Bar, D: 4μm
The observed requirement of VSV for transport to late endosomes does not seem to reflect a property unique to BHK cells, since infection of bovine cells (cattle is naturally infected by VSV) was also sensitive to microtubule depolymerization or endocytosed anti-LBPA antibodies (Fig 4C; see also HeLa cells in Fig 7A, F). Moreover, infection of BHK cells with an HIV-1-derived vector expressing GFP and pseudotyped with VSV G-protein 33 was also inhibited by endocytosed anti-LBPA antibodies (Fig 4D). Since HIV-1 normally fuses at the plasma membrane, these data show that the characteristic features of VSV-G protein are transferable and responsible for the observed phenotype. Hence, the requirement for transport to late endosomes seems to be dictated by the fusion and trafficking properties of the G-glycoprotein.
Figure 7. Hrs and PI3P.
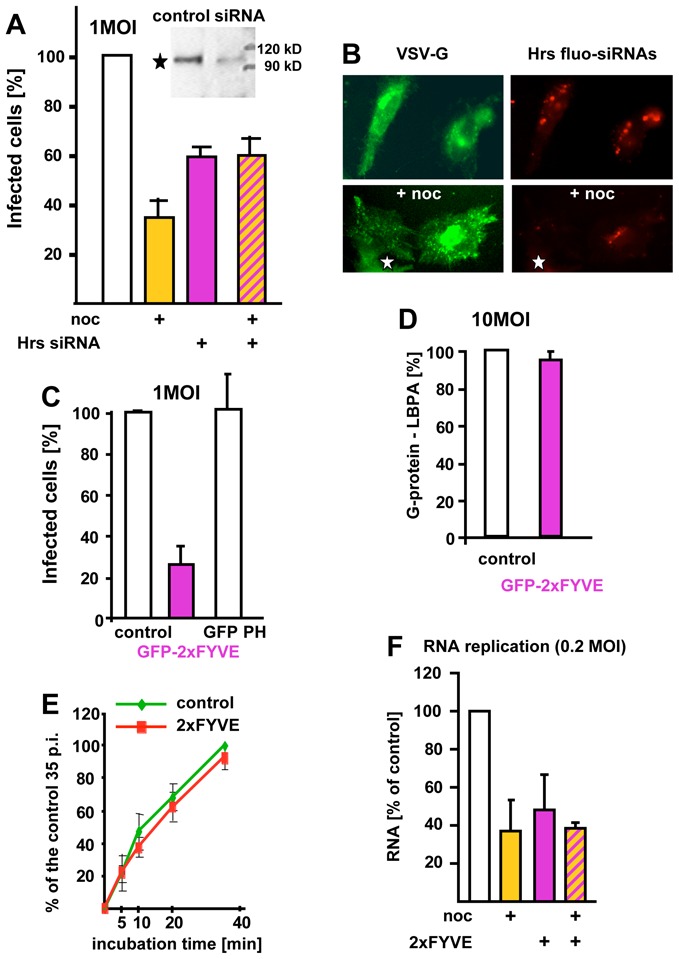
(A–B) The expression of Hrs was silenced using fluorescently labeled siRNAs (Hrs fluo-siRNAs), to identify transfected cells (B). The inset in (A) shows the Western blot of cells treated with Hrs siRNAs using anti-Hrs antibodies. Cells were then infected with VSV with or without nocodazole (noc), as in Fig 2D, and analyzed by fluorescence microscopy to identify cells containing labeled siRNAs and cells expressing VSV-G. The star shows an infected cell that did not contain siRNAs, for comparison. Infected and non-infected cells were quantified as in Fig 2D. (C) Cells were transfected with GFP-2xFYVE or GFP-PH for 36h, infected with VSV (1MOI), and analyzed by fluorescence microscopy to identify transfected cells. Infected and non-infected cells were quantified as in Fig 2D. (D) After transfection with GFP-2xFYVE, as in (C), excess VSV (50μg/1.3 × 107 cells) was bound to the cell surface. The virus was labeled with anti-VSV antibodies, followed by secondary antibodies and then endocytosed for 45 min, as in Fig 1E. Cells were fixed, labeled with anti-LBPA antibodies and analyzed by double-channel fluorescence. The number of cells where both markers colocalized was counted and is expressed as a percentage of the control. (E) Viral fusion was quantified as in Fig 1A–B in cells overexpressing GFP-2xFYVE, as in C. (F) RNA replication was quantified as in Fig 2E in HeLa cells overexpressing GFP-2xFYVE with or without nocodazole (noc) treatment. Number of experiments: A, C–F, 3. Bar, B: 4μm
To ensure that nocodazole or anti-LBPA antibodies did not interfere somehow with the G-protein biosynthetic pathway, we quantified the replication of viral RNA minus strand by TaqMan-RT-PCR. Microtubule depolymerization (Fig 2E) or endocytosed anti-LBPA antibodies (Fig 4B) inhibited the synthesis of viral RNA minus-strand (and plus-strand, Fig S4A, supplementary materials). Altogether, these experiments show that fusion of VSV envelope and nucleocapsid delivery to the cytosol occur sequentially, in ECV/MVBs and late endosomes, respectively.
Intra-luminal vesicles: PI3P signaling and Hrs
The unexpected findings that fusion and nucleocapsid release were sequential events, combined with the apparently crucial role of multivesicular endosomes, prompted us to investigate the possible role of endosome intraluminal vesicles in fusion and nucleocapsid delivery. During ECV/MVB biogenesis, both the formation of intra-luminal vesicles and the sorting of activated receptors within these invaginations are regulated by PI3P signaling via the PI3P effector Hrs 5,6,34. PI 3-kinase inhibition with wortmannin caused endosome vacuolation (Fig 5A, frame t = 35 min, black arrow), as expected 35–37, but did not significantly affect VSV fusion (Fig 5A–B). Interestingly, the drug increased the percentage of VSV infected cells (Fig 5C) and, to some extent, RNA replication (Fig 5D). But, in the presence of nocodazole and wortmannin, both infection (Fig 5C) and RNA replication (Fig 5D) were back to the levels measured in untreated control cells.
Figure 5. PI 3-kinase inhibition.
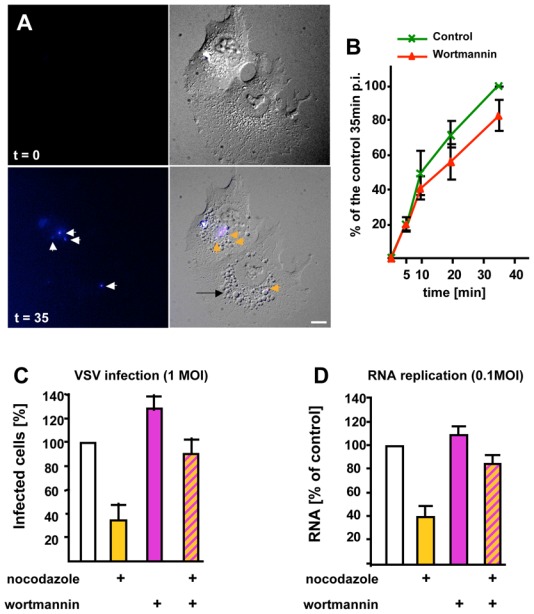
(A) Cells were treated with Dil-labeled VSV and analyzed as in Fig 1A, except that 100nM wortmannin was added. The figure shows frames captured at the indicated time. The black arrow points vacuoles appearing after wortmannin treatment. (B) Cells were treated as in (A) with or without wortmannin. The number of cells containing fused viruses was counted at the indicated times during the 37°C incubation, and is expressed as in Fig 2B. (C–D) VSV at the indicated MOI was endocytosed with or without nocodazole, as in Fig 2D, except that cells were treated with 100nM wortmannin 5 min after raising the temperature to 37°C. [Wortmannin was omitted during the initial 5 min, because the drug inhibits internalization.] In (C), cells were analyzed by fluorescence microscopy, and infected and non-infected cells were quantified as in Fig 2D, while in (D) replication of viral RNA was measured as in Fig 2E. Number of experiments: B–D, 4. Bar, A: 2,5μm.
We thus carried out an analysis by electron microscopy using large virus amounts — VSV could not be detected by electron microscopy at the low doses used for infection and RNA release. Intact viral particles (Fig 6A–B, arrows) can be identified in longitudinal sections by their characteristic bullet-like shape, and in cross section by the presence of G-protein spikes (Fig 6A–B, B′ small arrows). Such intact virions were very rarely seen 45 min after VSV internalization, and then only in endosomes with few internal vesicles, presumably early endosomes (Fig 6A). By contrast, smaller electron-dense structures without a visible spike-delineated envelope, presumably capsids (compare Fig 6B′ with D′, Fig 6C–G, arrows and inserts), were frequently observed within late endosomes identified with a pulse of endocytosed BSA-gold that had been chased for 45 min at 37°C (Fig 6F–G). These late endosomes exhibited a morphology characteristic for this compartment in BHK 24 and other cell types 38, including abundant internal membranes and large electron-lucent elements perhaps of lipidic origin. Capsids (Fig 6C–G, inserts) were present within an electron-lucent space separated from the lumen of the endosome itself (containing BSA-gold) by a membrane (small arrows), presumably intra-luminal vesicles. Vesicles of a similar diameter were seen in control cells that had not been treated with VSV (not shown), and similar ‘empty’ vesicles (without capsids) were clearly evident in the same endosomal structures (e.g. see Fig 6C). Moreover, in an independent set of experiments, endosomes were labeled in vivo (with endocytosed G-protein bound to anti-G antibodies and 10nm proteinA-gold)24, and endosomal fractions were prepared and incubated with 4nm BSA-gold at 4°C in vitro. Then, internal structures with a diameter similar to that of internal vesicles could be labeled with small gold (Fig 6I–J), suggesting that they represent cross-sections of membrane invaginations with a lumen equivalent to the cytoplasmic space, perhaps pre-fission or post-fusion intermediates. Finally, immunogold labeling of cryosections showed that VSV-G was indeed present within internal membranes (Fig 6H). Altogether, these observations are thus consistent with our findings that fusion occurs before late endosomes, and further suggest that, once fusion has occurred, capsids reside within the lumen of intra-endosomal vesicles. In cells treated with wortmannin, intact virions were occasionally detected in vesicles resembling early endosomes (Fig 6B–B′), much like in controls (Fig 6A), but capsids were not observed in endosomes, suggesting that, once fusion has occurred in wortmannin-treated cells, capsids are rapidly released into the cytoplasm.
Figure 6.
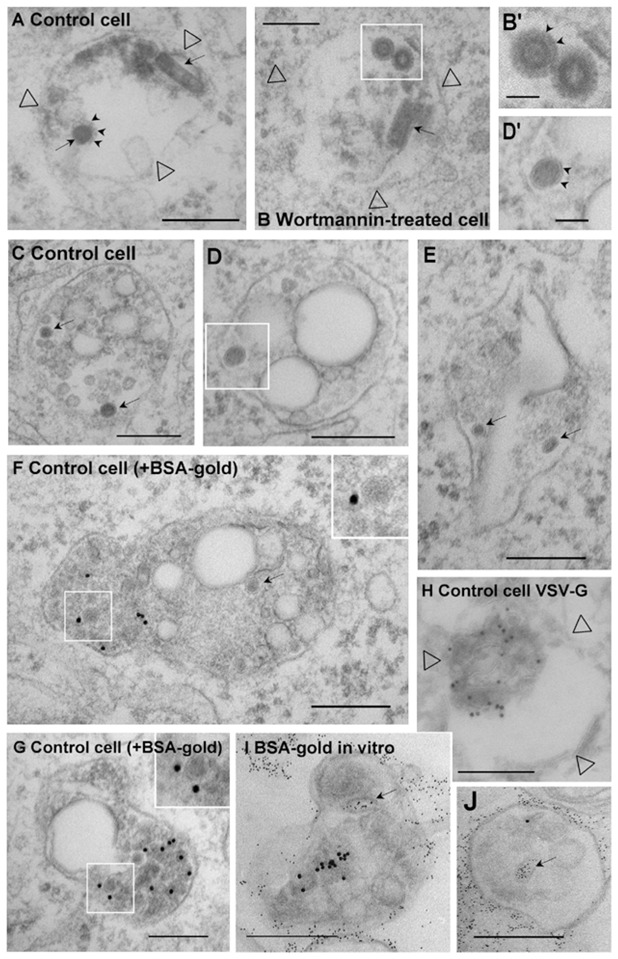
(A–H) After pre-binding to the surface (Fig 2D), VSV (100μg per 106 cells) was co-endocytosed with (F–G) or without (A-E and H) 10nm BSA-gold (OD = 1.7) for 10 min at 37°C, and then for 40 min without BSA-gold, in BHK cells treated (B) or not (A and C–H) with wortmannin. Cells were then processed for electron microscopy. (A–B) These panels show endosome (empty arrowheads point at the membrane) largely devoid of internal vesicles, presumably early endosomes, which contain both bullet-shaped virions and virions viewed in cross-section (arrows; small arrows point at G-protein spikes). (C–G) Electron-dense structures without a visible spike-delineated envelope, presumably capsids (arrows) are found within internal vesicles (see insets and the boxed area in D shown in D′; note clear membrane indicated by arrowheads in D′) of late endosomes containing gold particles in their lumen (F–G). (H) Experiment as in (A) and then the G-protein distribution was analyzed by immunogold labeling of cryo-sections using anti-VSV-G antibodies (empty arrowheads point at the membrane): note the characteristic electron-lucent space that is also observed in plastic embedded samples (C–G). (I–J) The G-protein was endocytosed for 45 min at 37°C into BHK cells 24 after labeling at the cell surface with a polyclonal antibody against G and then with 10nm proteinA-gold. Endosomal fractions were then prepared and incubated in vitro (as in Fig 8A) at 4°C with 4nm BSA-gold, and processed for electron microscopy. Note that BSA-gold labels apparently internal structures (arrows), which must have continuity with the limiting membrane (and are equivalent to cytoplasmic space) out of the plane of section. Bars: A–J: 0.2μm; B′-D′: 0.05μm).
Hrs is probably the most important PI3P effector responsible for both membrane invagination within ECV/MVBs and ubiquitinylated receptor sorting within these invaginations. Indeed, treatment with siRNAs against Hrs reduces the number of intraluminal vesicles in endosomes and receptor sorting, as also observed in yeast and Drosophila mutants with impaired Hrs function 5,39,40. Silencing Hrs expression with siRNAs (Fig 7A, inset) decreased VSV infection (Fig 7A), not surprisingly since Hrs downexpression causes major alterations in endocytic trafficking 39,40. But, even if somewhat reduced, infection still occurred efficiently despite Hrs knockdown. This is consistent with findings that VSV infection does not depend on an ubiquitin-dependent sorting mechanism, in contrast to influenza virus, which may use a receptor that is a target for ubiquitylation 41. Interestingly, however, as observed for the wortmannin treatment, viral infection was no longer sensitive to microtubule depolymerization in Hrs siRNA-treated cells (Fig 7A–B). Since both PI 3-kinase inhibition 37 and Hrs down-expression 40 inhibit the formation of intraluminal vesicles within ECV/MVBs, our data strongly suggest that these intraluminal vesicles are required for proper delivery of infectious VSV particles to late endosomes (see Model Fig S1 supplementary materials, and Discussion).
A dual role for PI3P
To further explore the possible role of PI3P, we investigated whether infection was sensitive to the expression of the PI3P binding domain FYVE, using a GFP-tagged tandem FYVE construct (GFP-2xFYVE) 42, which we have shown to inhibit receptor sorting, but not bulk transport to late endosomes 43. In marked contrast to PI 3-kinase inhibition, we found that 2xFYVE efficiently inhibited infection (Fig 7C), without affecting G-protein transport to late endosomes containing LBPA (quantification in Fig 7D) or viral fusion (Fig 7E), and did not render infection insensitive to microtubule depolymerization (Fig 7F). The effects of the tandem FYVE were specific for PI3P, since overexpression of the PH domain of phospholipase C delta, which binds PI(4,5)P2 44, had no effect on VSV infection (Fig 7C). We thus reasoned that PI3P, in addition to its role in the Hrs-ESCRT pathway, is perhaps also involved in nucleocapsid release from late endosomes, consistently with the presence of PI3P on late endosomes 42, where it may serve as a substrate for the PI3P 5-kinase Fab1/PIKfyve 45. We thus designed an assay that monitors nucleocapsid release in vitro to study the possible role of PI3P in the process.
RNA export in vitro
After binding VSV to the cell surface at 4°C, the virus was endocytosed at 37°C in the absence of microtubules, and then chased into late endosomes by allowing microtubule re-polymerization. Using this pulse-chase protocol, dextran and VSV accumulated in late endosomes, and VSV RNA minus strands then co-fractionated with late endosomes (Fig S4B supplementary materials). The viral RNA present in the fractions was not released by trypsin treatment of the membranes, indicating that capsids were present within endosomes, and not peripherally associated (Fig S4C, supplementary materials). Endosomal fractions were prepared and incubated in the assay with ATP and cytosol. Then, endosomes were separated from the cytosol (presumably containing the released viral RNA) by floatation in sucrose gradients, and RNA was quantified by RT-PCR in cytosol and endosomes. Viral RNA export from late endosomes occurred efficiently (30% of the amounts originally present in endosomes) at 37°C, but not at 4°C, and required the presence of ATP and cytosol (Fig 8A). Endosomes remained latent during the assay (90% of endocytosed HRP, used as a marker of the endosomal content, remained entrapped within endosomes), indicating that RNA was not released because of some damage caused to endosomes during the in vitro incubation. Moreover, viral RNA export was inhibited by the addition of excess purified recombinant Alix or by cytosol prepared from cells overexpressing Alix (Fig 8F), consistently with our previous in vivo observations 15. Altogether, these observations show that our assay measuring nucleocapsid release is valid. They also indicate that Alix controls the process directly, presumably by regulating the dynamics of late endosome internal membranes 15.
Figure 8. Nucleocapsid release in vitro.
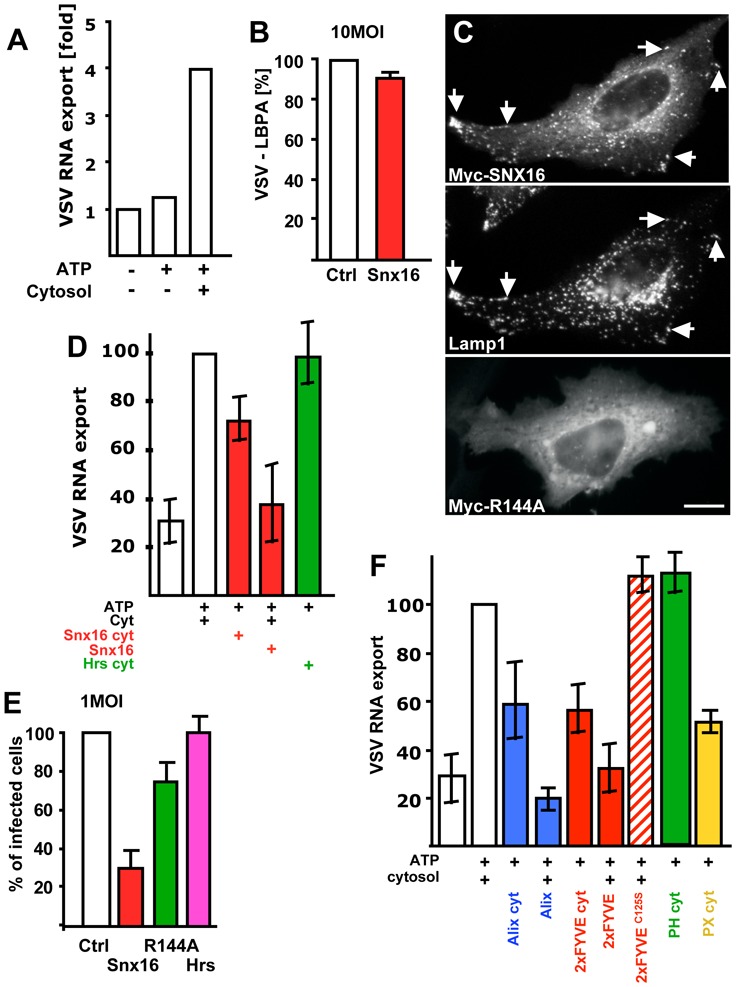
(A) Late endosomes were loaded with VSV in vivo, and endosomal fractions were prepared. These fractions [18μg] were incubated in vitro with an ATP-regenerating (ATP), with or without cytosol for 20 min at 37°C. Then, free (cytosolic) RNA was separated from endosome-associated RNA by floatation in a sucrose gradient, and VSV RNA minus strand was quantified by TaqMan-RT-PCR. RNA export is expressed as the ratio of free RNA released in the presence of ATP and cytosol over the negative controls without cytosol and ATP. (B) VSV endocytosed for 45 min (as in Fig 1E) in cells expressing Snx16-myc was analyzed by fluorescence microscopy; VSV-G colocalization with LBPA was quantified as in Fig 7D. (C) After expression of WT Snx16-myc or Snx16R144A-myc, HeLa cells were processed for immunofluorescence using antibodies against myc and Lamp1 (upper panels, double immuno-fluorescence) or myc alone (lower panel). (D) The assay was as (A) with cytosol prepared from cells overexpressing Snx16 (Snx16 cyt) or Hrs (Hrs cyt), or with control cytosol supplemented with 0.5μg purified recombinant Snx16 (Snx16). (E) Cells expressing or not Snx16, Snx16R144A or Hrs were infected with VSV (1MOI), and analyzed by fluorescence microscopy; infected and non-infected cells were quantified as in Fig 2D. (F) The assay was as in (A) with cytosol prepared from cells overexpressing GFP-2xFYVE (2xFYVE cyt), Alix (Alix cyt), the GFP-PH domain of PLC™ (PH cyt) or the GFP-PX domain of p40phox (PX cyt). Alternatively, the assay was carried out with control cytosol supplemented with 0.5μg purified, recombinant Alix, GST-2xFYVE (2xFYVE) or GST-2xFYVEC125S (2xFYVEC125S). VSV RNA− export was expressed as a percentage of the positive control, to facilitate comparison between different experiments. Number of experiments: A, 6; B–E 3; F, 4. Bar, C: 2,5μm.
SNX16 acts as PI3P effector in nucleocapsid export
Since GFP-2xFYVE inhibited viral infection, we tested whether the tandem FYVE also interfered with nucleocapsid release in vitro. Indeed, RNA export from late endosomes was efficiently inhibited by cytosol prepared from cells overexpressing GFP-2xFYVE or, even more so, by purified, recombinant GST-2xFYVE (Fig 8F). Effects were specific, since no inhibition was observed with the purified recombinant GST-2xFYVEC125S mutant (Fig 8F), which does not bind PI3P 42, or the PI(4,5)P2-binding domain PH of PLCδ (Fig 8F), much like in vivo (Fig 7C). In addition, we confirmed that the inhibition we observed was not due to some indirect effects of the FYVE domain, since RNA export was also inhibited by the addition of cytosol prepared from cells overexpressing the PI3P-binding domain PX of p40Phox (Fig 8F).
To identify a possible PI3P effector that may regulates RNA export from late endosomes, we screened the distribution of proteins containing PI3P-binding domains, and found that SNX-16 was an interesting candidate. This protein is a member of the sorting nexin family and contains the PI3P-binding motive PX 46,47. Previous studies showed that overexpressed SNX16 localizes to early and late endosomes and that the protein plays a role in EGF receptor sorting and EGF-induced signaling 48,49. Consistently, myc-tagged SNX16 (expressed at low levels) colocalized with Lamp1 (Fig 8C), an abundant protein of both late endosomes and lysosomes 8,27, and to a lesser extent with the early endosomal marker EEA1 (Fig S5A, supplementary materials). Strikingly, overexpression of SNX16-GFP strongly reduced VSV infection in vivo (Fig 8E), without affecting VSV transport to late endosomes containing LBPA (Fig 8B), consistently with findings that overexpressed SNX16 did not prevent, but increased, EGFR degradation 49. By contrast, the SNX16R144A mutant, with a single point mutation in the predicted PI3P binding motive, was largely cytosolic (Fig 8C), as expected, and had little effects on VSV infection (Fig 8E). Similarly, RNA export was inhibited in vitro by the addition of cytosol from cells overexpressing SNX16, or by purified recombinant SNX16 (Fig 8D). Effects were specific, since cytosol prepared from cells overexpressing Hrs had no effect on RNA export in vitro (Fig 8D), in agreement with our in vivo observations that Hrs overexpression had no effect on viral infection (Fig 8E). These observations suggest that SNX16 may act as PI3P effector in regulating viral RNA export from late endosomes.
DISCUSSION
Here, we show that the fusion of the enveloped virus VSV and the subsequent release of the viral nucleocapsid into the cytosol occur sequentially, at successive steps of the endocytic pathway. While viral fusion already occurs in ECV/MVBs, the nucleocapsid is delivered to the cytosol from late endosomes, in a process that depends on the late endosomal lipid LBPA, and its putative effector Alix/AIP1. Moreover, our data show that the delivery process is regulated by PI3P signaling via the PI3P effector SNX16. We conclude that, upon fusion of the viral envelope, the nucleocapsid is first delivered within the lumen of vesicles contained within ECV/MVBs— a volume topologically equivalent to, but not continuous with, the cytosol — where the nucleocapsid remains hidden. Then, the nucleocapsid travels within intraluminal vesicles to reach and penetrate late endosomes. There, the back-fusion of internal vesicles with the endosome limiting membrane delivers the nucleocapsid to the cytoplasm. Our data thus argue that such back-fusion events depend on LBPA under the control of Alix and PI3P signaling via SNX16.
Entry into the multivesicular endosome: an early one-way traffic step?
Our data show that viral fusion already occurred in ECV/MVBs, before VSV reached late endosomes. Why isn’t the nucleocapsid concomitantly delivered to the cytoplasm? And why is transport to late endosomes then needed? All endosomes along the so-called degradation pathway, including ECV/MVBs 24,26,27, accumulate high amounts of internal membranes, and thus show a characteristic multivesicular appearance, which sometimes becomes multilamellar at late stages of the pathway 9. Without evoking any specific mechanism, one may envision that virions present in the endosomal lumen are more likely to interact with these internal membranes than with the limiting membrane, particularly at low (0.1 to 1.0) MOI, simply because the latter membranes are far more abundant. Alternatively, VSV bound to a putative VSV receptor, perhaps glycolipids 50,51 consistently with the recent findings that VSV infection does not depend on protein ubiquitinylation 41, may be selectively incorporated into internal membranes. In either situation, fusion would cause the release of the nucleocapsid within an internal volume, which is topologically equivalent to — but not continuous with — the cytoplasm. Nucleocapsid release into the cytoplasm and infection would then require the back-fusion of these intra-endosomal vesicles with the limiting membrane.
Evidence shows that the fate of internal vesicles and their cargo, including in particular downregulated receptors, is to be degraded in lysosomes. Both in yeast and mammalian cells, the same molecular mechanisms operate on early endosomes to sort the downregulated receptors that are destined for lysosomes into these internal membranes, and to drive the biogenesis of these internal membranes 5,6. And, intraluminal vesicles accumulate in the vacuole of yeast degradation mutants 5. Nucleocapsid delivery into the lumen of internal vesicles would then be a dead-end, leading to degradation in lysosomes, rather than infection.
Late endosome membrane dynamics in animal cells
Several lines of evidence indicate that, in late endosomes of animal cells, internal membranes also contain proteins and lipids that are not destined for the lysosomes. LBPA, which is poorly degradable, accumulates within internal membranes of late endosomes in animal cells — whether the lipid is present in yeast is not known. Moreover, MHC class II molecules are predominantly found within internal membranes of late endosomes (MIICs) in dendritic cells. Upon cell activation, these molecules are rapidly transported to the cell surface demonstrating that back-transport from late endosomes internal membranes can occur, at least in these cells, presumably via tubules 52 that may form at the expense of internal membranes via back-fusion 9. Moreover, cycling molecules of the mannose-6-phosphate receptor (MPR) in transit are found within late endosome internal membranes 8, where they accumulate in cells containing endocytosed antibodies against LBPA 11. Similarly, these antibodies inhibit cholesterol 31 and nucleocapsid export (this study) from late endosomes. Finally, anthrax toxin pore formation occurs relatively early in the endocytic pathway, but the delivery of the lethal factor to the cytoplasm requires transport to late endosomes and is inhibited by Alix downregulation 53.
We conclude that the nucleocapsid makes use of internal vesicles to reach late endosomes, from where it is efficiently released, through the back-fusion of internal vesicles with the limiting membrane (see Model, Fig S1 supplementary materials). It thus appears that, in addition to the downregulation pathway conserved from yeast to man, animal cells have evolved a more elaborate membrane system in late endosomes for more efficient re-utilization and sorting of specialized lipid and protein. VSV may have highjacked this pathway to overcome the diffusion barrier imposed by the cortical actin cytoskeleton, and reach more efficiently the perinuclear region of the cell.
Mechanisms that control nucleocapsid delivery
Clearly, the precise mechanism that leads to nucleocapsid delivery, presumably via back-fusion of endosome internal vesicles with the limiting membranes, remains to be unraveled. However, some speculations are already possible. We find that the process depends on LBPA and its effector Alix, as well as on PI3P and SNX-16. The yeast Alix homologue Bro1p/Vps31p belongs to the same Class E sub-group of VPS genes as Vps27p/Hrs and ESCRT proteins in yeast, and Alix interacts with ESCRTs during HIV budding at the surface of T helper cells. But, most of the infectious HIV produced by primary macrophages is assembled on late endocytic membranes 54, where the virus presumably uses the same machinery. These observations are consistent with our previous findings that LBPA itself may trigger the invagination process in late endosomes, and that Alix is a likely LBPA effector in this process 15.
Interestingly, we find that Alix is required for efficient nucleocapsid release, suggesting that invagination and back-fusion are coupled mechanistically via Alix. Clearly, the precise mechanism of Alix action remains to be elucidated. As do the molecular events that regulate fission and fusion within the endosomal lumen. Indeed, it is far from clear how fission and fusion can be controlled by cytosolic machineries from the opposite side of the membrane – as opposed to the role of coat proteins and SNAREs in intracellular transport. Our previously published data argue that these intraluminal fission and fusion events may depend, at least in part, on the intrinsic properties of the bilayer itself, via LBPA 15,55. LBPA-rich membranes may have a high propensity to interact spontaneously with the limiting bilayer, involving some sort of kiss-and-run fission and fusion events. However, proteins are likely to control the process, since, in particular, fission may remain frustrated if uncontrolled fusion occurs concomitantly, and vice-versa. Our previous data suggested that Alix negatively controls the invagination process by binding LBPA-rich membranes 55. Thus, a simple and naïve view is that Alix traps the membrane intermediate in fission or fusion, by interacting with LBPA as the lipid appears on the limiting bilayer, and thereby controls the rates of both vesicle formation (invagination) and consumption (fusion). Both excess Alix and the lack of Alix are likely to deregulate this balanced process, and eventually inhibit fusion. This view is attractive, because it provides a reasonable mechanistic explanation for the coupling, which must exist between invagination and back-fusion. Indeed, if uncoupled, the internal membrane pool would disappear (uncontrolled back–fusion), or the organelle collapse (uncontrolled fission).
Further support for the existence of some molecular coupling between invagination and back-fusion comes from observations that PI3P signaling seems to regulate both the invagination process, via Hrs at earlier steps of the pathway 5,39,40, and back fusion, presumably via the PI3P effector SNX16 (this study). Members of the SNX family were recently shown to contain a BAR domain 56, which was proposed to function as a sensor of membrane curvature, based on the crystal structure of the BAR-containing protein amphyphysin 57, and which may also act as a GTPase binding motive 56. Whether SNX-16 also contains a BAR domain is not known. However, one may speculate that the protein functions as a sensor or a regulator of membrane curvature at sites where invagination or back fusion occurs. It is attractive to believe that Alix and SNX16 act in tandem to integrate the invagination and back-fusion processes so that proper homeostasis of internal membranes is maintained within the endosomal lumen.
MATERIALS AND METHODS
Cells, antibodies and reagents
Baby Hamster Kidney cells (BHK) were grown and maintained as described 24, as was the production and purification of vesicular stomatitis virus (VSV Indiana serotype) 24. Recombinant Sendaï virus expressing RedFP was a gift from Laurent Roux (Geneva, Switzerland), anti-Hrs antibodies from Harald Stenmark (Oslo, Norway), MDBK cells from Ernst Peterhans (Bern, Switzerland), and HIV-1-derived vector expressing GFP and pseudotyped with VSV-G from Didier Trono (Lausanne, Switzerland). For video-microscopy, BHK cells were grown in chambered coverglass (MatTeck Corporation, Ashland, MA). The monoclonal antibodies against LBPA (6C4) 11, BHK Lamp1 (4A1) 27 and VSV-G (17.2.21.4 and P5D4) 32 have been described, as well as the polyclonal antibody against VSV-G 272/2 24. Reagents were obtained from the following sources: anti-GFP monoclonal antibody from Roche Diagnostics (Rotkreuz, Switzerland); anti-myc antibodies from Santa-Cruz Biotechnology, Inc. (San Diego, CA); anti-EEA1 monoclonal antibody from BD Transduction Laboratories (Lexington, KY); fluorescently labeled secondary antibodies from Jackson Immunoresearch Laboratories (West Grove, PA); rhodamine-dextran, BCECF-Dextran, DiL (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocabocyanine perchlorate), from Molecular Probes, Inc. (Eugene, OR); nocodazole, PMSF (phenylmethanesulfonyl fluoride), Wortmannin, Bafilomycin A1, pepstatinA, apyrase, ATP, ProteinaseK, TPCK-treated trypsin and soybean trypsin inhibitor from Sigma Chemical Co. (St. Louis, MO); FuGene6 from Roche Diagnostics; Oligofectamine from Invitrogen, (Basel, CH), Silencer siRNA labeling Kit–cy3 from Ambion, Inc. (Huntingdon, UK). Oligonucleotides and TaqMan probes were synthesized by Eurogentec SA (Seraing, BE).
Plasmids, RNA interference and transfection
The plasmid containing Hrs-myc was obtained from Harald Stenmark (Oslo, Norway). The production of GFP-2xFYVE and the purification of recombinant GST-2xFYVE and GST-2xFYVEC215S were described 42,43. The PX domains from pET32a-p40phox (Marie-Claire Dagher, Grenoble, France) was amplified by PCR and introduced into pEGFPC2 after EcoRI-SalI digestion. PGEX4T-1-Snx16 and pDmyc-SNX16 were obtained from Wajin Hong (Singapore); Snx16 cDNA was amplified by PCR and introduced into pEGFPC2 after KpnI-BamH1 digestion. Arginine 144, corresponding to arginine 58 in p40phox PX domain, was mutated to alanine (R144A) by site-directed mutagenesis. Hrs expression was silenced by RNAi 40 in HeLa cells transfected with oligofectamine, according to 58, as described 15,59, using annealed 21-nucleotide RNA duplexes with 2-nucleotide 3′-(2-deoxy)thymidine overhangs (obtained from Xeragon Inc., Huntsville, Al). BHK cells were transfected 24 h after seeding with FuGene6 using 1μg cDNA and incubated for 36h.
VSV fusion
DiL-labeled VSV 23 was prepared by incubating 200μl of 2mg/ml VSV in TNE-sucrose supplemented with 2μL of 25mM DiL in DMSO, for 2h at room temperature with gentle mixing. The unbound dye was removed by centrifugation on a step sucrose gradient; the sample in 10% sucrose was layered onto 30% and then 55% sucrose cushions in TNE buffer, and the gradient was centrifuged at 55000×g for 90 min at 4°C. The labeled virus was collected on top of the 55% sucrose cushion. Immediately before the experiments, possible viral aggregates were removed by filtration (0.22 μm pore size), and viral aggregates were never observed in our experiments (see Fig 1). This labeling procedure barely affected the virus infectious titer (Fig S5B, supplementary materials). Sub-confluent BHK cells in chambered coverglass were incubated for 15 min at 4°C with 0.3 MOI DiL-labeled VSV in MEM, pH 7.4. Then, cells were warmed up to 37°C and immediately analyzed by video microscopy, using a Leica AS MDW microscope with a CO2- and temperature-controlled stage (excitation: 549nm with a 75W Xe lamp (Leica) during 400ms). The fluorescent emission was collected by a glycerol-immersion objective with a 1.3 numerical aperture (Leica) and imaged onto a charge-coupled device (CCD) camera (Roper Scientific, CoolSnap HQM). DIC optics (Leica) was used to capture cell images before each fluorescence picture. Captured frames (exposure time, 400ms; one frame/5 min; sequence: 45 min) were processed and converted into avi movies with ImageJ software, and quantification was done manually using the ImageJ software.
Infection and replication
To monitor infection, sub-confluent BHK cells grown on glass coverslips were incubated for 1h at 4°C with VSV (0.1–1.0 MOI) in MEM, pH 7.4, and then for 3h at 37°C 24. Cells were analyzed by immunofluorescence, and infection was quantified by counting the number of cells expressing newly synthesized VSV-G-protein. When indicated, microtubules were predepolymerised with 10μM nocodazole for 2h, and the drug remained present during infection, or cells were treated with 100nM wortmannin as in 43 or with the V-ATPase inhibitors bafilomycinA1 or concanamycinB 60. Alternatively, cells were pretreated overnight at 37°C with 5 or 50μg/ml 6C4 antibody. To quantify VSV replication, cells were infected with 0.1MOI as above. A PNS was then prepared, diluted twice in PK buffer (200mM NaCl, 200mM Tris pH 9.0, 20mM EDTA, 2% SDS), and incubated with 2μg/μl proteinaseK. Total RNA was extracted with phenol-chloroform and precipitated with 2M NaCl and 100% ethanol. Then 0.5μg total RNA was used for retrotranscription with Superscript™RT (Invitrogen, Scotland, UK). To retrotranscribe the genomic VSV-G RNA, we chose the oligo (5′TTACCATTATTGGCCCGTCAAGCT3′). Then, the transcribed DNA was subjected to TaqMan PCR using two primers and a fluorescent probe: forward primer: 5′-AGG CAC AGC CAT ACA AGT CAA A-3′, reverse primer: 5′-TTT GGA AGC ATG ACA CAT CCA-3′ probe: 5′-CCG TCT GCT TGA ATA GCC TTG TGA CTC TTG-3′. The probe was modified by addition in 5′ of 6-FAM and in 3′ of TAMRA. For TaqMan real-time PCR we used the ICycler.IQ™ (Bio-Rad laboratory, PA). When indicated, BHK cells, pre-treated or not with 50μg/ml anti-LBPA antibodies, were incubated for 1h at 4°C with an HIV-1-derived vector expressing GFP and pseudotyped with VSV-G protein (3μl of a 109 pfu/ml stock for 106 cells) 33, and then for 20h at 37°C with or without the antibody. GFP expression was then analyzed by indirect immunofluorescence or SDS gel electrophoresis and Western blotting using a monoclonal anti-GFP antibody. Alternatively, cells were infected with 1 MOI (105 pfu/ml) recombinant Sendaï virus expressing RedFP for 14 hrs at 37°C.
pH measurements
Subconfluent BHK cells grown on glass coverslips and treated with 10μM nocodazole as above, were incubated with 10μg/ml BCECF-dextran (a pH-sensitive dye) for 15 min at 37°C followed by a 30 min chase to accumulate the dye in ECV/MVBs. The endosomal pH was measured by ratio fluorescence imaging 61. Briefly, the fluorescence emitted by single labeled vesicles was measured, and the signal was calibrated after nigericin addition to neutralize the pH. We used the Nipkow QLC100 Real-time confocal system (Visitron systems GmbH, Puchheim, D). Coverslips were inserted into a perfusion chamber with a controlled temperature. Calibration and image processing were described 62.
Nucleocapsid release in vitro
VSV (1μg VSV/1.3 ×107 cells) was bound for 1h at 4°C to the surface of BHK cells, which had been pretreated for 2h with 10μM nocodazole. Cells were then incubated for 45 min at 37°C in GMEM containing 10μM nocodazole, washed 3X with PBS at 37°C, and re-incubated first for 10 min in medium without drugs and then for 30 min with 100nM of the reversible V-ATPase inhibitor concanamycin B, to ensure that viral fusion, if at all, would not occur in late endosomes. ConcanamycinB was omitted during the first incubation step to limit the danger that it might interfere with transport itself 60,63. Cells were washed 3X at 4°C with PBS, and fractions containing both early and late endosomes prepared 64. In the assay, 60μl fraction was mixed with 4.5μl concentrated salt solution (0.625M Hepes pH 7.0; 75mM MgOAC2, 50mM DTT), 18μl 1M KCl, 120μg HeLa cytosol 65 and 6U apyrase or ATP regenerating system 27. After 20 min at 37°C, the mixture were loaded at the bottom of a step sucrose gradient 27, and centrifuged at 100.000×g for 1h (TLS 55 rotor). Then, the load (containing released viral RNA), and the interface (containing endosomes with non-released viral RNA) were collected. RNA was extracted from each fraction and quantified by Real Time PCR, as above. Cytosol was prepared from PNSs of untransfected or transfected HeLa cell, which had been complemented with protease inhibitors (10μM leupeptin, 1μM, aprotinin, 1μM pepstatin), by centrifugation at 100,000×g for 1h. The supernatant (cytosol) was collected, aliquoted, flash frozen and stored at −90°C.
Other methods
Protein quantification 66, gel electrophoresis 67, Western blotting 43, electron microscopy after plastic embedding 68, and immunofluorescence 69 were as described previously. For in vitro incubation of endosomes with 4nm BSA-gold, endosomal fractions (prepared as above) were incubated with highly concentrated BSA-gold (OD520 = 40) for 30 min at 4°C as described for nucleocapsid release.
Acknowledgments
We would like to thank Brigitte Viaccoz, Marie-Claire Velluz and Marie-Hélène Beuchat for expert technical assistance, Uli Laemmli and Christoph Bauer (Bioimaging platform, Frontiers in Genetics NCCR, Geneva) for help with light microscopy and imaging. We are very grateful to Kiyoshi Miwa and Didier Trono for providing us with concanamycin B, and HIV-1-derived vector pseudotyped with VSV-G, respectively. We also wish to thank Gisou van der Goot and Marc Fivaz for critical reading of the manuscript. This work was supported by the Swiss National Science Foundation (J.G.), the International Human Frontier Science Program (J.G and RGP), and the Australian National Health and Medical Research Council (RGP). ILB was supported by the award of the Bettencourt Foundation price (Prix des Jeunes Chercheurs), by the French Cancer Research Association (ARC) and by the Roche Foundation. VP was supported by the Roche Foundation and EMBO.
LIST OF REFERENCES
- 1.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee S, Maxfield FR. Role of Membrane Organization and Membrane Domains in Endocytic Lipid Trafficking. Traffic. 2000;1:203–211. doi: 10.1034/j.1600-0854.2000.010302.x. [DOI] [PubMed] [Google Scholar]
- 3.Sannerud R, Saraste J, Goud B. Retrograde traffic in the biosynthetic-secretory route: pathways and machinery. Curr Opin Cell Biol. 2003;15:438–45. doi: 10.1016/s0955-0674(03)00077-2. [DOI] [PubMed] [Google Scholar]
- 4.Gruenberg J. The endocytic pathway: a mosaic of domains. Nature Reviews Molecular Cell Biology. 2001;2:721–30. doi: 10.1038/35096054. [DOI] [PubMed] [Google Scholar]
- 5.Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 6.Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5:317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- 7.Escola JM, et al. Selective Enrichment of Tetraspan Proteins on the Internal Vesicles of Multivesicular Endosomes and on Exosomes Secreted by Human B-lymphocytes. J Biol Chem. 1998;273:20121–7. doi: 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths G, Hoflack B, Simons K, Mellman I, Kornfeld S. The mannose-6-phosphate receptor and the biogenesis of lysosomes. Cell. 1988;52:329–341. doi: 10.1016/s0092-8674(88)80026-6. [DOI] [PubMed] [Google Scholar]
- 9.Murk JL, Stoorvogel W, Kleijmeer MJ, Geuze HJ. The plasticity of multivesicular bodies and the regulation of antigen presentation. Semin Cell Dev Biol. 2002;13:303–11. doi: 10.1016/s1084952102000605. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi T, Gu F, Gruenberg J. Lipids, lipid domains and lipid-protein interactions in endocytic membrane traffic. Semin Cell Dev Biol. 1998;9:517–26. doi: 10.1006/scdb.1998.0257. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi T, et al. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392:193–7. doi: 10.1038/32440. [DOI] [PubMed] [Google Scholar]
- 12.Galve-de Rochemonteix B, et al. Interaction of anti-phospholipid antibodies with late endosomes of human endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:563–74. doi: 10.1161/01.atv.20.2.563. [DOI] [PubMed] [Google Scholar]
- 13.Incardona JP, Gruenberg J, Roelink H. Sonic hedgehog induces the segregation of patched and smoothened in endosomes. Curr Biol. 2002;12:983–95. doi: 10.1016/s0960-9822(02)00895-3. [DOI] [PubMed] [Google Scholar]
- 14.Lebrand C, et al. Late endosome motility depends on lipids via the small GTPase Rab7. Embo J. 2002;21:1289–300. doi: 10.1093/emboj/21.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuo H, et al. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science. 2004;303:531–4. doi: 10.1126/science.1092425. [DOI] [PubMed] [Google Scholar]
- 16.Strack B, Calistri A, Craig S, Popova E, Göttlinger HG. AIP1/ALIX Is a Binding Partner for HIV-1 p6 and EIAV p9 Functioning in Virus Budding. Cell. 2003;114:689–699. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 17.von Schwedler UK, et al. The Protein Network of HIV Budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 18.Whitney JA, Gomez M, Sheff D, Kreis TE, Mellman I. Cytoplasmic coat proteins involved in endosome function. Cell. 1995;83:703–713. doi: 10.1016/0092-8674(95)90183-3. [DOI] [PubMed] [Google Scholar]
- 19.Daro E, Sheff D, Gomez M, Kreis T, Mellman I. Inhibition of endosome function in CHO cells bearing a temperature-sensitive defect in the coatomer (COPI) component epsilon-COP. J Cell Biol. 1997;139:1747–59. doi: 10.1083/jcb.139.7.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simons K, Garoff H, Helenius A. How an animal virus gets into and out of its host cell. Sci Am. 1982;246:58–66. doi: 10.1038/scientificamerican0282-58. [DOI] [PubMed] [Google Scholar]
- 21.Marsh M, Bolzau E, Helenius A. Penetration of Semliki Forest virus from acidic prelysosomal vacuoles. Cell. 1983;32:931–40. doi: 10.1016/0092-8674(83)90078-8. [DOI] [PubMed] [Google Scholar]
- 22.Pak CC, Puri A, Blumenthal R. Conformational changes and fusion activity of vesicular stomatitis virus glycoprotein: [125I]iodonaphthyl azide photolabeling studies in biological membranes. Biochemistry. 1997;36:8890–6. doi: 10.1021/bi9702851. [DOI] [PubMed] [Google Scholar]
- 23.Lakadamyali M, Rust MJ, Babcock HP, Zhuang X. Visualizing infection of individual influenza viruses. Proc Natl Acad Sci U S A. 2003;100:9280–5. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gruenberg J, Griffiths G, Howell KE. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J Cell Biol. 1989;108:1301–16. doi: 10.1083/jcb.108.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffiths G, Back R, Marsh M. A quantitative analysis of the endocytic pathway in baby hamster kidney cells. J Cell Biol. 1989;109:2703–20. doi: 10.1083/jcb.109.6.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bomsel M, Parton R, Kuznetsov SA, Schroer TA, Gruenberg J. Microtubule and motor dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell. 1990;62:719–731. doi: 10.1016/0092-8674(90)90117-w. [DOI] [PubMed] [Google Scholar]
- 27.Aniento F, Emans N, Griffiths G, Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol. 1993;123:1373–87. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gruenberg J, Maxfield F. Membrane transport in the endocytic pathway. Curr Op Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- 29.Pesonen M, Ansorge W, Simons K. Transcytosis of the G protein of vesicular stomatitis virus after implantation into the apical plasma membrane of Madin-Darby canine kidney cells. I. Involvement of endosomes and lysosomes. J Cell Biol. 1984;99:796–82. doi: 10.1083/jcb.99.3.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsh M, Bron R. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J Cell Sci. 1997;110 ( Pt 1):95–103. doi: 10.1242/jcs.110.1.95. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi T, et al. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat Cell Biol. 1999;1:113–8. doi: 10.1038/10084. [DOI] [PubMed] [Google Scholar]
- 32.Gruenberg J, Howell KE. Immuno-isolation of vesicles using antigenic sites either located on the cytoplasmic or the exoplasmic domain of an implanted viral protein. A quantitative analysis. Eur J Cell Biol. 1985;38:312–21. [PubMed] [Google Scholar]
- 33.Naldini L, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 34.Simonsen A, Wurmser AE, Emr SD, Stenmark H. The role of phosphoinositides in membrane transport. Curr Opin Cell Biol. 2001;13:485–92. doi: 10.1016/s0955-0674(00)00240-4. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Borja M, et al. Multivesicular body morphogenesis requires phosphatidyl-inositol 3-kinase activity. Curr Biol. 1999;9:55–8. doi: 10.1016/s0960-9822(99)80048-7. [DOI] [PubMed] [Google Scholar]
- 36.Reaves BJ, Bright NA, Mullock BM, Luzio JP. The effect of wortmannin on the localisation of lysosomal type I integral membrane glycoproteins suggests a role for phosphoinositide 3-kinase activity in regulating membrane traffic late in the endocytic pathway. J Cell Sci. 1996;109:749–762. doi: 10.1242/jcs.109.4.749. [DOI] [PubMed] [Google Scholar]
- 37.Futter CE, Collinson LM, Backer JM, Hopkins CR. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J Cell Biol. 2001;155:1251–1264. doi: 10.1083/jcb.200108152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tooze J, Hollinshead M. Tubular early endosomal networks in AtT20 and other cells. J Cell Biol. 1991;115:635–53. doi: 10.1083/jcb.115.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lloyd TE, et al. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell. 2002;108:261–9. doi: 10.1016/s0092-8674(02)00611-6. [DOI] [PubMed] [Google Scholar]
- 40.Bache KG, Brech A, Mehlum A, Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol. 2003;162:435–42. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khor R, McElroy LJ, Whittaker GR. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic. 2003;4:857–68. doi: 10.1046/j.1398-9219.2003.0140.x. [DOI] [PubMed] [Google Scholar]
- 42.Gillooly DJ, et al. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. Embo J. 2000;19:4577–88. doi: 10.1093/emboj/19.17.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petiot A, Faure J, Stenmark H, Gruenberg J. PI3P signaling regulates receptor sorting but not transport in the endosomal pathway. J Cell Biol. 2003;162:971–9. doi: 10.1083/jcb.200303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sbrissa D, Ikonomov OC, Shisheva A. Phosphatidylinositol 3-phosphate-interacting domains in PIKfyve. Binding specificity and role in PIKfyve. Endomenbrane localization. J Biol Chem. 2002;277:6073–9. doi: 10.1074/jbc.M110194200. [DOI] [PubMed] [Google Scholar]
- 46.Haft CR, de la Luz Sierra M, Barr VA, Haft DH, Taylor SI. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol Cell Biol. 1998;18:7278–87. doi: 10.1128/mcb.18.12.7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Worby CA, Dixon JE. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol. 2002;3:919–931. doi: 10.1038/nrm974. [DOI] [PubMed] [Google Scholar]
- 48.Hanson BJ, Hong W. Evidence for a role of SNX16 in regulating traffic between the early and later endosomal compartments. J Biol Chem. 2003;278:34617–30. doi: 10.1074/jbc.M300143200. [DOI] [PubMed] [Google Scholar]
- 49.Choi JH, et al. Sorting nexin 16 regulates EGF receptor trafficking by phosphatidylinositol-3-phosphate interaction with the Phox domain. J Cell Sci. 2004;117:4209–18. doi: 10.1242/jcs.01233. [DOI] [PubMed] [Google Scholar]
- 50.Marsh M, Helenius A. Virus entry into animal cells. Adv Virus Res. 1989;36:107–51. doi: 10.1016/S0065-3527(08)60583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conti C, Mastromarino P, Orsi N. Role of membrane phospholipids and glycolipids in cell-to-cell fusion by VSV. Comp Immunol Microbiol Infect Dis. 1991;14:303–13. doi: 10.1016/0147-9571(91)90136-2. [DOI] [PubMed] [Google Scholar]
- 52.Chow A, Toomre D, Garrett W, Mellman I. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature. 2002;418:988–94. doi: 10.1038/nature01006. [DOI] [PubMed] [Google Scholar]
- 53.Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J Cell Biol. 2004 doi: 10.1083/jcb.200312072. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pelchen-Matthews A, Kramer B, Marsh M. Infectious HIV-1 assembles in late endosomes in primary macrophages. J Cell Biol. 2003;162:443–55. doi: 10.1083/jcb.200304008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kobayashi T, et al. Separation and characterization of late endosomal membrane domains. J Biol Chem. 2002;277:32157–64. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 56.Habermann B. The BAR-domain family of proteins: a case of bending and binding? EMBO Rep. 2004;5:250–5. doi: 10.1038/sj.embor.7400105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peter BJ, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–9. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 58.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 59.Mayran N, Parton RG, Gruenberg J. Annexin II regulates multivesicular endosome biogenesis in the degradation pathway of animal cells. Embo J. 2003;22:3242–53. doi: 10.1093/emboj/cdg321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clague MJ, Urbe S, Aniento F, Gruenberg J. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J Biol Chem. 1994;269:21–4. [PubMed] [Google Scholar]
- 61.Piguet V, et al. Nef-induced CD4 degradation: a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell. 1999;97:63–73. doi: 10.1016/s0092-8674(00)80715-1. [DOI] [PubMed] [Google Scholar]
- 62.Demaurex N, Furuya W, D’Souza S, Bonifacino JS, Grinstein S. Mechanism of acidification of the trans-Golgi network (TGN). In situ measurements of pH using retrieval of TGN38 and furin from the cell surface. J Biol Chem. 1998;273:2044–51. doi: 10.1074/jbc.273.4.2044. [DOI] [PubMed] [Google Scholar]
- 63.Aniento F, Gu F, Parton RG, Gruenberg J. An endosomal beta COP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol. 1996;133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huber LA, et al. Both calmodulin and the unconventional myosin Myr4 regulate membrane trafficking along the recycling pathway of MDCK cells. Traffic. 2000;1:494–503. doi: 10.1034/j.1600-0854.2000.010607.x. [DOI] [PubMed] [Google Scholar]
- 65.Cavalli V, et al. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol Cell. 2001;7:421–32. doi: 10.1016/s1097-2765(01)00189-7. [DOI] [PubMed] [Google Scholar]
- 66.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 67.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 68.Parton RG, Schrotz P, Bucci C, Gruenberg J. Plasticity of early endosomes. J Cell Sci. 1992;103:335–348. doi: 10.1242/jcs.103.2.335. [DOI] [PubMed] [Google Scholar]
- 69.Rojo M, et al. Involvement of the transmembrane protein p23 in biosynthetic protein transport. J Cell Biol. 1997;139:1119–35. doi: 10.1083/jcb.139.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]