

Abstract
α1-antitrypsin (A1AT) deficiency is an autosomal co-dominant disease that can cause chronic liver disease, cirrhosis, and hepatocellular carcinoma (HCC) in children and adults and increases risk for emphysema in adults. The development of symptomatic disease varies—some patients have life-threatening symptoms in childhood whereas others remain asymptomatic and healthy into old age. As a result of this variability, patients present across multiple disciplines, including pediatrics, adult medicine, hepatology, genetics, and pulmonology. This can give physicians the mistaken impression that the condition is less common than it actually is, and can lead to fragmented care that omits critical interventions commonly performed other specialists. We sought to present a rational approach for hepatologists to manage adult patients with A1AT deficiency.
Keywords: fibrosis, hepatocellular center, autophagy, therapy, diagnosis
Introduction
The most common form of alpha-1 antitrypsin (A1AT) deficiency occurs in individuals who carry a homozygous variant of the A1AT gene (SERPINA1) that causes a Glu342Lys substitution in the gene product, also called ZZ or PIZZ in World Health Organization nomenclature. Approximately 2% of the US population is heterozygous for the Z allele and .05% are homozygous (ZZ). The most common allele of the A1AT gene, called M, produces normal levels of A1AT. A1AT is highly expressed by hepatocytes and the protein is secreted into the blood; it inhibits neutrophil proteases to protect host tissues from non-specific injury associated with episodes of inflammation. The product of ZZ folds abnormally to form homopolymers and insoluble aggregates within the endoplasmic reticulum of hepatocytes, instead of being secreted. The aggregates can lead to hepatocyte cell death and liver inflammation, regeneration, and progressive fibrosis.
Signs or symptoms of liver dysfunction occur in as many as 50% of ZZ children (although many are undiagnosed), whereas cirrhosis and life-threatening disease only occur in about 5% before the age of 18. The risk of cirrhosis appears to increase with age in adults. The peak ages for the incidence of liver and lung disease differ; liver disorders usually occur during childhood and old age and lung disease develops in middle age. Clinicians therefore have the impression that involvement of 1 organ excludes the other, although there is good evidence that the risks of lung and liver disease are independent. Individuals with 1 M and 1 Z allele (MZ) are generally healthy, but appear to be more susceptible to liver injury; they are typically overrepresented among cirrhotic patients in adult liver clinics. The S variant of A1AT, which causes the Glu288Val substitution, was named for the slow migration of the protein in starch gel electrophoresis. Carriers of 1 allele of S and 1 of Z (SZ) are common, but generally develop only lung or liver disease.
A range of advocacy and research activities are available to patients with this disease and their families. The NIH-sponsored Children’s Liver Disease Research and Education Network is a study that is enrolling patients up 25 years old to collect data on the natural history and genetic and environmental modifers of the disease. Patients of all ages and disease manifestations can participate in the Alpha-1 Registry, a self-report database based at the Medical University of South Carolina that provides a contact point for many kinds of research studies. Several nationwide organizations, including The Alpha-1 Foundation, The Alpha-1 Association, and AlphaNet encourage advocacy, supply educational materials, support research, and provide medical and pharmacy services to this patient community. Increasing the interaction among hepatologists, other medical disciplines, and these community resources will improve the care of patients with this disease.
Liver Disease in Pediatric Patients
Although most cases of A1AT deficiency are associated with a single variant (such as ZZ), the clinical presentation of liver disease varies greatly (1). Some children develop cholestatic jaundice and hepatitis shortly after birth, called neonatal hepatitis syndrome; it is indistinguishable from the early phases of extrahepatic biliary atresia, cystic fibrosis, congenital infection, and many other conditions. Because A1AT deficiency is relatively more common than the other disorders, serum testing for A1AT deficiency is performed early in the evaluation of infants with cholestatic jaundice. This approach can avoid more invasive tests, such as the cholangiogram performed by open laparotomy, which is usually required to diagnose biliary atresia. However, many infants with ZZ-associated A1AT deficiency do not develop cholestasis, for unknown reasons. Some children present later in infancy or childhood with unexplained increases in levels of amino transferases, hepatomegaly, or, on rare occasions, cirrhosis or acute liver failure. These patients are usually tested for A1AT deficiency during early stages of evaluation. In rare cases, infants with A1AT deficiency do not eat or grow well, classified as failure to thrive.
The emphysema associated with A1AT deficiency takes decades to develop, so it is not observed in children. However, children with A1AT deficiency have an increased risk for developing asthma. Pulmonary evaluation of a Swedish birth cohort did not identify any respiratory disorders other than asthma among 18 year-olds with A1AT deficiency; they had normal results from pulmonary function tests, except for mild changes among smokers. At least 50% of children with A1AT deficiency are healthy throughout childhood and remain undiagnosed. Newborns are not screened for A1AT deficiency in North America.
Liver Disease in Adult Patients
Most adult patients with A1AT deficiency have fairly normal results from liver tests and minimal or no symptoms of liver disease. They can present with asymptomatic, abnormal levels of liver enzymes, clinical manifestations of advanced cirrhosis, or hepatocellular carcinoma (HCC) (2); liver disease appears to be the next most common manifestation of A1AT deficiency, after lung disease (3). The best information for the effects of A1AT deficiency on liver disease comes from an analysis of autopsy data from Sweden, in which 43% of patients with the disease had developed cirrhosis and 28% had developed HCC (4). A1AT deficiency is therefore a significant risk factor for the development of cirrhosis in adults. However, there is limited information on many important factors—it is not clear how many adults with A1AT deficiency have liver disease, what the best markers of liver disease are in these patients, how liver disease progresses, or what factors contribute to or prevent it.
The reported prevalence of cirrhosis in ZZ adults varies; estimates range from 2%–43% (3). The only longitudinal study ever conducted was performed in Sweden and followed individuals identified at birth with A1AT deficiency (ZZ) into young adulthood (5). Within the first 6 months of life, more than 50% of the children had either clinical signs of liver disease or abnormal results from liver function tests. Remarkably, the abnormal results from liver function tests decreased with time; by the time patients were 26 years old, 5.7% of those tested had minor abnormalities in their level of alanine aminotransferase (ALT). A follow-up report analyzed the prevalence of liver test abnormalities in A1AT-deficient individuals at age 30 (6). Blood samples were obtained from 89 ZZ, 40 SZ, and 84 MM (control) individuals. Although the mean values of all liver enzymes were within the normal range for all groups, the mean level of aspartate aminotransferase (AST) was higher in the ZZ and SZ groups, and the mean level of ALT was higher in the ZZ group, compared with controls. Interestingly, women with the ZZ alleles who took oral contraceptives had higher mean levels of AST and ALT than women in the control group who took oral contraceptives. The authors were unable to identify any factors in children that predicted adult defects in liver function, such as liver disease or increased levels of liver enzymes. From these results, some investigators have concluded that liver disease is not a significant problem for adults with the ZZ alleles of A1AT. However, this conclusion is that it is based on the premise that abnormal serum levels of liver enzymes accurately diagnose liver disease, but this cohort has not been followed into late adulthood. Levels are liver enzymes can be normal in individuals with liver disease, including cirrhosis.
A recent analysis of 647 ZZ individuals in the US found that they have ongoing, subclinical liver injuries that escape detection by routine liver tests (7). The mean level of AST was 43 IU/L (range 7–325 IU/L), which is just above the reported upper limit of normal (0–40 IU/L). The mean level of ALT for men (33 IU/L, range 7–110 IU/L) and women (24 IU/L, range 6–301 IU/L) fell to within normal limits, and the overall prevalence of abnormal levels of ALT was only 7.7%. However, if a stricter cutoff for the normal level of ALT was used (≤19 IU/L for women and ≤30 IU/L for men, the prevalence of an abnormal level of ALT in the population is 49%. Despite this, only 7.9% of the entire cohort reported any history of liver disease. These findings indicate that a significant proportion of ZZ individuals have small increases in levels of ALT, in the presence or absence of clinically observable liver disease. Our lack of knowledge about the amounts of liver injury and stage of disease in these patients poses an obstacle to treating, modifying risk factors, and screening these patients for HCC.
Pathophysiology
Strategies to degrade the aggregates of A1AT in heptocytes might be used to reduce or prevent liver disease. Soluble monomers of A1AT in the endoplasmic reticulum could be degraded by proteasomes, or polymerized aggregates could be degraded by autophagosomes (see Figure 1).
Figure 1.
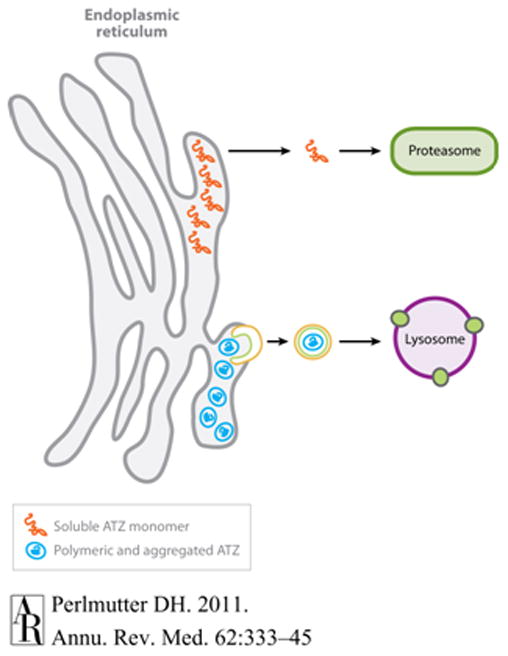
Pathway for the degradation of PIZZ that accumulates in the endoplasmic reticulum. (Reproduced with Permission from Perlmutter DH, Annu. Rev. Med. 2011. 62:4.1–4.13).
The genetic and environmental factors that predispose some patients with A1AT deficiency to liver disease are unknown. A post-mortem study in Sweden reported that adults with ZZ-associated A1AT deficiency who died from causes unrelated to this disease often had asymptomatic cirrhosis that increased with age (8). A1AT-deficient individuals who were more than 50 years old and never smoked had the greatest risk for cirrhosis and a significant risk for HCC (9). Men with A1AT deficiency who were 51–60 years old had an increased risk of developing liver disease, compared to other patients with A1AT deficiency (10).
A study that investigated the relationship between A1AT variants and manifestation of liver disease found a significant degree of discordance between sibling pairs in the severity of liver disease (11). Progression of liver damage appears to vary and involve environmental and genetic factors, beyond A1AT genotype. Single nucleotide polymorphisms in genes that regulate protein degradation pathways have been associated with some types of liver disease, but other factors have not been identified (12, 13). In a study of primary cell lines from ZZ patients with and without liver disease, the cells from the patients with liver disease had reduced intracellular degradation of the Z protein. Genetic differences that affect protein degradation pathways (such as the proteasome or autophagic processes) might determine which individuals develop liver disease (14, 15)
Many different factors, including age, sex, alcohol consumption, and obesity have been reported to affect risk for liver disease (16). The nonsteroidal anti-inflammatory drug (NSAID) indomethacin increases the level of liver damage in transgenic mice that express the Z form of a1AT (Za1AT). Therefore, environmental factors such as medications could potentiate the liver injury associated with hepatic accumulation of the Z protein. NSAIDs might be especially injurious to patients with A1AT deficiency, increasing the expression and accumulation of the hepatotoxic form of the protein (17). The few studies to identify risk factors for liver disease in adults have relied on patient self reporting of liver disease, rather than histologic analysis. These studies did not verify the absence of other common causes of liver disease and had inconsistent reporting of the degree of liver injury, so it was difficult to make conclusions about risk factors. Although ZZ-associated liver disease is likely under-diagnosed in adults, it has been increasingly recognized in patients with cryptogenic cirrhosis, alcoholic liver disease, and HCC. A1AT deficiency might therefore increase the severity of liver disorders such as hepatitis B and C, autoimmune hepatitis, or hemachromatosis (18).
Heterozygous Variants of A1AT.
The affects of heterozygous variants of A1AT on risk for liver disease are controversial. Several studies have reported that there is a higher prevalence the MZ genotype among patients with cirrhosis, compared with control populations or patients with non-cirrhotic liver diseases (19–23), and also among patients with crytogenic cirrhosis (19–22). However, a more recent study did not associate the MZ genotype with chronic liver disease overall, although a significantly greater number of patients with severe liver disease were MZ, compared to those with less severe liver disease (24). The MZ genotype was also associated with increased severity of liver disease and the need for transplantation among patients with hepatitis C virus infection (24).
Liver cells of healthy individuals with A1AT MZ do not contain periodic acid Schiff-positive globules, but livers of those with fibrosis do, along with diastase-resistant inclusions of A1AT, which vary from sheets of hepatocytes to scattered periportal fibroblasts. The Z form of A1AT protein can be identified by immunohistochemical analysis (23). Although the MZ alleles have been associated with cirrhosis, MS and SS have not been associated with liver disease (22).
Rationale and Options for A1AT testing
Despite its relatively high prevalence, A1AT deficiency is under-recognized and under-diagnosed (25). Although it is considered to be a rare condition, there are approximately 20 million individuals in the United States who carry at least 1 allele of A1AT associated with the disease, and the prevalence of homozygosity for variants associated with A1AT deficiency is at least 100,000 in the US (26). An estimated fewer than 10% of people with this disorder have been properly diagnosed, with an average 6-year delay in diagnosis from the time that symptoms are apparent (27, 28). It is therefore important to test all patients with liver disease of unexplained etiology, and those with a family history of liver disease, for A1AT deficiency. Other populations that should be tested include those with a diagnosis of chronic obstructive pulmonary disease, adults with asthma that does not respond to maximal medical therapy, and all family members of those diagnosed with A1AT deficiency.
A1AT deficiency can be diagnosed by measuring serum or plasma levels of A1AT, phenotype analysis of the protein, or genotype analysis. Plasma levels of A1AT are usually measured by rocket immunoelectropheresis, radial immunodiffusion, or nephelometry. The assay to measure the concentration of A1AT in plasma is a reasonable initial test; abnormal or borderline results indicate the need for genetic testing (29). A blood level of A1AT less than 50% of the lower limit of normal provides clear evidence for A1AT deficiency. However, because A1AT is an acute-phase reactant (its synthesis and release from hepatocytes are increased by systemic inflammation), levels can increase under certain conditions, making it a challenge to always detect its deficiency (30). Despite this, cells from ZZ individuals rarely produce enough A1AT to obscure a deficiency. Therefore, even though concentration assays are routinely used for the initial step in screening, they should not be used to exclude A1AT deficiency, because they have low levels of sensitivity and specificity (31). A1AT proteins are characterized by phenotype analysis, using isoelectric focusing, which separates proteins based on their isoelectric point (32). Phenotype analysis alone cannot distinguish an individual that is homozygous for alleles that produce an altered protein from individuals that carry 1 allele that produces and altered protein and 1 allele that expresses no protein. Results from phenotyping assays are subjective and vary, so we do not recommend their use in diagnosis of patients with liver disease.
Genotype analysis can be used to make a definitive diagnosis, and determine specific phenotypic variations. Genotyping uses allele-specific amplification that allows the variants to be identified, such as S or Z, and also identify less-common variants that affect the structure or expression level of the gene product. Although the optimal algorithm for laboratory testing for A1AT deficiency has not been defined, a combination of measuring serum levels of A1AT and genotype analysis seems to be the most useful.
Management
Because A1AT deficiency is an inherited disease, family members are at risk and should be considered for testing. Siblings of individuals with A1AT deficiency are the only family members for whom genetic testing is recommended, because they have 25% or greater risk of also having the condition (33). Although the risk of A1AT deficiency in parents, offspring, and second-degree relatives of patients is low, but greater than that of the general population, it is common practice to recommend that they be referred to healthcare professional to discuss the benefits and risks of A1AT testing.
Although studies have not demonstrated that increased detection of A1AT deficiency improves health outcomes, once an individual with A1AT deficiency and liver disease is identified, specific treatments and preventive measures can be implemented. Educating newly diagnosed patients about A1AT deficiency, disease mechanisms, and risk can help them obtain appropriate treatment. After the diagnosis of A1AT deficiency is confirmed, patients should be referred to specialists in lung and liver disease with expertise in managing their disease. Patients should first be assessed for liver and pulmonary function (spirometry), then referred to a pulmonologist for respiratory monitoring and management—even when they have normal pulmonary function. Since cigarette smoking promotes the development of lung disease in patients with A1AT deficiency, smoking cessation should be recommended or smoking initiation discouraged. A liver biopsy is not typically required to diagnose A1AT-related liver disease, but can be used to rule out other causes of liver disease and to assess disease severity. Histologic analysis of liver samples from patients with A1AT deficiency usually reveal accumulation of A1AT (as polymers) within hepatocytes and intracytoplasmic inclusion bodies in periportal hepatocytes (Figure 2).
Figure 2.
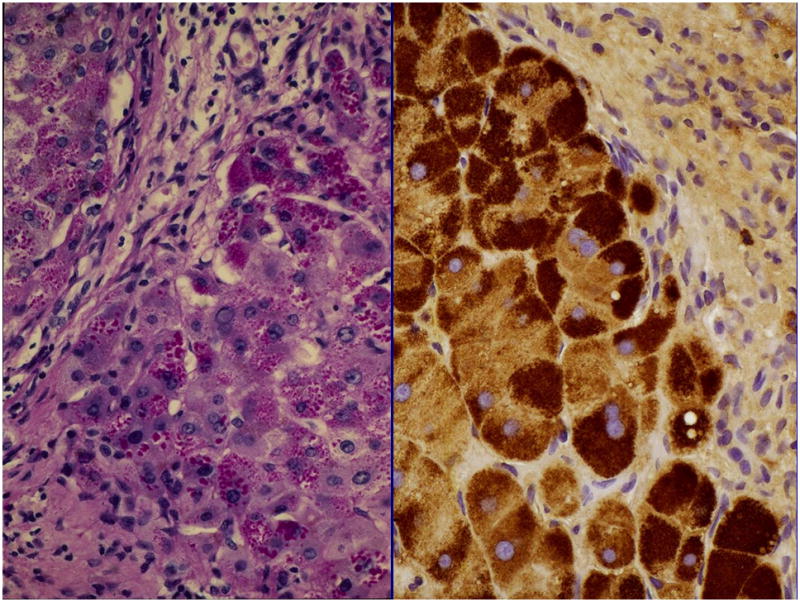
Liver from patient with PIZZ alpha 1 antitrypsin deficiency. (Left) Periodic acid Schiff-positive diastase resistant globules. (Right) Immunohistochemistry for alpha 1 antitrypsin identifies globules.
An assessment for advanced liver disease (ultrasound or other type of imaging analysis) is also recommended, to identify portal hypertension and cancer. Patients with cirrhosis or portal hypertension from A1AT deficiency are managed the same as those with liver disease from any other cause: using cancer surveillance, tests for and management of esophageal varices, and disease-modification strategies (limiting alcohol consumption, avoiding exposure to hepatotoxic agents, and controlling obesity or weight management, etc). Vaccination against hepatitis A and B viruses is recommended for all A1AT-deficient patients, who might have an increased risk for chronic liver disease if they become infected (34).
Liver injury from A1AT deficiency is believed to result from hepatic accumulation of polymerized A1AT Z protein (35). Although no therapies have been approved to reduce this accumulation or prevent the development of liver disease (other than transplantation), there are a number of new therapeutic and preventative strategies. Augmentation therapy can increase serum levels of A1AT to levels that reduce pulmonary disease, although this strategy is not likely to affect liver disease. In a positive feedback loop, α-elastase complexes bind to the serpin-enzyme complex receptor, which upregulates A1AT synthesis. Therefore, augmentation therapy might exacerbate disease in ZZ individuals.
Research to develop therapeutics for A1AT-associated liver disease has therefore focused on preventing protein polymerization, increasing excretion of the Z protein, increasing the intracellular degradation of the Z protein; nonspecific approaches are also being developed to reduce liver inflammation and fibrosis. The most promising strategy for treating liver disease in patients with A1AT deficiency is to decrease the accumulation of polymerized A1AT by stimulating its autophagy in hepatocytes (36, 37). Carbamazepine (38) and rapamycin (39) stimulated autophagy and reduced A1AT (Z-form)-associated liver fibrosis in transgenic mice.
Liver Transplantation
For patients who develop decompensated cirrhosis or early-stage HCC, liver transplantation replaces the diseased liver and corrects the underlying metabolic disorder. Rates of survival of grafts and patients with this metabolic disease are similar to those of patients with other indications who receive liver transplants. Rates of 1-, 3-, and 5-year survival after liver transplantation were reported to be 89%, 85%, and 83% among adults with A1AT deficiency (40). Interestingly, livers of transplant recipients maintain the donor’s phenotype and recipients maintain normal serum levels of A1AT (40, 41). Preliminary results indicate that liver transplantation also prevents or slows the progression of pulmonary disease in patients with A1AT deficiency (42). There are many uncertainties about development of liver disease in adults with A1AT deficiency and challenges to treating it. Large studies are needed to determine the prevalence, natural history, and best treatment options.
HCC
Individuals with the A1AT deficiency caused by the Z allele who develop cirrhosis are at great risk for HCC (43)—especially middle-age or older men, although rare cases have been reported in children (44). However the risk has been difficult to quantify and is often over-shadowed or even confounded by the association with chronic viral hepatitis (45).
A retrospective, case-control, autopsy study conducted in Sweden reported an odds ratio of 20 for the development of HCC among individuals with A1AT deficiency—comparable with the risk from genetic hemochromatosis (43). However, a subsequent study by the same authors (4) calculated an odds ratio of approximately 0.5 (16.1% of ZZ individuals were found to have HCC in autopsies). Among a cases series of 19 adult patients with A1AT deficiency-associated liver disease, 2 had HCC (10.5%, both ZZ individuals) (46); 2 of the 8 ZZ individuals (25%) in this case series had HCC (46). Interestingly, in a case series of 21 children undergoing liver transplantation for A1AT deficiency, none had HCC, supporting the concept that risk increases with age (47). All reports seemed to confirm the association with cirrhosis—all cases with HCC (even children) had cirrhosis.
The risk for HCC in individuals with 1 Z allele is less well defined than for homozygous individuals; heterozygosity for the Z allele might modify risk or increase it in patients with other causes of liver diseases, such as chronic hepatitis C (4). However, at least 1 Z allele is thought to be necessary for HCC to develop; the combination of the MZ, SZ, and even the rare ZMmalton (Mmalton causes a deletion at Phe52 of A1AT)alleles (48, 49) have been associated with HCC in individuals with other forms of liver disease.
Pathogenesis of HCC
The process by which A1AT deficiency results in HCC is not clear. Intracellular accumulation of the Z protein in hepatocytes causes cell damage that can lead to a condition that resembles chronic hepatitis and then cirrhosis. Cirrhosis is a risk factor for HCC, at a rate of approximately 1%–4% per year (50). Studies have indicated that hepatocytes with accumulated Z protein are cancer-prone, surviving with intrinsic damage and chronically stimulating, in trans, adjacent, relatively undamaged hepatocytes, which then begin to proliferate. Interestingly, HCC cells only rarely contain globules of Z protein, which are usually abundant in surrounding, non-tumor tissues (51). Also, large-cell dysplasia is often found in A1AT-deficient livers. This dysplasia is thought to increase the risk for HCC, although it does not seem to be an intermediate step in the pathway to malignancy (52). Transgenic expression of the Z protein in livers of mice causes accumulation of globules in a pattern similar to that of humans with A1AT deficiency, making this a mouse model of the disease. By 16 to 19 months of age, nearly 70% of these mice develop tumors (53). Z protein accumulation was found to alter the regulation of several genes involved in cell proliferation and tumorigenesis.
Recommendations for Screening
The American Association for the Study of Liver Disease recommends that patients with cirrhosis from A1AT deficiency be screened for HCC. This practice guideline bases its recommendations for surveillance on estimates of the risk for HCC. For groups of patients where the threshold incidence for efficacy of screening is estimated to be >1.5% per year, surveillance is recommended. For patients with A1AT deficiency, it is acknowledged that the exact incidence of HCC is unknown, but is thought to be >1.5% per year (50). Thus, this practice guidelines call for regular screening that includes ultrasound examination of the liver every 6 months. If abnormalities or new defects are identified, they can be further evaluated with dynamic imaging studies or even liver biopsy analysis. There does not appear to be a basis for recommending routine surveillance for patients with A1AT deficiency who do not have significant liver disease or cirrhosis.
It is clear that patients with liver disease associated with A1AT deficiency are at risk for HCC. This risk needs to be more accurately quantified, as do the roles of other associated risk factors. Those with cirrhosis need to be entered into routine surveillance programs for HCC, to identify tumors at an early stage, when they are most treatable.
Patient Education
A range of advocacy and research activities are available to patients with this disease and to their families. The NIH sponsored Children’s Liver Disease Research and Education Network is a study enrolling patients who are up to 25 years old in a prospective cohort study, to gather data on natural history and to investigate genetic and environmental modifers of disease. Patients can also be offered contact with the Alpha-1 Registry, a self-report database for all ages and disease manifestations, based at the Univesity of South Carolina; it provides a contact point for many kinds of research studies. Several nation wide organizations, The Alpha-1 Foundation, The Alpha-1 Association, and AlphaNet encourage advocacy, supply educational materials, support research, and provide medical and pharmacy services to this patient community. Increased interactation among hepatologists, other specialists, and these organizations will improve the care of patients with this disease.
Acknowledgments
Grant Support: This study was supported by grants from the National Institute of Health (NIH RO1 DK090962-01, NIH RO1 DK072237-06, NIH RO1 GM041804-24, NIH P50 AA011999-13) from David A. Brenner.
Abbreviations
- a1AT
Alpha-1-antitrypsin
- HCC
hepatocelluar carcinoma
- NSAID
nonsteroidal anti-inflammatory drug
Footnotes
No conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blank CA, Brantly M. Clinical features and molecular characteristics of alpha 1-antitrypsin deficiency. Ann Allergy. 1994;72:105–120. quiz 120–102. [PubMed] [Google Scholar]
- 2.Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N Engl J Med. 1986;314:736–739. doi: 10.1056/NEJM198603203141202. [DOI] [PubMed] [Google Scholar]
- 3.American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168:818–900. doi: 10.1164/rccm.168.7.818. [DOI] [PubMed] [Google Scholar]
- 4.Elzouki AN, Eriksson S. Risk of hepatobiliary disease in adults with severe alpha 1-antitrypsin deficiency (PiZZ): is chronic viral hepatitis B or C an additional risk factor for cirrhosis and hepatocellular carcinoma? Eur J Gastroenterol Hepatol. 1996;8:989–994. doi: 10.1097/00042737-199610000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294:1316–1321. doi: 10.1056/NEJM197606102942404. [DOI] [PubMed] [Google Scholar]
- 6.Bernspang E, Carlson J, Piitulainen E. The liver in 30-year-old individuals with alpha(1)-antitrypsin deficiency. Scand J Gastroenterol. 2009;44:1349–1355. doi: 10.3109/00365520903296669. [DOI] [PubMed] [Google Scholar]
- 7.Clark V, Brantly M, Dhanasekaran R, Schreck P, Rouhani F, Nelson D. ALT abnormalities in adults with alpha-1 antitrypsin deficiency. Hepatology. 2011 doi: 10.1016/j.cgh.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eriksson S. Alpha 1-antitrypsin deficiency and liver cirrhosis in adults. An analysis of 35 Swedish autopsied cases. Acta Med Scand. 1987;221:461–467. [PubMed] [Google Scholar]
- 9.Larsson C. Natural history and life expectancy in severe alpha1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204:345–351. doi: 10.1111/j.0954-6820.1978.tb08452.x. [DOI] [PubMed] [Google Scholar]
- 10.Cox DW, Smyth S. Risk for liver disease in adults with alpha 1-antitrypsin deficiency. Am J Med. 1983;74:221–227. doi: 10.1016/0002-9343(83)90615-0. [DOI] [PubMed] [Google Scholar]
- 11.Hinds R, Hadchouel A, Shanmugham NP, Al-Hussaini A, Chambers S, Cheeseman P, Mieli-Vergani G, et al. Variable degree of liver involvement in siblings with PiZZ alpha-1-antitrypsin deficiency-related liver disease. J Pediatr Gastroenterol Nutr. 2006;43:136–138. doi: 10.1097/01.mpg.0000226370.09085.39. [DOI] [PubMed] [Google Scholar]
- 12.Pan S, Huang L, McPherson J, Muzny D, Rouhani F, Brantly M, Gibbs R, et al. Single nucleotide polymorphism-mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in alpha1-antitrypsin deficiency. Hepatology. 2009;50:275–281. doi: 10.1002/hep.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chappell S, Hadzic N, Stockley R, Guetta-Baranes T, Morgan K, Kalsheker N. A polymorphism of the alpha1-antitrypsin gene represents a risk factor for liver disease. Hepatology. 2008;47:127–132. doi: 10.1002/hep.21979. [DOI] [PubMed] [Google Scholar]
- 14.Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter DH. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci U S A. 1994;91:9014–9018. doi: 10.1073/pnas.91.19.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teckman JH, Perlmutter DH. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J Biol Chem. 1996;271:13215–13220. doi: 10.1074/jbc.271.22.13215. [DOI] [PubMed] [Google Scholar]
- 16.Bowlus CL, Willner I, Zern MA, Reuben A, Chen P, Holladay B, Xie L, et al. Factors associated with advanced liver disease in adults with alpha1-antitrypsin deficiency. Clin Gastroenterol Hepatol. 2005;3:390–396. doi: 10.1016/s1542-3565(05)00082-0. [DOI] [PubMed] [Google Scholar]
- 17.Rudnick DA, Shikapwashya O, Blomenkamp K, Teckman JH. Indomethacin increases liver damage in a murine model of liver injury from alpha-1-antitrypsin deficiency. Hepatology. 2006;44:976–982. doi: 10.1002/hep.21326. [DOI] [PubMed] [Google Scholar]
- 18.Stolk J, Seersholm N, Kalsheker N. Alpha1-antitrypsin deficiency: current perspective on research, diagnosis, and management. Int J Chron Obstruct Pulmon Dis. 2006;1:151–160. doi: 10.2147/copd.2006.1.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodges JR, Millward-Sadler GH, Barbatis C, Wright R. Heterozygous MZ alpha 1-antitrypsin deficiency in adults with chronic active hepatitis and cryptogenic cirrhosis. N Engl J Med. 1981;304:557–560. doi: 10.1056/NEJM198103053041001. [DOI] [PubMed] [Google Scholar]
- 20.Carlson J, Eriksson S. Chronic 'cryptogenic' liver disease and malignant hepatoma in intermediate alpha 1-antitrypsin deficiency identified by a Pi Z-specific monoclonal antibody. Scand J Gastroenterol. 1985;20:835–842. doi: 10.3109/00365528509088831. [DOI] [PubMed] [Google Scholar]
- 21.Bell H, Schrumpf E, Fagerhol MK. Heterozygous MZ alpha-1-antitrypsin deficiency in adults with chronic liver disease. Scand J Gastroenterol. 1990;25:788–792. doi: 10.3109/00365529008999216. [DOI] [PubMed] [Google Scholar]
- 22.Graziadei IW, Joseph JJ, Wiesner RH, Therneau TM, Batts KP, Porayko MK. Increased risk of chronic liver failure in adults with heterozygous alpha1-antitrypsin deficiency. Hepatology. 1998;28:1058–1063. doi: 10.1002/hep.510280421. [DOI] [PubMed] [Google Scholar]
- 23.Fischer HP, Ortiz-Pallardo ME, Ko Y, Esch C, Zhou H. Chronic liver disease in heterozygous alpha1-antitrypsin deficiency PiZ. J Hepatol. 2000;33:883–892. doi: 10.1016/s0168-8278(00)80119-1. [DOI] [PubMed] [Google Scholar]
- 24.Regev A, Guaqueta C, Molina EG, Conrad A, Mishra V, Brantly ML, Torres M, et al. Does the heterozygous state of alpha-1 antitrypsin deficiency have a role in chronic liver diseases? Interim results of a large case-control study. J Pediatr Gastroenterol Nutr. 2006;43 (Suppl 1):S30–35. doi: 10.1097/01.mpg.0000226387.56612.1e. [DOI] [PubMed] [Google Scholar]
- 25.Luisetti M, Seersholm N. Alpha1-antitrypsin deficiency. 1: epidemiology of alpha1-antitrypsin deficiency. Thorax. 2004;59:164–169. doi: 10.1136/thorax.2003.006494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Serres FJ, Blanco I, Fernandez-Bustillo E. Genetic epidemiology of alpha-1 antitrypsin deficiency in southern Europe: France, Italy, Portugal and Spain. Clin Genet. 2003;63:490–509. doi: 10.1034/j.1399-0004.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- 27.Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med. 2009;360:2749–2757. doi: 10.1056/NEJMcp0900449. [DOI] [PubMed] [Google Scholar]
- 28.Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest. 2005;128:1989–1994. doi: 10.1378/chest.128.4.1989. [DOI] [PubMed] [Google Scholar]
- 29.Campbell EJ. Alpha1-antitrypsin deficiency: incidence and detection program. Respir Med. 2000;94 (Suppl C):S18–21. doi: 10.1053/rmed.2000.0854. [DOI] [PubMed] [Google Scholar]
- 30.Borawski J, Naumnik B, Mysliwiec M. Serum alpha1-antitrypsin but not complement C3 and C4 predicts chronic inflammation in hemodialysis patients. Ren Fail. 2003;25:589–593. doi: 10.1081/jdi-120022550. [DOI] [PubMed] [Google Scholar]
- 31.Fairbanks KD, Tavill AS. Liver disease in alpha 1-antitrypsin deficiency: a review. Am J Gastroenterol. 2008;103:2136–2141. doi: 10.1111/j.1572-0241.2008.01955.x. quiz 2142. [DOI] [PubMed] [Google Scholar]
- 32.Rachelefsky G, Hogarth DK. Issues in the diagnosis of alpha 1-antitrypsin deficiency. J Allergy Clin Immunol. 2008;121:833–838. doi: 10.1016/j.jaci.2007.12.1183. [DOI] [PubMed] [Google Scholar]
- 33.Hogarth DK, Rachelefsky G. Screening and familial testing of patients for alpha 1-antitrypsin deficiency. Chest. 2008;133:981–988. doi: 10.1378/chest.07-1001. [DOI] [PubMed] [Google Scholar]
- 34.Hashemi M, Alavian SM, Ghavami S, de Serres FJ, Salehi M, Doroudi T, Fard AH, et al. High prevalence of alpha 1 antitrypsin phenotypes in viral hepatitis B infected patients in Iran. Hepatol Res. 2005;33:292–297. doi: 10.1016/j.hepres.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 35.Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol. 2004;286:G851–862. doi: 10.1152/ajpgi.00175.2003. [DOI] [PubMed] [Google Scholar]
- 36.Perlmutter D. Alpha-1 Antitrypsin Deficiency: Importance of Proteasomal and Autophagic Degradative Pathways in Disposal of Liver Disease-Associated Protein Aggregates. Annu Rev Med. doi: 10.1146/annurev-med-042409-151920. [DOI] [PubMed] [Google Scholar]
- 37.Marciniak SJ, Lomas DA. Alpha1-antitrypsin deficiency and autophagy. N Engl J Med. 363:1863–1864. doi: 10.1056/NEJMcibr1008007. [DOI] [PubMed] [Google Scholar]
- 38.Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 39.Kaushal S, Annamali M, Blomenkamp K, Rudnick D, Halloran D, Brunt EM, Teckman JH. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp Biol Med (Maywood) 235:700–709. doi: 10.1258/ebm.2010.009297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kemmer N, Kaiser T, Zacharias V, Neff GW. Alpha-1-antitrypsin deficiency: outcomes after liver transplantation. Transplant Proc. 2008;40:1492–1494. doi: 10.1016/j.transproceed.2008.02.075. [DOI] [PubMed] [Google Scholar]
- 41.Hood JM, Koep LJ, Peters RL, Schroter GP, Weil R, 3rd, Redeker AG, Starzl TE. Liver transplantation for advanced liver disease with alpha-1-antitrypsin deficiency. N Engl J Med. 1980;302:272–275. doi: 10.1056/NEJM198001313020505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain AB, Patil V, Sheikh B, Apostolakos M, Ryan C, Kashyap R, Orloff M. Effect of liver transplant on pulmonary functions in adult patients with alpha 1 antitrypsin deficiency: 7 cases. Exp Clin Transplant. 8:4–8. [PubMed] [Google Scholar]
- 43.Eriksson SG. Liver disease in alpha 1-antitrypsin deficiency. Aspects of incidence and prognosis. Scand J Gastroenterol. 1985;20:907–911. doi: 10.3109/00365528509088844. [DOI] [PubMed] [Google Scholar]
- 44.Hadzic N, Quaglia A, Mieli-Vergani G. Hepatocellular carcinoma in a 12-year-old child with PiZZ alpha1-antitrypsin deficiency. Hepatology. 2006;43:194. doi: 10.1002/hep.21009. [DOI] [PubMed] [Google Scholar]
- 45.Vergalla J, Jones EA, Kew MC. Alpha-1-antitrypsin deficiency and hepatocellular carcinoma. Determination of Pi phenotypes using iso-electric focusing. S Afr Med J. 1983;64:950–951. [PubMed] [Google Scholar]
- 46.Rakela J, Goldschmiedt M, Ludwig J. Late manifestation of chronic liver disease in adults with alpha-1-antitrypsin deficiency. Dig Dis Sci. 1987;32:1358–1362. doi: 10.1007/BF01296661. [DOI] [PubMed] [Google Scholar]
- 47.Prachalias AA, Kalife M, Francavilla R, Muiesan P, Dhawan A, Baker A, Hadzic D, et al. Liver transplantation for alpha-1-antitrypsin deficiency in children. Transpl Int. 2000;13:207–210. doi: 10.1007/s001470050688. [DOI] [PubMed] [Google Scholar]
- 48.Francalanci P, Santorelli FM, Saccani S, Bonetti MF, Medicina D, Coni P, Faa G, et al. Z and Mmalton-1-antitrypsin deficiency-associated hepatocellular carcinoma: a genetic study. Liver Int. 2009;29:1593–1596. doi: 10.1111/j.1478-3231.2009.02091.x. [DOI] [PubMed] [Google Scholar]
- 49.Kok KF, Wahab PJ, Houwen RH, Drenth JP, de Man RA, van Hoek B, Meijer JW, et al. Heterozygous alpha-I antitrypsin deficiency as a co-factor in the development of chronic liver disease: a review. Neth J Med. 2007;65:160–166. [PubMed] [Google Scholar]
- 50.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 51.Rudnick DA, Perlmutter DH. Alpha-1-antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology. 2005;42:514–521. doi: 10.1002/hep.20815. [DOI] [PubMed] [Google Scholar]
- 52.Reid CL, Wiener GJ, Cox DW, Richter JE, Geisinger KR. Diffuse hepatocellular dysplasia and carcinoma associated with the Mmalton variant of alpha 1-antitrypsin. Gastroenterology. 1987;93:181–187. doi: 10.1016/0016-5085(87)90332-5. [DOI] [PubMed] [Google Scholar]
- 53.Marcus NY, Brunt EM, Blomenkamp K, Ali F, Rudnick DA, Ahmad M, Teckman JH. Characteristics of hepatocellular carcinoma in a murine model of alpha-1-antitrypsin deficiency. Hepatol Res. 2010;40:641–653. doi: 10.1111/j.1872-034X.2010.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]