

Abstract
We investigated whether Kif3a in osteoblasts has a direct role in regulating postnatal bone formation. We conditionally deleted Kif3a in osteoblasts by crossing osteocalcin (Oc; also known as Bglap)–Cre with Kif3aflox/null mice. Conditional Kif3a-null mice (Kif3aOc-cKO) had a 75% reduction in Kif3a transcripts in bone and osteoblasts. Conditional deletion of Kif3a resulted in the reduction of primary cilia number by 51% and length by 27% in osteoblasts. Kif3aOc-cKO mice developed osteopenia by 6 weeks of age unlike Kif3aflox/+ control mice, as evidenced by reductions in femoral bone mineral density (22%), trabecular bone volume (42%) and cortical thickness (17%). By contrast, Oc-Cre;Kif3aflox/+ and Kif3aflox/null heterozygous mice exhibited no skeletal abnormalities. Loss of bone mass in Kif3aOc-cKO mice was associated with impaired osteoblast function in vivo, as reflected by a 54% reduction in mineral apposition rate and decreased expression of Runx2, osterix (also known as Sp7 transcription factor 7; Sp7), osteocalcin and Dmp1 compared with controls. Immortalized osteoblasts from Kif3aOc-cKO mice exhibited increased cell proliferation, impaired osteoblastic differentiation, and enhanced adipogenesis in vitro. Osteoblasts derived from Kif3aOc-cKO mice also had lower basal cytosolic calcium levels and impaired intracellular calcium responses to fluid flow shear stress. Sonic hedgehog-mediated Gli2 expression and Wnt3a-mediated β-catenin and Axin2 expression were also attenuated in Kif3aOc-cKO bone and osteoblast cultures. These data indicate that selective deletion of Kif3a in osteoblasts disrupts primary cilia formation and/or function and impairs osteoblast-mediated bone formation through multiple pathways including intracellular calcium, hedgehog and Wnt signaling.
Key words: Primary cilia, Conditional deletion, Kif3a, Bone formation, Osteoblast functions, Signaling
Introduction
Primary cilia are present on most mammalian cells, including osteoblasts and osteocytes (Berbari et al., 2009; Davis et al., 2006; Xiao et al., 2006), but their function is not known. Primary cilia are dynamic organelles in which the kinesin-2 motor mediates the anterograde transport of intraflagellar transport (IFT) rafts to the distal tip of the cilium and the dynein motor facilitates the retrograde transport of IFT cargos to the basal body of the cilium (Davis et al., 2006; Gerdes et al., 2009). Kinesin-2 forms a heterotrimeric complex of two motor subunits, kinesin-2 family protein A (Kif3a) and B (Kif3b) and a nonmotor subunit, kinesin-associated protein (Kap3).
Primary cilia house many signaling pathways that might be involved in osteoblast development and postnatal osteoblast functions (Christensen et al., 2007; Gerdes et al., 2009; Han et al., 2008; Huangfu and Anderson, 2005; Kovacs et al., 2008; Serra, 2008; Veland et al., 2009; Wong et al., 2009), including polycystin-1 (Pkd1) and 2 (Pkd2), which form a mechanosensing signaling complex that localizes to primary cilia. The C-terminal region of Kif3a has recently been shown to bind to the C-terminus of PC2 (also known as Pkd2), resulting in its localization and function in primary cilia (Li et al., 2006). Other studies have reported that Kif3b, the other motor subunit of kinesin-2, serves as a linker between PC2 and fibrocystin (also known as polyductin), the gene product of PKHD1, the gene responsible for autosomal recessive polycystic kidney disease (ARPKD) (Wu et al., 2006). Polycystins and primary cilia have interdependent functions in renal epithelial cells, and mutations in either of the polycystin genes, as well as in other genes required for ciliogenesis, lead to a common PKD phenotype. We have recently identified an important role of Pkd1 in osteoblastic development and mechanosensing in responses to skeletal loading (Xiao et al., 2011; Xiao, Z. et al., 2010; Xiao et al., 2008; Xiao et al., 2006). This suggests that primary cilia also have a role in regulating osteoblast development and function.
There is additional evidence supporting the role of primary cilia in bone development. Global deletion and/or mutations of IFT88 (also known as Tg737 and polaris) or Kif3a, which blocks cilia formation (Goetz and Anderson, 2010; Pedersen et al., 2008; Serra, 2008) are associated with abnormalities of the skeleton. Disruption of gene products involved in cilium assembly and IFT also results in abnormalities of skeletal development (Serra, 2008), including left–right asymmetry (Marszalek et al., 1999), limb patterning (Haycraft and Serra, 2008; Zhang et al., 2003), endochondral bone formation (Haycraft et al., 2007), postnatal growth plate (McGlashan et al., 2007; Song et al., 2007) and craniofacial development (Kolpakova-Hart et al., 2007; Zhang et al., 2003). However, global deletion of Ift88 or Kif3a in mice, which disrupts ciliogenesis, is embryonic lethal, making it difficult to determine the role of primary cilia in postnatal osteoblast function (Marszalek et al., 1999; Murcia et al., 2000).
There is strong evidence that primary cilia regulate embryonic endochondral bone formation through hedgehog (Hh) signaling during both embryonic and postnatal endochondral bone formation. Mice with conditional deletion of Ift88 or Kif3a resulting from the use of both Prx1–Cre and Dermo1–Cre, that are expressed in limb mesenchyme (including chondrocytes and perichondrium) have shorter bones in the limbs because of alterations in embryonic endochondral bone formation, similar to that seen in mice with germline mutations in indian hedgehog (Ihh) (Haycraft et al., 2007; Kolpakova-Hart et al., 2007; St-Jacques et al., 1999). By contrast, mice with conditional deletion of Ift88 or Kif3a from the use of Col2a–Cre, which is expressed in the chondrocyte lineage, show no skeletal abnormalities during the embryonic stages, but develop postnatal dwarfism because of a progressive loss of the growth plate between postnatal day (P) 7 and P15 (Haycraft et al., 2007; Koyama et al., 2007; Song et al., 2007), similar to mice with conditional deletion of Ihh induced in postnatal cartilage (Col2a-CreER;Ihhflox/flox) (Maeda et al., 2007). So far, the focus on mice with mutations in IFT- and cilia-related genes has been on embryonic skeletal development and chondrocyte function; the role of primary cilia in osteoblast function and postnatal bone homeostasis remains to be studied.
In the study reported here we used genetic approaches to conditionally delete Kif3a (disruption of ciliogenesis) in osteoblasts of mice to gain insights into whether IFT and primary cilia have a direct function in regulating postnatal bone formation. We demonstrated that conditional deletion of Kif3a from osteoblasts of heterozogyous Kif3anull/flox mutant mice resulted in defective osteoblast function in vivo and in vitro and osteopenia, a condition in which bone mineral density is lower than normal. In addition, Kif3a-deficient osteoblasts exhibited a gene dose-dependent reduction in response to fluid flow shear stress in vitro and an impairment of many cilia-related pathways including Hh and Wnt signaling. These results indicate that IFT and cilia have a direct role in regulating osteoblast function and skeletal homeostasis.
Results
Osteocalcin (Oc)-Cre-mediated conditional deletion of Kif3a in different tissues
The four genotypes from the breeding strategy crossing osteocalcin (Oc; also known as Bglap)–Cre with Kif3aflox/null mice (Oc-Cre;Kif3aflox/null or Kif3aOc-cKO, Oc-Cre;Kif3aflox/+, Kif3aflox/null and Kif3aflox/+; see Materials and Methods) were born at the expected Mendelian frequency, and the survival of all Kif3a-deficient mice was indistinguishable from that of control mice (Kif3aflox/+, equivalent to wild type). The normal survival of conditional Kif3aOc-cKO null mice (Oc-Cre;Kif3aflox/null) contrasts with the perinatal lethality of homozygous Kif3anull/null mice (Marszalek et al., 1999). To confirm that the Kif3a-floxed allele was selectively deleted in bone, we performed PCR analysis by using a combination of primers that specifically detect floxed Kif3a alleles (Kif3aflox) and the excised floxed Kif3a alleles (Kif3aΔflox), as well as wild-type alleles (Kif3a+) in Oc-Cre;Kif3aflox/+ mice (Fig. 1A). Oc-Cre expression is limited to mature osteoblasts (bone surface osteoblasts that synthesize new bone and osteocytes embedded in bone that regulate bone remodeling) with onset of expression just before birth (E18.5) (Zhang et al., 2002). We demonstrated that Oc-Cre-mediated floxed recombination occurred exclusively in tissues that contain osteoblasts, whereas nonskeletal tissues retained the intact floxed Kif3a alleles (Kif3aflox; Fig. 1B). Consistent with the lack of Cre expression in the kidney, Kif3aOc-cKO, Oc-Cre;Kif3aflox/+ and Kif3aflox/null mice had no cysts in the kidney (data not shown).
Fig. 1.

Oc-Cre-mediated conditional deletion of Kif3a from the floxed Kif3a allele (Kif3aflox) in different tissues. (A) Schematic illustration of the wild-type (Kif3a+), mutant (Kif3aΔflox or Kif3anull) and floxed Kif3a allele before (Kif3aflox) and after deletion (Kif3aΔflox) of the loxP cassette containing exon 2 through Cre-mediated recombination. // indicates all the introns and exons omitted after exon 3. (B) Genotyping PCR analysis of different tissues that were harvested from 6-week-old Oc-Cre;Kif3aflox/+ mice showed bone-specific deletion of the Kif3a gene. Oc-Cre-mediated recombination of excised floxed Kif3a (Kif3aΔflox) allele occurred exclusively in bone, whereas nonskeletal tissues retained the floxed Kif3a allele (Kif3aflox). (C,D) Real-time RT-PCR analysis of total Kif3a transcripts in bone and cultured osteoblasts. Total Kif3a transcripts was expressed using Kif3a-allele-specific primers as described in Materials and Methods. Data are expressed as the percentage of normal (wild-type Kif3a+ and Kif3aflox) and mutant (Kif3aΔflox and Kif3anull) Kif3a allele expressions in Kif3aflox/+ control, single Oc-Cre;Kif3aflox/+ and Kif3aflox/null heterozygous mice, and conditional Kif3aOc-cKO null mice. (E,F) Immunofluorescence analysis of primary cilia in immortalized osteoblasts. Immunostaining of primary cilia (red) was performed with acetylated α-tubulin antibody as described in Materials and Methods. Counterstaining with a nuclear marker (DAPI blue) was used to calculate the percentage presence of primary cilia in immortalized osteoblasts. There were no obvious differences in the number of primary cilia in single Oc-Cre;Kif3aflox/+ and Kif3aflox/null heterozygous osteoblasts; however, a marked reduction of cilia formation was observed in conditional Kif3aOc-cKO null osteoblasts compared with control cells. *Significant difference from control (Kif3aflox/+); #significant difference from single heterozygous Oc-Cre;Kif3aflox/+ and Kif3aflox/null mice (P<0.05).
To quantify the effect of combined use of the floxed Kif3aflox allele with the nonfunctional Kif3anull allele to increase the net efficiency of Kif3a inactivation by Cre recombinase, we examined the percentage of Kif3a, conditional deleted (Kif3aΔflox) and null (Kif3anull) allele expressions in bone tissues from 6-week-old mice by real-time RT-PCR. As expected, Kif3aflox/null mice expressed 50% of the Kif3anull null allele, whereas Oc-Cre;Kif3aflox/+ mice exhibited approximately 25% excision of the floxed exon 2 from Kif3a, indicating that Oc-Cre-mediated bone-specific deletion of the floxed Kif3a allele was incomplete (Fig. 1C). The combined effect of Kif3aΔflox and Kif3anull alleles in Oc-Cre;Kif3aflox/null mice resulted in a net reduction of Kif3a expression by ~75% in bone (Fig. 1C). Unlike a time-dependent increase of total Pkd1 transcripts during osteogenic culture (Xiao et al., 2011), we observed no further increase of total Kif3a transcripts in control Kif3aflox/+ osteoblasts. Consistent with the lower expression of Kif3a expression in bone, conditional Kif3aOc-cKO null osteoblasts showed more than 75% inactivation of Kif3a transcripts by real-time RT-PCR during 18 days of osteogenic culture (Fig. 1D). In addition, whereas in both Oc-Cre;Kif3aflox/+ and Kif3aflox/null heterozygous mice there was no change in the appearance of primary cilia, in conditional Kif3aOc-cKO null mice there was substantially fewer primary cilia in osteoblast cultures (Fig. 1E,F), in association with a 75% lower expression of Kif3a transcript in osteoblasts derived from conditional Kif3aOc-cKO null mice (Fig. 1D).
A gene dose-independent role of global and Oc-Cre-mediated conditional deletion of Kif3a in postnatal bone formation
We observed no differences in body weight, lean body mass or fat mass in any of the three Kif3a-deficient mice compared with controls at 6 weeks of age (data not shown). Consistent with our previous report (Qiu et al., 2010), we found that single heterozygous Oc-Cre;Kif3aflox/+ or Kif3aflox/null mice had no demonstrable bone abnormalities (Fig. 2). Bone mineral density (BMD) and bone structure (Fig. 2A,B), as well as mineral apposition rates (MAR; Fig. 2C) were not different in single heterozygous Oc-Cre;Kif3aflox/+, Kif3aflox/null and age-matched control mice (Kif3aflox/+). In addition, bone samples from Oc-Cre;Kif3aflox/+ and Kif3aflox/null mice had no detectable changes in markers of either osteoblast or osteoclast gene expression (Table 1). However, Kif3aflox/null mice had significantly (P<0.05) lower expression of adipocyte-related marker genes, including peroxisome proliferator-activated receptor gamma (Pparg), adipocyte-specific fatty acid binding protein (aP2; also known as Fabp4) and lipoprotein lipase (Lpl) in long-bone samples (Table 1).
Fig. 2.

Oc-Cre-mediated somatic deletion in a Kif3a-deficient background leads to osteopenia. Effects of global and/or Oc-Cre-mediated deletion of Kif3a on (A) bone mineral density (BMD), (B) bone structure of femurs and (C) bone mineral apposition rate (MAR) at 6 weeks of age. There was no bone loss in either female or male single heterozygous Oc-Cre;Kif3aflox/+ and Kif3aflox/null mice, evidenced by normal BMD, BV/TV, Ct.Th and MAR compared with age-matched control mice (Kif3aflox/+). By contrast, there was a significant reduction in BMD in both female and male conditional Kif3aOc-cKO null mice compared with age-matched control mice (Kif3aflox/+). μCT analysis revealed that the lower bone mass in male conditional Kif3aOc-cKO null mice was the result of a reduction in trabecular BV/TV and cortical Ct.Th. These reductions in bone mass and structure were associated with a 50% decrease in MAR in male conditional Kif3aOc-cKO null mice compared with age-matched control mice. Values are means ± s.d. from five or six individual mice. *Significant difference from control (Kif3aflox/+) and #significant difference from single heterozygous Oc-Cre;Kif3aflox/+ and Kif3aflox/null mice (P<0.05).
Table 1.
Gene expression profiles in 6-week-old mice

We observed a significantly (P<0.05) lower BMD, of 11~21%, in both female and male conditional Kif3aOc-cKO mice at 6 weeks of age compared with age-matched control mice (Kif3aflox/+; Fig. 2A). MicroCT (μCT) analysis revealed that the lower bone mass in male conditional Kif3aOc-cKO null mice was caused by reduced trabecular bone volume (BV/TV, 42%) and cortical bone thickness (Ct.Th, 17%; Fig. 2B). These reductions in bone volume and cortical thickness were associated with a 50% decrease in periosteal MAR in male conditional Kif3aOc-cKO null mice compared with age-matched control mice (Fig. 2C). To investigate the effects of the combined Kif3aΔflox and Kif3anull deficiency on gene expression profiles in bone, we examined, by real-time RT-PCR, the expression levels of a panel of osteoblast lineage-, osteoclast- and adipocyte-related mRNAs from the tibias of 6-week-old male control and conditional Kif3aOc-cKO null mice (Table 1). Bone derived from conditional Kif3aOc-cKO null mice had significantly lower levels of osteoblast-lineage gene transcripts, including Runx2, osterix (also known as Sp7 transcription factor 7; Sp7), osteocalcin, rank ligand (RankL; also known as tumor necrosis factor ligand superfamily member 11; Tnfsf11) and Dmp1 mRNA, but no obvious differences were found in osteoprotegerin (Opg; Tnfrsf11b) expression compared with that in control mice. In this regard, the Opg/RankL gene expression ratio was higher in conditional Kif3aOc-cKO null mice (Table 1). Consistent with a ratio of Opg/RankL genes that favored the lower osteoclastogenesis, bone expression of Trap and Mmp9, markers of bone resorption, were also lower in conditional Kif3aOc-cKO null mice (Table 1). By contrast, the gene encoding Pparγ, an adipocyte transcription factor, and adipocyte markers such as aP2 and Lpl were significantly (P<0.05) higher in tibias of conditional Kif3aOc-cKO null mice compared with single heterozygous Kif3aflox/null mice, and the expression of Lpl was higher in conditional Kif3aOc-cKO null mice than that in control Kif3aflox/+ mice (Table 1).
Changes in gene expression in bone correlated with in serum biomarkers. In this regard, control Kif3aflox/+ mice had reduced osteoblastic and osteoclastic markers as a function of age, consistent with an age-dependent decrease in bone formation and resorption. At 6 weeks of age, Kif3aOc-cKO null mice had lower levels of certain markers than age-matched control Kif3aflox/+ mice: the osteoblast markers osteocalcin (42±8 vs 59±10 g/ml) and RankL (79±28 vs 122±24 pg/ml), as well as the osteoclastic marker tartrate-resistant acid phosphatase (TRAP; 4.5±0.43 vs 5.6±0.29 IU/l). At 24 weeks of age, there was no longer a difference in osteocalcin, but RankL (54±16 vs 95±22 pg/ml) and TRAP (0.8±0.11 vs 1.7±0.32 IU/l) remained lower than in the age-matched control Kif3aflox/+ mice. Although there was no difference in the OPG level, the RankL/OPG ratio, which is an indicator of osteoclastogenesis, was lower by approximately 29% at 6 weeks of age and 47% at 24 weeks of age in Kif3aOc-cK null mice compared with age-matched control Kif3aflox/+ mice. These data suggest that conditional deletion of Kif3a in osteoblasts results in diminished osteoblast-mediated bone formation and osteoclast-mediated bone resorption, resulting in low-turnover osteopenia. Kif3aOc-cKO null mice had no change in serum urea nitrogen (BUN), calcium or phosphorus levels at either 6 or 24 weeks (data not shown).
Age-dependent effects of global and Oc-Cre-mediated conditional deletion of Kif3a on bone mass, structure, geometry and mechanical properties
We observed no differences in bone mass and bone structure between single heterozygous Oc-Cre; Kif3aflox/+, Kif3aflox/null and control Kif3aflox/+ mice from 6 to 24 weeks of age. However, we observed an age-dependent partial recovery of BMD from 21% lower at 6 weeks of age to 7% lower at 24 weeks of age in male conditional Kif3aOc-cKO null mice compared with age-matched control mice, indicating age-dependent effects that attenuate the effects of deleted Kif3a on bone mass (Fig. 3A). μCT analysis revealed that the increase in bone mass was caused by a recovery in cortical bone thickness. Indeed, the differences in cortical bone thickness at 6 weeks of age were no longer significant in the four genotypes at 24 weeks of age (Fig. 3B). By contrast, there was less recovery of BV/TV with age in Kif3aOc-cKO mice. In this regard, BV/TV remained significantly (P<0.05) lower in Kif3aOc-cKO mice compared with control mice at 24 weeks of age (Fig. 3C), a finding that suggests a site-specific interaction between Kif3a deficiency and age-dependent changes in bone structure. Moreover, we found that the mechanism of BMD and cortical bone thickness recovery resulted from alterations in bone geometry and led to a compensatory increase in bone mechanical properties between 6 and 24 weeks in Kif3aOc-cKO mice (Table 2). Also, conditional Kif3aOc-cKO null mice showed a greater total bone area, moment of inertia (Ix) and distance from the neutral axis to the plane where the load is applied (c) at 6 weeks of age. However, there was no difference in cortical bone area compared with age-matched control mice (Table 2), but the marrow cavity was larger, resulting in a greater midshaft diameter compared with that in control mice. However, these structural differences were no longer evident in 24-week-old Kif3aOc-cKO mice because of a recovery in femoral midshaft diameter and the size of the marrow cavity with age (Table 2).
Fig. 3.

Age-dependent effects of global and/or Oc-Cre-mediated conditional deletion of Kif3a on bone mass and structure. Age-dependent effects of global and Oc-Cre-mediated somatic deletion of Kif3a on (A) femoral BMD, (B) trabecular bone volume of distal femoral metaphyses, (C) cortical bone thickness of femoral midshaft diaphyses. Compared with control (Kif3aflox/+) mice, there was an age-dependent partial recovery of BMD in male conditional Kif3aOc-cKO null mice from 6 to 24 weeks of age. μCT analysis revealed that the increase in bone mass resulted from a complete recovery in cortical bone thickness, but BV/TV remained significantly lower in male conditional Kif3aOc-cKO null mice at 24 weeks of age. However, single heterozygous Oc-Cre;Kif3aflox/+ and Kif3aflox/null mice had normal bone mass and bone structure compared with age-matched controls. Values are means ± s.d. from five or six individual mice. *Significant difference from control (Kif3aflox/+) and #significant difference from single heterozygous Oc-Cre;Kif3aflox/+ and Kif3aflox/null mice (P<0.05).
Table 2.
Femur bone geometry and biomechanical properties in 6- and 24-week-old mice

To examine whether changes of femoral bone geometry might affect bone mechanical properties, we used these femurs to perform three-point bending experiments (Table 2). At 6 weeks of age, conditional Kif3aOc-cKO null mice had a lower elastic modulus in three-point bending experiments but no significant differences in bending stiffness or maximum force compared with age-matched control mice (Table 2), indicating that the changes in bone geometry and bone structure at 6 weeks of age had an impact on bone strength in conditional Kif3aOc-cKO null mice. Again, the recovery of bone geometry and structure in 24-week-old Kif3aOc-cKO normalized these mechanical properties (Table 2). Both the alterations in geometry and mechanical properties in Kif3aOc-cKO are similar to observations in Dmp1–Cre-mediated conditional Pkd1Dmp1-cKO-null mice, consistent with the known interdependent functions of polycystins and primary cilia (Xiao et al., 2011).
Effect of conditional deletion of Kif3a on osteoblastic function ex vivo
To determine the impact of conditional deletion of Kif3a on osteoblast function ex vivo, we used immortalized osteoblasts derived from E17.5 control Kif3aflox/+ mice, single heterozygous Oc-Cre;Kif3aflox/+ mice, single heterozygous Kif3aflox/null mice and conditional Kif3aOc-cKO null mice. Immortalized osteoblasts in culture undergo progressive alterations in cell proliferation and osteoblastic differentiation that recapitulates the osteoblastic developmental program (Xiao et al., 2004). Consistent with our previous report (Qiu et al., 2010), we found that heterozygous Kif3a-deficient mice had no abnormalities in cell proliferation or osteoblastic differentiation. In this regard, the osteoblasts from Oc-Cre;Kif3aflox/+ mice and heterozygous Kif3aflox/null mice exhibited a time-dependent increase of total BrdU incorporation during 48 hours of osteogenic culture (Fig. 4A) as well as increased alkaline phosphatase (ALP) activity (a marker of differentiated osteoblasts) and calcium deposition in long-term cultures similar to controls (Fig. 4B,C), However, we found that conditional Kif3aOc-cKO null osteoblasts had a higher BrdU incorporation than the other three groups, indicating a greater proliferation rate in Kif3aOc-cKO null osteoblasts (Fig. 4A). In addition, Kif3aOc-cKO null osteoblasts had impaired osteoblastic differentiation and maturation, as evidenced by culture duration-dependent reductions in ALP activity (Fig. 4B), less calcium deposition in extracellular matrix (Fig. 4C), and lower expression of osteoblastic differentiation markers, including Runx2-II, osterix and osteocalcin, compared with control osteoblasts (Fig. 4D–F). The immortalized primary calvarial osteoblasts derived from Kif3aOc-cKO mice also exhibited evidence for divergence from the osteoblastic development program, analogous to the higher levels of adipogenic markers observed in vivo. Indeed, under osteogenic culture condition, Kif3aOc-cKO-derived osteoblasts exhibited a marked increase of adipocyte markers such as Pparg2 and the aP2 gene (Fig. 4G,H), suggesting that impairment of osteogenesis was associated with enhancement of adipogenesis in conditional Kif3aOc-cKO null osteoblast cultures.
Fig. 4.

Effects of global and/or Oc-Cre-mediated conditional deletion of Kif3a on osteoblastic proliferation and maturation, as well as gene expression profiles ex vivo. (A) BrdU incorporation. There was a time-dependent increase of total BrdU incorporation during 48 hours in osteogenic culture in osteoblasts from each of the four genotypes. However, a higher BrdU incorporation was observed in Kif3aOc-cKO null osteoblasts compared with the other three groups of osteoblasts at the indicated times. (B) ALP activity. All osteoblasts from each of the four genotypes displayed time-dependent increases in ALP activity during 14 days in culture, but ALP activity was significantly lower in Kif3aOc-cKO null osteoblasts compared with the other three groups of osteoblasts at day 14. (C) Quantification of mineralization. Alizarin Red-S was extracted with 10% cetylpyridinium chloride and quantified as described in Materials and Methods. All osteoblasts from each of the four genotypes had time-dependent increases in Alizarin Red-S accumulation during the 21 days in culture, but the accumulation was significantly lower in Kif3aOc-cKO null osteoblasts compared with the other three groups of osteoblasts at day 21. (D–H) Gene expression profiles by real-time RT-PCR. Immortalized Kif3aOc-cKO null osteoblasts in osteogenic medium showed time-dependent increases in osteogenesis, evident from increases in osteoblastic markers (Runx2, osterix and osteocalcin) during 18 days in culture which were significantly lower than in control (Kif3aflox/+) osteoblasts at 12 and 18 days (D–F). By contrast there was a marked increase in adipocyte markers (Pparg2; PPARγ2; and aP2) in the Kif3aOc-cKO null osteoblasts compared with control osteoblasts. Values are means ± s.d. from three independent experiments. *Significant difference from control (Kif3aflox/+), and #significant difference from single heterozygous Oc-Cre;Kif3aflox/+ and Kif3aflox/null mice (P<0.05).
Effects of conditional deletion of Kif3a on intracellular calcium, Wnt and Hh signaling in immortalized osteoblasts
Loss of Kif3a in a gene-dose-dependent fashion resulted in a reduction in the length of primary cilia in primary cultured osteoblasts and altered responses to flow shear stress, consistent with alterations of cilia functions. Primary cilia were approximately 27% shorter in Kif3aOc-cKO null osteoblasts (Fig. 5A). In addition, we found that Kif3aOc-cKO null osteoblasts had a significantly (P<0.05) lower basal intracellular calcium ([Ca2+]i) concentration compared with control Kif3aflox/+ cells, whereas [Ca2+]i levels in single heterozygous Oc-Cre;Kif3aflox/+ and Kif3aflox/null osteoblasts were similar to controls (Fig. 5B). These findings are consistent with previously reported effects of primary cilia and/or polycystic complex regulation of intracellular calcium signaling in renal epithelial cells (Nauli et al., 2003; Nauli et al., 2006; Praetorius et al., 2003; Praetorius and Spring, 2001; Shiba et al., 2005; Yoder, 2007). The change in intracellular calcium was proportional to the magnitude of the fluid flow shear stress (FFSS, 0.69~9.5 dynes/cm2) in control Kif3aflox/+ osteoblasts (Fig. 5C), with the highest peak-flow-induced intracellular calcium response of 6.24 dynes/cm2. No further significant increase in intracellular calcium was observed with 9.5 dynes/cm2, indicating that 6.24 dynes/cm2 is the optimal FFSS to induce intracellular calcium responses in immortalized osteoblasts under these experimental conditions. Applying this amount of FFSS to heterozygous Oc-Cre;Kif3aflox/+, Kif3aflox/null and Kif3aOc-cKO null osteoblasts demonstrated a gene-dose-dependent reduction in flow-induced intracellular calcium response compared with controls (Fig. 5D), indicating that FFSS is a more sensitive measure of primary cilia abnormalities than the assessment of cilia length or number. Indeed, an intermediate calcium response was observed in single heterozygous cells whereas minimal calcium influx was observed in conditional Kif3aOc-cKO null osteoblasts in response to FFSS (Fig. 5D). However, 10 mM caffeine resulted in normal calcium influx in conditional Kif3aOc-cKO null cells after flow stimulus (data not shown), indicating the viability of the conditional Kif3aOc-cKO null cells. Finally, we assessed the effects of FFSS on expression of ‘mechanosensing’ genes by real-time RT-PCR and protein expression by western blot analysis using RNA and cytoplasmic proteins isolated from control and Kif3aOc-cKO null cells with or without FFSS. FFSS increased both the mRNA and protein of Cox2, a mechanoresponsive gene, in control osteoblasts (Fig. 5E,F), whereas these changes were less in Kif3aOc-cKO null cells exposed to identical FFSS (Fig. 5E,F).
Fig. 5.

Effects of global and/or Oc-Cre-mediated conditional deletion of Kif3a on baseline and flow-induced intracellular calcium ([Ca2+]i) response, as well as mechanoresponsive gene expression in osteoblasts. (A) Length of primary cilia (n=15~20). A gene dose-dependent reduction of cilium length was observed in the Kif3a-deficient osteoblasts compared with control Kif3aflox/+ cells. (B) Basal [Ca2+]i levels. Only Kif3aOc-cKO null osteoblasts (n=53) showed a significantly lower basal [Ca2+]i levels compared with the other three group of osteoblasts (n=53). (C) Flow-induced [Ca2+]i response curves using different flow rates. Control Kif3aflox/+ osteoblasts exhibited an FFSS-dependent intracellular Ca2+ response. Of the four flow rates (n=4), 6.24 dynes/cm2 was the optimal FFSS to induce the intracellular Ca2+ response in the immortalized osteoblasts. There was no significant difference between 6.24 dynes/cm2 and 9.50 dynes/cm2 loading. (D) Flow-induced [Ca2+]i response curves obtained using cells of different genotypes (n=4). A gene dose-dependent reduction of flow-induced [Ca2+]i responses was observed in the Kif3a-deficient osteoblasts compared with control Kif3aflox/+ cells, which is in agreement with the length of primary cilia in Kif3a-deficient osteoblasts. There was no significant difference between Oc-Cre;Kif3aflox/+ and Kif3aflox/null cells. (E–F) Expression of mechanoresponsive genes. Real-time RT-PCR and western blot analyses from control Kif3aflox/+ and Kif3aOc-cKO null osteoblasts showed that mRNA and protein levels of Cox-2 were markedly increased in the loaded control cells but the increase in the loaded Kif3aOc-cKO null cells was much less. Values are means ± s.d. from three independent experiments. *Significant difference from control Kif3aflox/+ cells (P<0.05); #significant difference from Oc-Cre;Kif3aflox/+ or Kif3aflox/null cells at P<0.05; @significant difference from 9.50 dynes/cm2 and 6.24 dynes/cm2 (P<0.05); &significant difference from 1.56 dynes/cm2 at P<0.05. Values sharing the same superscript are not significantly different, P<0.05.
To explore additional mechanisms whereby loss of Kif3a leads to alterations in osteoblast functions, we examined other signaling molecules associated with primary cilia, including the patched (Ptch1)–smoothened (Smo)–hedgehog (Hh) and Wnt–β-catenin pathways. Using total RNA from Kif3aOc-cKO null tibias and cultured osteoblasts, we found that expression of Gli2, a downstream gene of Hh signaling, was significantly lower in Kif3aOc-cKO-derived bone samples compared with Kif3aflox/+ controls (Fig. 6A). Addition of sonic hedgehog (Shh; 1 μg/ml) to immortalized osteoblast cultures also resulted in significant (P<0.05) increases in Gli-responsive-promoter luciferase activity, Gli2 mRNA expression and Gli2 protein levels in Kif3aflox/+ control cells (Fig. 6B–D). By contrast, Shh failed to stimulate any of these parameters in the Kif3aOc-cKO null osteoblasts (Fig. 6B–D), suggesting that either loss of Kif3a and/or disruption of primary cilium function impairs Hh signaling in osteoblasts.
Fig. 6.

Effects of global and/or Oc-Cre-mediated conditional deletion of Kif3a on Hh and Wnt signaling in bone and osteoblasts. (A–D) Hh–Gli2 pathway. Both tibias and cultured osteoblasts exhibited significant downregulation of Gli2 mRNA in the Kif3aOc-cKO group compared with Kif3aflox/+ controls. An administration of Shh (1 μg/ml) resulted in significant increases in 8xGli-responsive luciferase (p8xGli-Luc) activity and mRNA and protein levels of Gli2 in Kif3aflox/+ control cells, whereas less or no stimulation with Shh was observed in the Kif3aOc-cKO null osteoblasts. (D–H) Wnt–β-catenin pathway. Both tibias and cultured osteoblasts showed significantly lower expression of Axin2 mRNA in the Kif3aOc-cKO group compared with Kif3aflox/+controls. Wnt3a-induced Super 8xTOPFlash luciferase (p8xTOPFlash-Luc) activity, the level of cytoplasmic β-catenin protein and Axin2 mRNA expression were much higher in the control cells than in the Kif3aOc-cKO null osteoblasts. Values are means ± s.d. from five to six individual mice in three independent experiments. *Significant difference from control (Kif3aflox/+; P<0.05). Values with the same superscript are not significantly different (P<0.05).
To examine the effect of conditional deletion of Kif3a we examined the expression of Axin2, a direct downstream gene of the Wnt–β-catenin pathway, in bone and immortalized osteoblasts. We found that Axin2 was significantly (P<0.05) lower in Kif3aOc-cKO null bone and osteoblasts compared with Kif3aflox/+controls (Fig. 6E). Wnt3a–β-catenin transcriptional activity was also lower, by approximately 35%, in Kif3aOc-cKO null osteoblasts. In this regard, Wnt3a induced a respective 45- and 12-fold increase in Super 8xTOPFlash promoter luciferase activity and Axin2 expression in control osteoblasts (Fig. 6F,G), but only a 28- and 8-fold increase in these parameters in Kif3aOc-cKO null osteoblasts (Fig. 6F,G). In agreement with the changes of Wnt–β-catenin downstream signaling described above, Wnt3a-induced accumulation of cytoplasmic β-catenin protein was lower in Kif3aOc-cKO null osteoblasts compared with controls (Fig. 6H), indicating that conditional deletion of Kif3a significantly attenuates Wnt–β-catenin signaling in osteoblasts.
Discussion
Our studies show that the selective deletion of Kif3a, a subunit of kinesin II, an anterograde ciliary motor protein, from osteoblasts in mice causes defects in osteoblast-mediated bone formation and abnormalities of multiple cilia-related signaling pathways, resulting in osteopenia. We found that Kif3aOc-cKO mice had a 75% less Kif3a transcripts in bone. Loss of Kif3a resulted in reductions in both the number and length of primary cilia in osteoblasts, consistent with the known role of Kif3a in primary cilia formation and function (Lin et al., 2003; Marszalek et al., 1999). Adult Kif3aOc-cKO null mice exhibited lower bone mineral density, trabecular bone volume, cortical thickness, bone formation rate and osteoblast-related gene expression, and had impaired bone mechanical properties. Because Oc-Cre directs expression of the recombinase to postnatal osteoblasts, bone abnormalities in Kif3aOc-cKO mice reflect the function of Kif3a in differentiated osteoblasts. A direct effect of Kif3a on postnatal osteoblast function was also demonstrated in ex vivo experiments in primary osteoblast cultures. These osteoblasts displayed impairment of intracellular calcium signaling, Shh-induced upregulation of Hh and Gli2 signaling, and Wnt–β-catenin and Axin2 activation in osteoblasts.
There are several potential mechanisms whereby disruption of Kif3a in mature osteoblasts leads to abnormal function in the adult mouse. Because the C-terminal region of Kif3a binds to the C-terminus of PC2 and mediates polycystin complex colocalization to primary cilia (Li et al., 2006), the effects of Kif3a on osteoblast function might be mediated through polycystins. This possibility is supported by several observations. First, Kif3a and the polycystins, Pkd1 and Pkd2, are known to be functionally interrelated in other organs, such as the kidney, where loss of Kif3a or polycystins cause a common phenotype (i.e. polycystic kidney disease) (Hou et al., 2002; Lin et al., 2003; Pazour et al., 2002; Ward et al., 2003; Yoder et al., 2002a; Yoder et al., 2002b). Second, conditional Kif3aOc-cKO null mice exhibit similar bone phenotypes to conditional Pkd1Oc-cKO and Pkd1Dmp1-cKO-null mice (Xiao et al., 2011; Xiao, Z. et al., 2010), consistent with the possibility that Kif3a regulates osteoblast function through its mediation of the localization of polycystins to primary cilia in osteoblasts and osteocytes in bone (Xiao et al., 2008; Xiao et al., 2006). Third, both immortalized conditional Kif3aOc-cKO- and Pkd1Dmp1-cKO-null primary osteoblasts exhibit increased cell proliferation and impaired osteoblast gene expression (Qiu et al., 2010; Xiao et al., 2011; Xiao, Y. et al., 2010; Xiao, Z. et al., 2010). Fourth, Kif3aOc-cKO- and Pkd1Dmp1-cKO-null primary osteoblasts exhibit similar defects in flow-induced changes in intracellular calcium, consistent with the fact that mutations in Pkd1 and Pkd2 result in impaired intracellular calcium response to fluid flow in renal epithelial cells (Nauli et al., 2003; Nauli et al., 2006). Finally, Cox2 expression, Shh-induced Gli2 expression and Wnt3a-stimulated Axin2 transcription are abnormal in both Kif3aOc-cKO and Pkd1Dmp1-cKO mice (Qiu et al., 2010; Xiao et al., 2011; Xiao, Z. et al., 2010).
Our studies do not assess the role of Kif3a in the early stage of the osteoblast lineage or in skeletal development. Indeed, we found no evidence for abnormalities in skeletogenesis, because Oc-Cre is expressed in mature osteoblasts at E18.5, after completion of bone development. Additional studies in which Kif3a is ablated earlier in the osteoblast lineage, during embryogenesis, will be needed to establish a role of Kif3a in osteoblast development and skeletogenesis. Although global Kif3a-null mice die before skeletal development, global deletion of Pkd1 and Pkd2 results in severe abnormalities of skeletogenesis, probably because of their effect on primary cilia and polycytins in the bone development process. However, in this study, we focused on the function of Kif3a in mature osteoblasts, and we cannot exclude the possibility that disruption of Kif3a through polycystin signaling might affect the differentiation state of osteoblast as previously reported in renal epithelia cells (Rankin et al., 1992). Our current results show that lower osteoblast-specific gene expression concomitant with higher adipocyte-specific gene expression in bones of 6-week-old mice is similar to the data from the osteoblast cultures in vitro, and probably reflects abnormalities in the bone remodeling process and alterations in marrow fat during the process of postnatal bone generation, which recapitulates aspects of embryonic bone development (Griffith et al., 2009).
Other signaling pathways that are known to regulate osteoblast function, including Hh–Gli (Han et al., 2008; Huangfu and Anderson, 2005; Kovacs et al., 2008; Serra, 2008; Veland et al., 2009; Wong et al., 2009) and Wnt–β-catenin (Christensen et al., 2007; Corbit et al., 2008; Gerdes et al., 2009) are also present in primary cilia. We found that expression of Gli2, a downstream gene of Hh signaling, was significantly decreased in bone and osteoblasts from conditional Kif3aOc-cKO null mice compared with Kif3aflox/+ controls. We also found that Shh-induced expression of Gli2 mRNA and protein were markedly attenuated in osteoblasts from conditional Kif3aOc-cKO null mice. With regards to Wnt signaling, we observed that the expression of Axin2, a direct downstream gene of the Wnt–β-catenin signaling, was significantly lower in bone and osteoblasts from conditional Kif3aOc-cKO null mice compared with Kif3aflox/+ controls. In addition, Wnt3a-induced accumulation of cytoplasmic β-catenin protein and β-catenin transcriptional activity were significantly attenuated in osteoblasts from conditional Kif3aOc-cKO null mice. These findings suggest that the skeletal phenotype in Kif3aOc-cKO mice results from abnormalities in multiple cilia-related pathways including Pkd1, Pkd2, Ca2+, hedgehog and Wnt signaling. Our studies do not separate specific functions of Kif3a from more global effects of primary cilium dysfunction caused by loss of Kif3a (Praetorius and Spring, 2001; Praetorius and Spring, 2003; Yoder, 2007). Comparative analysis of the bone phenotype in additional mouse genetic models in which ciliogenesis is selectively disrupted by targeting genes other than Kif3a, such as Ift88, as well as investigations of specific downstream signaling pathways, will be necessary to determine the precise mechanisms whereby Kif3a regulates osteoblast functions.
Because Oc-Cre-mediated deletion of Kif3a in osteoblasts affects the terminally differentiated osteocytes, we cannot exclude a possible role of the osteocyte in the bone phenotype of Kif3aOc-cKO null mice. There is growing support for a central role of osteocytes in bone mechanosensing (Bonewald and Johnson, 2008; Galli et al., 2010; Rochefort et al., 2010; Santos et al., 2009; Tatsumi et al., 2007). Using a 6.24 dynes/cm2 FFSS regime, we observed that global and/or conditional deletion of Kif3a produced a gene-dose effect on flow-induced intracellular calcium response in immortalized Kif3a-deficient osteoblasts, consistent with reports by others of cilia-dependent calcium signaling in renal epithelial cells (Nauli et al., 2003; Nauli et al., 2006). Our findings differ from those of Malone et al. who found that disruption of primary cilia in osteoblasts and osteocytes impairs mechanosensing through mechanisms independent of Ca2+ flux in a high flow shear stress (12 dynes/cm2) (Malone et al., 2007). The reason for these different results is not clear. Mechanical loading experiments in vivo, are needed to establish the mechanosensing function of Kif3a in osteoblasts and osteocytes.
In conclusion, the disruption of Kif3a in postnatal osteoblasts either directly, through Kif3a regulation of specific signaling pathways, or indirectly, through the broader effects of Kif3a through disruption of primary cilium formation and/or function, results in osteopenia. Disruption of Kif3a ex vivo affects cilia-related pathways, such as intracellular calcium, Hedgehog and Wnt signaling.
Materials and Methods
Mice
We obtained the floxed Kif3a mice from Lawrence S. B. Goldstein at the University of California San Diego (Marszalek et al., 1999) and osteocalcin (Oc)-Cre mice from Thomas Clemens at the University of Alabama (Zhang et al., 2002). The Kif3anull/+ heterozygous mice were from our laboratory stock, as previously described (Qiu et al., 2010). These mice were bred and maintained on a C57BL/6J background. Because Cre-recombinase-mediated deletion of a single flox/null allele reduces the risk of mosaicism that could occur because of the less than 100% efficiency of Cre recombinase to excise two floxed alleles (flox/flox) (Kwan, 2002), we created double heterozygous Oc-Cre;Kif3anull/+ mice and homozygous Kif3aflox/flox mice. Double heterozygous Oc-Cre;Kif3anull/+ mice were mated with homozygous Kif3aflox/flox mice to generate excised floxed Kif3a heterozygous (Oc-Cre;Kif3aflox/+) and null mice (Oc-Cre;Kif3aflox/null or Kif3aOc-cKO), as well as Kif3a heterozygous mice (Kif3anull/flox) and Oc-Cre-negative control mice (Kif3aflox/+, equivalent to wild type). These last four genotypes were used for skeletal phenotype analysis and primary osteoblast cultures. All animal research was conducted according to guidelines provided by the National Institutes of Health and the Institute of Laboratory Animal Resources, National Research Council. The University of Tennessee Health Science Center's Animal Care and Use Committee approved all animal studies (Protocol number 1885 and 1889).
Genotyping polymerase chain reaction (PCR) to detect bone-specific deletions
Genomic DNA was prepared from tail and other tissue specimens using standard procedures (Xiao et al., 2005). PCR genotyping was performed using the following primers (Fig. 1A) (Lin et al., 2003; Qiu et al., 2010): Kif3a wild-type (Kif3a+) and floxed (Kif3aflox) alleles, F1, 5′-AGGGCAGACGGAAGGGTGG-3′, R1, 5′-TCTGTGAGTTTGTGACCAGCC-3′; Kif3a null (Kif3anull) and conditional null (Kif3aΔflox) alleles, F1, 5′-AGGGCAGACGGAAGGGTGG-3′, R2, 5′-TGGCAGGTCAATGGACGCAG-3′. The Kif3a floxed (Kif3aflox), wild-type (Kif3a+) and Kif3a null (or conditional null; Kif3anull or Kif3aΔflox) alleles were identified, using 2% agarose gels as 490 bp, 360 bp and 200 bp bands, respectively (Fig. 1B) (Lin et al., 2003; Qiu et al., 2010).
Bone densitometry, histomorphometric and microcomputed tomography analyses
BMD of femurs was assessed at 6 and 24 weeks of age with a LUNARPIXIMUS bone densitometer (Lunar Corp., Madison, WI). Calcein (Sigma, St. Louis, MO) double labeling of bone and histomorphometric analyses of periosteal MAR in tibias were performed using the osteomeasure analysis system (OsteoMetrics, Decatur, GA) (Glass et al., 2005; Xiao et al., 2005). The distal femoral metaphyses were also scanned with a Scanco μCT 40 (Scanco Medical AG, Brüttisellen, Switzerland). Three-dimensional images were analyzed to determine bone volume, trabecular volume and cortical thickness as previously described (Xiao et al., 2005).
Bone geometry and mechanical property testing
For each femur, μCT data (1520 slices per femur) were imported into the imaging software Amira (Pro Medicus Limited, Richmond, Australia), and saved in a DICOM file format. Measurements of the inner and outer diameters in both the x and y directions were taken using a DICOM viewer (Santa DICOM Viewer FREE, Santesoft, Athens, Greece). To increase the accuracy of the measurements, a threshold on the luminosity was applied with a lower limit of 1000 and an upper limit of 1500. For each specimen, three slices were measured and the averages were used for the calculations. From these data, the moment area of inertia (I) was calculated using the formula 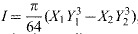 , where X1 and Y1 are the outer diameters and X2 and Y2 are the inner diameters. After I was calculated, it was combined with the force–displacement data from the three-point bending tests to calculate the apparent elastic modulus, Eapp, using the following expression:
, where X1 and Y1 are the outer diameters and X2 and Y2 are the inner diameters. After I was calculated, it was combined with the force–displacement data from the three-point bending tests to calculate the apparent elastic modulus, Eapp, using the following expression: 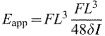 , where F is the applied force, L is the span and δ is the deflection.
, where F is the applied force, L is the span and δ is the deflection.
Eight femurs from 6-week-old male mice and eight femurs from 24-week-old male were used for the three-point bend testing. Femurs were stored at −20°C prior to data acquisition from the three-point bending experiments. On the day of testing, the bones were thawed on ice and rehydrated with 1× phosphate-buffered saline (PBS) at room temperature for 5 minutes before testing. The distance between the supports was held constant for all femurs, at 6 mm, and the radius of supports was 0.5 mm. The femurs were tested using an Instron 33R (Instron, Norwood, MA) at a rate of 2 mm/second to a 40% decline in maximum load. Load magnitude and displacement data were collected using Bluehill® Materials Testing Software (Instron).
Biochemistry
Serum osteocalcin levels were measured a mouse Osteocalcin EIA (Biomedical Technologies, Inc., Stoughton, MA). Serum blood urea nitrogen (BUN) was determined using a BUN diagnostic kit from Pointe Scientific, Inc. (Canton, MI). Serum calcium was measured by the colorimetric cresolphthalein binding method, and phosphorus was measured by the phosphomolybdate–ascorbic acid method (Stanbio Laboratory, Boerne, TX). Serum OPG and RankL were measured using mouse ELISA kits (Quantikine®, R&D Systems, Minneapolis, MN), and serum TRAP was assayed with the ELISA-based SBA Sciences mouse TRAP™ assay (Immunodiagnostic Systems, Fountain Hills, AZ).
Bone RNA isolation and real-time RT-PCR
For quantitative real-time RT-PCR, 1.0 μg total RNA isolated from whole tibias of 6-week-old control and Kif3a-deficient mice was reverse transcribed as previously described (Xiao et al., 2004). PCR reactions contained 20 ng template (cDNA or RNA), 375 nM each forward and reverse primers and 1× SsoFast™ EvaGreen® supermix (Bio-Rad, Hercules, CA) in a total of 10 μl reaction volume. The threshold cycle (Ct) of tested gene product from the indicated genotype was normalized to the Ct for cyclophilin A. Expression of total Kif3a transcripts was performed using the following Kif3a-allele-specific primers in exon 2: forward primer of normal Kif3a+ transcript (Kif3a+ plus Kif3aflox): 5′-GCTATAGACAGGCCGTCAGC-3′ and reverse primer: 5′-GTCTTTGGAGGTTCGTTGGA-3′. The normal Kif3a+ versus cyclophilin A was normalized to the mean ratio of five control mice, which was set to 1. The percentage of Kif3a null (Kif3anull) and/or conditional deleted (Kif3aΔflox) transcripts was calculated from the relative levels of the normal Kif3a+ transcripts in different Kif3a-deficient mice (Xiao, Z. et al., 2010). All primer information of other genes used in real-time RT-PCR can be found in our previous report (Xiao et al., 2011).
Cell proliferation, osteoblastic differentiation and gene expression profiles in immortalized osteoblast cultures
Calvaria from E17.5 control and Kif3a-deficient embryos were used to isolate primary osteoblasts by sequential collagenase digestion at 37°C. To engineer immortal osteoblast cell lines, isolated primary osteoblasts were infected using a retroviral vector carrying SV40 large and small T antigen as previously described (Borton et al., 2001; Xiao et al., 2004). Briefly, cells were grown in 100-mm plates at 50–60% confluency the day before infection. On the day of infection, the medium was removed and medium containing SV40 large and small T antigen-helper-free viral supernatant was added in the presence of 4 mg/ml polybrene (Sigma, St. Louis, MO) for 48 hours. The cells were allowed to recover for 72 hours followed by selection with 1 mg/ml puromycin (Sigma) for up to 15 days. The immortalized osteoblasts were cultured in α-MEM containing 10% FBS and 1% penicillin and streptomycin (P/S) and characterized following the protocols below. Cell proliferation was detected by BrdU incorporation assays following the manufacturer's directions (QIA58, Calbiochem, Gibbstown, NJ). To induce differentiation, the immortalized osteoblasts were plated at a density of 2×104 cells per well in a 12-well plate and 4×04 cells per well in a 6-well plate and grown for 21 days in α-MEM containing 10% FBS supplemented with 5 mM β-glycerophosphate and 25 μg/ml ascorbic acid. ALP activity and Alizarin Red-S histochemical staining for mineralization were performed as previously described (Xiao et al., 2006; Xiao et al., 2004). Total DNA content was measured with a PicoGreen® dsDNA quantitation kit (Molecular Probes, Eugene, OR). Protein concentrations of the supernatant were determined with a Bio-Rad protein assay kit. For gene expression profiles, 1.0 μg total RNA were isolated from primary osteoblasts cultured for 3, 12 and 18 days in differentiation medium. The cDNAs were generated using an iScript reverse transcription kit (Bio-Rad). PCR reactions contained 20 ng template (cRNA or cDNA), 375 nM each forward and reverse primer, 1× SsoFast EvaGreen® supermix (Bio-Rad), in a total of 10 μl reaction volume. The Ct of tested gene product from the indicated genotype was normalized to the Ct for cyclophilin A as previously described (Xiao et al., 2008; Xiao et al., 2006; Xiao et al., 2004).
Immunofluorescence
The immortalized control and Kif3a-deficient osteoblasts were grown on collagen-coated 4-well chambers at 1×105 cells per well and kept at confluence for at least 3 days. The end of the culture, the cells were washed three times with PBS, fixed with cold 4% paraformaldehyde, 0.2% Triton X-100 for 10 minutes at room temperature, and washed with PBS three times. The cells were incubated for 30 minutes in 1% BSA before incubation with primary acetylated α-tubulin antibody (1:4000; T6793, Sigma) for 1 hour at room temperature. After washing three times in PBS, cells were treated with secondary Texas-Red-labeled anti-mouse IgG (1:400; 715-076-150, Jackson ImmunoResearch, West Grove, PA) in 1% BSA for 1 hour at room temperature and washed three times in PBS before mounting with ProLong® Gold antifade reagent (P36935, Invitrogen). Nuclei were counterstained with DAPI blue. Photographs were taken under a microscope with magnification of 40× and 100× to count the number and measuring the length of primary cilia, respectively, in these osteoblasts as previously described (Xiao et al., 2006).
Intracellular calcium measurements in vitro
We measured basal intracellular calcium ([Ca2+]i) concentration and flow-induced intracellular calcium response in immortalized control and Kif3a-deficient osteoblasts as previously described (Xiao et al., 2011). Briefly, the immortalized cells were cultured on type I rat tail collagen-coated 40-mm diameter glass slides at 80–90% confluency in α-MEM containing 2% FBS and 1% P/S for 3 days. The cells were loaded with 3 μM Fura2-AM (Molecular Probes, Eugene, OR), a fluorescent Ca2+ probe, in Hank's balanced salt solution (HBSS) that contained 2% FBS and 20 mM HEPES for 30 minutes at 37°C. Loaded cells were incubated for an additional 45 minutes with HBSS alone to ensure complete de-esterification of the fluorescent molecule. A glass slide was then placed in an FCS2 parallel plate flow chamber (Bioptechs, Inc., Butler, PA), 0.25×14×22 mm. A fresh bolus of flow medium was added to the chamber and the cells were left undisturbed for 30 minutes. The flow medium consisted of phenol-free α-MEM and 2% FBS equilibrated with 5% CO2, 95% air at 37°C. The chamber was mounted on the stage of an inverted microscope with a CCD camera to allow real-time recording of fluorescence intensity (F340/F380 ratio) to generate ratiometric video images of individual static cells or cells exposed to pulsatile laminar fluid flow (Intracellular Imaging, Inc., Cincinnati, OH). To obtain the optimal FFSS to induce response of intracellular calcium in individual osteoblasts, the immortalized control cells were exposed to various pulsatile laminar fluid flow rates resulting in shear stresses of 0.69, 1.56, 6.24 and 9.5 dynes/cm2. To assess mechanoresponsive gene expression, total RNA was harvested and the cells subjected to FFSS (6.24 dynes/cm2) for 30 minutes and then returned to static culture for 30 minutes (post-FFSS), which was based on previous studies in osteoblasts (Mehrotra et al., 2006).
Transient transfection and western blot analysis
The immortalized control and conditional Kif3a null osteoblasts were cultured in α-MEM containing 10% FBS and 1% P/S. To examine whether conditional deletion of Kif3a (disruption of ciliogenesis) has an impact on Hh signaling in osteoblasts, 1.5×106 cells were transfected with 3.0 μg Gli-responsive luciferase reporter construct (8xGli-Luc) (Zhao et al., 2009), 3.0 μg pcDNA3.1 empty vector and 0.6 μg Renilla luciferase-null (RL-null) as an internal control plasmid by electroporation using a Cell Line Nucleofector Kit R according to the manufacturer's protocol (Amaxa, Inc., Gaithersburg, MD). The cells were cultured in α-MEM supplemented with 1% FBS and the relative luciferase activity of cell lysates was measured using a luciferase assay kit (Promega, Madison, WI) 72 hours after transfection in the presence or absence of 1 μg/ml of recombinant mouse Shh N-terminus (Shh-N) for the last 8 hours (Qiu et al., 2010; Xiao, Z. et al., 2010). Total RNA was also isolated for real-time RT-PCR analysis.
To explore potential abnormalities of the Wnt pathway in conditional Kif3a null mice, control (Kif3aflox/+) and conditional Kif3a null (Kif3aOc-cko) osteoblasts were transiently cotransfected with 3.0 μg Super 8xTOPFlash luciferase reporter plasmid (8xTOPFlash-Luc), 3.0 μg pcDNA3.1 empty vector or 0.6 μg Renilla luciferase-null (RL-null; Promega) as an internal control, by electroporation. Promoter activity was assessed by measuring luciferase activity 48 hours after transfection in the presence or absence of 100 ng/ml recombinant Wnt3a for the last 8 hours (Qiu et al., 2010; Xiao, Z. et al., 2010). Total RNA was also isolated for real-time RT-PCR analysis.
To examine the amounts of cytoplasmic Cox-2, Gli-2 and β-catenin, the cells were prepared using 1× passive lysis buffer for 30 minutes at 4°C (Promega) and centrifuged at 100,000 g for 45 minutes at 4°C. Protein concentrations of the supernatant were determined with a Bio-Rad protein assay kit. Equal quantities of protein were applied to a NuPAGE™ 4–12% Bis-Tris gel (Invitrogen, Carlsbad, CA) and analyzed with standard western blot protocols (HRP-conjugated secondary antibodies from Santa Cruz Biotechnology, CA, and ECL from Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Antibody against Cox2 (4842S) was from Cell Signaling Technology (Beverly, MA). Antibody against Gli2 (ab7195, 80 kDa) was purchased from Abcam (San Francisco, CA). Anti-β-catenin (sc-7199) and anti-β-actin (sc-47778) antibodies were from Santa Cruz Biotechnology.
Statistical analysis
We evaluated differences between two groups using unpaired t-tests and multiple groups using one-way analysis of variance. All values are expressed as means ± s.d. All computations were performed using GraphPad Prism5 (GraphPad Software Inc., La Jolla, CA, USA).
Footnotes
Funding
This work was supported by the National Institutes of Health [grant numbers R21-AR056794 to Z.S.X. and R01-DK083303 to L.D.Q.]. Deposited in PMC for release after 12 months.
References
- Berbari N. F., O'Connor A. K., Haycraft C. J., Yoder B. K. (2009). The primary cilium as a complex signaling center. Curr. Biol. 19, R526-R535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonewald L. F., Johnson M. L. (2008). Osteocytes, mechanosensing and Wnt signaling. Bone 42, 606-615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borton A. J., Frederick J. P., Datto M. B., Wang X. F., Weinstein R. S. (2001). The loss of Smad3 results in a lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J. Bone Miner. Res. 16, 1754-1764 [DOI] [PubMed] [Google Scholar]
- Christensen S. T., Pedersen L. B., Schneider L., Satir P. (2007). Sensory cilia and integration of signal transduction in human health and disease. Traffic 8, 97-109 [DOI] [PubMed] [Google Scholar]
- Corbit K. C., Shyer A. E., Dowdle W. E., Gaulden J., Singla V., Chen M. H., Chuang P. T., Reiter J. F. (2008). Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 10, 70-76 [DOI] [PubMed] [Google Scholar]
- Davis E. E., Brueckner M., Katsanis N. (2006). The emerging complexity of the vertebrate cilium: new functional roles for an ancient organelle. Dev. Cell 11, 9-19 [DOI] [PubMed] [Google Scholar]
- Galli C., Passeri G., Macaluso G. M. (2010). Osteocytes and WNT: the mechanical control of bone formation. J. Dent. Res. 89, 331-343 [DOI] [PubMed] [Google Scholar]
- Gerdes J. M., Davis E. E., Katsanis N. (2009). The vertebrate primary cilium in development, homeostasis, and disease. Cell 137, 32-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass D. A., 2nd, Bialek P., Ahn J. D., Starbuck M., Patel M. S., Clevers H., Taketo M. M., Long F., McMahon A. P., Lang R. A., et al. (2005). Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8, 751-764 [DOI] [PubMed] [Google Scholar]
- Goetz S. C., Anderson K. V. (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith J. F., Yeung D. K., Ahuja A. T., Choy C. W., Mei W. Y., Lam S. S., Lam T. P., Chen Z. Y., Leung P. C. (2009). A study of bone marrow and subcutaneous fatty acid composition in subjects of varying bone mineral density. Bone 44, 1092-1096 [DOI] [PubMed] [Google Scholar]
- Han Y. G., Spassky N., Romaguera-Ros M., Garcia-Verdugo J. M., Aguilar A., Schneider-Maunoury S., Alvarez-Buylla A. (2008). Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci. 11, 277-284 [DOI] [PubMed] [Google Scholar]
- Haycraft C. J., Serra R. (2008). Cilia involvement in patterning and maintenance of the skeleton. Curr. Top. Dev. Biol. 85, 303-332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft C. J., Zhang Q., Song B., Jackson W. S., Detloff P. J., Serra R., Yoder B. K. (2007). Intraflagellar transport is essential for endochondral bone formation. Development 134, 307-316 [DOI] [PubMed] [Google Scholar]
- Hou X., Mrug M., Yoder B. K., Lefkowitz E. J., Kremmidiotis G., D'Eustachio P., Beier D. R., Guay-Woodford L. M. (2002). Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J. Clin. Invest. 109, 533-540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D., Anderson K. V. (2005). Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA 102, 11325-11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpakova-Hart E., Jinnin M., Hou B., Fukai N., Olsen B. R. (2007). Kinesin-2 controls development and patterning of the vertebrate skeleton by Hedgehog- and Gli3-dependent mechanisms. Dev. Biol. 309, 273-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs J. J., Whalen E. J., Liu R., Xiao K., Kim J., Chen M., Wang J., Chen W., Lefkowitz R. J. (2008). Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 320, 1777-1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama E., Young B., Nagayama M., Shibukawa Y., Enomoto-Iwamoto M., Iwamoto M., Maeda Y., Lanske B., Song B., Serra R., et al. (2007). Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 134, 2159-2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan K. M. (2002). Conditional alleles in mice: practical considerations for tissue-specific knockouts. Genesis 32, 49-62 [DOI] [PubMed] [Google Scholar]
- Li Q., Montalbetti N., Wu Y., Ramos A., Raychowdhury M. K., Chen X. Z., Cantiello H. F. (2006). Polycystin-2 cation channel function is under the control of microtubular structures in primary cilia of renal epithelial cells. J. Biol. Chem. 281, 37566-37575 [DOI] [PubMed] [Google Scholar]
- Lin F., Hiesberger T., Cordes K., Sinclair A. M., Goldstein L. S., Somlo S., Igarashi P. (2003). Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. USA 100, 5286-5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y., Nakamura E., Nguyen M. T., Suva L. J., Swain F. L., Razzaque M. S., Mackem S., Lanske B. (2007). Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. USA 104, 6382-6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone A. M., Anderson C. T., Tummala P., Kwon R. Y., Johnston T. R., Stearns T., Jacobs C. R. (2007). Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proc. Natl. Acad. Sci. USA 104, 13325-13330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek J. R., Ruiz-Lozano P., Roberts E., Chien K. R., Goldstein L. S. (1999). Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc. Natl. Acad. Sci. USA 96, 5043-5048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlashan S. R., Haycraft C. J., Jensen C. G., Yoder B. K., Poole C. A. (2007). Articular cartilage and growth plate defects are associated with chondrocyte cytoskeletal abnormalities in Tg737orpk mice lacking the primary cilia protein polaris. Matrix Biol. 26, 234-246 [DOI] [PubMed] [Google Scholar]
- Mehrotra M., Saegusa M., Voznesensky O., Pilbeam C. (2006). Role of Cbfa1/Runx2 in the fluid shear stress induction of COX-2 in osteoblasts. Biochem. Biophys. Res. Commun. 341, 1225-1230 [DOI] [PubMed] [Google Scholar]
- Murcia N. S., Richards W. G., Yoder B. K., Mucenski M. L., Dunlap J. R., Woychik R. P. (2000). The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development 127, 2347-2355 [DOI] [PubMed] [Google Scholar]
- Nauli S. M., Alenghat F. J., Luo Y., Williams E., Vassilev P., Li X., Elia A. E., Lu W., Brown E. M., Quinn S. J., et al. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33, 129-137 [DOI] [PubMed] [Google Scholar]
- Nauli S. M., Rossetti S., Kolb R. J., Alenghat F. J., Consugar M. B., Harris P. C., Ingber D. E., Loghman-Adham M., Zhou J. (2006). Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J. Am. Soc. Nephrol. 17, 1015-1025 [DOI] [PubMed] [Google Scholar]
- Pazour G. J., San Agustin J. T., Follit J. A., Rosenbaum J. L., Witman G. B. (2002). Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr. Biol. 12, R378-R380 [DOI] [PubMed] [Google Scholar]
- Pedersen L. B., Veland I. R., Schrøder J. M., Christensen S. T. (2008). Assembly of primary cilia. Dev. Dyn. 237, 1993-2006 [DOI] [PubMed] [Google Scholar]
- Praetorius H. A., Spring K. R. (2001). Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol. 184, 71-79 [DOI] [PubMed] [Google Scholar]
- Praetorius H. A., Spring K. R. (2003). Removal of the MDCK cell primary cilium abolishes flow sensing. J. Membr. Biol. 191, 69-76 [DOI] [PubMed] [Google Scholar]
- Praetorius H. A., Frokiaer J., Nielsen S., Spring K. R. (2003). Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J. Membr. Biol. 191, 193-200 [DOI] [PubMed] [Google Scholar]
- Qiu N., Cao L., David V., Quarles L. D., Xiao Z. (2010). Kif3a deficiency reverses the skeletal abnormalities in Pkd1 deficient mice by restoring the balance between osteogenesis and adipogenesis. PLoS ONE 5, e15240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin C. A., Grantham J. J., Calvet J. P. (1992). C-fos expression is hypersensitive to serum-stimulation in cultured cystic kidney cells from the C57BL/6J-cpk mouse. J. Cell. Physiol. 152, 578-586 [DOI] [PubMed] [Google Scholar]
- Rochefort G. Y., Pallu S., Benhamou C. L. (2010). Osteocyte: the unrecognized side of bone tissue. Osteoporos. Int. 21, 1457-1469 [DOI] [PubMed] [Google Scholar]
- Santos A., Bakker A. D., Klein-Nulend J. (2009). The role of osteocytes in bone mechanotransduction. Osteoporos. Int. 20, 1027-1031 [DOI] [PubMed] [Google Scholar]
- Serra R. (2008). Role of intraflagellar transport and primary cilia in skeletal development. Anat. Rec. (Hoboken) 291, 1049-1061 [DOI] [PubMed] [Google Scholar]
- Shiba D., Takamatsu T., Yokoyama T. (2005). Primary cilia of inv/inv mouse renal epithelial cells sense physiological fluid flow: bending of primary cilia and Ca2+ influx. Cell Struct. Funct. 30, 93-100 [DOI] [PubMed] [Google Scholar]
- Song B., Haycraft C. J., Seo H. S., Yoder B. K., Serra R. (2007). Development of the post-natal growth plate requires intraflagellar transport proteins. Dev. Biol. 305, 202-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B., Hammerschmidt M., McMahon A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072-2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi S., Ishii K., Amizuka N., Li M. Q., Kobayashi T., Kohno K., Ito M., Takeshita S., Ikeda K. (2007). Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 5, 464-475 [DOI] [PubMed] [Google Scholar]
- Veland I. R., Awan A., Pedersen L. B., Yoder B. K., Christensen S. T. (2009). Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 111, 39-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C. J., Yuan D., Masyuk T. V., Wang X., Punyashthiti R., Whelan S., Bacallao R., Torra R., LaRusso N. F., Torres V. E., et al. (2003). Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 12, 2703-2710 [DOI] [PubMed] [Google Scholar]
- Wong S. Y., Seol A. D., So P. L., Ermilov A. N., Bichakjian C. K., Epstein E. H., Jr, Dlugosz A. A., Reiter J. F. (2009). Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 15, 1055-1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Dai X. Q., Li Q., Chen C. X., Mai W., Hussain Z., Long W., Montalbetti N., Li G., Glynne R., et al. (2006). Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum. Mol. Genet. 15, 3280-3292 [DOI] [PubMed] [Google Scholar]
- Xiao Y., Lv X., Cao G., Bian G., Duan J., Ai J., Sun H., Li Q., Yang Q., Chen T., et al. (2010). Overexpression of Trpp5 contributes to cell proliferation and apoptosis probably through involving calcium homeostasis. Mol. Cell. Biochem. 339, 155-161 [DOI] [PubMed] [Google Scholar]
- Xiao Z., Awad H. A., Liu S., Mahlios J., Zhang S., Guilak F., Mayo M. S., Quarles L. D. (2005). Selective Runx2-II deficiency leads to low-turnover osteopenia in adult mice. Dev. Biol. 283, 345-356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z., Zhang S., Mahlios J., Zhou G., Magenheimer B. S., Guo D., Dallas S. L., Maser R., Calvet J. P., Bonewald L., et al. (2006). Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. J. Biol. Chem. 281, 30884-30895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z., Zhang S., Magenheimer B. S., Luo J., Quarles L. D. (2008). Polycystin-1 regulates skeletogenesis through stimulation of the osteoblast-specific transcription factor RUNX2-II. J. Biol. Chem. 283, 12624-12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z., Zhang S., Cao L., Qiu N., David V., Quarles L. D. (2010). Conditional disruption of Pkd1 in osteoblasts results in osteopenia due to direct impairment of bone formation. J. Biol. Chem. 285, 1177-1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z., Dallas M., Qiu N., Nicolella D., Cao L., Johnson M., Bonewald L., Quarles L. D. (2011). Conditional deletion of Pkd1 in osteocytes disrupts skeletal mechanosensing in mice. FASEB J. 25, 2418-2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z. S., Hjelmeland A. B., Quarles L. D. (2004). Selective deficiency of the “bone-related” Runx2-II unexpectedly preserves osteoblast-mediated skeletogenesis. J. Biol. Chem. 279, 20307-20313 [DOI] [PubMed] [Google Scholar]
- Yoder B. K. (2007). Role of primary cilia in the pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 18, 1381-1388 [DOI] [PubMed] [Google Scholar]
- Yoder B. K., Hou X., Guay-Woodford L. M. (2002a). The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 13, 2508-2516 [DOI] [PubMed] [Google Scholar]
- Yoder B. K., Tousson A., Millican L., Wu J. H., Bugg C. E., Jr, Schafer J. A., Balkovetz D. F. (2002b). Polaris, a protein disrupted in orpk mutant mice, is required for assembly of renal cilium. Am. J. Physiol. Renal Physiol. 282, F541-F552 [DOI] [PubMed] [Google Scholar]
- Zhang M., Xuan S., Bouxsein M. L., von Stechow D., Akeno N., Faugere M. C., Malluche H., Zhao G., Rosen C. J., Efstratiadis A., et al. (2002). Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J. Biol. Chem. 277, 44005-44012 [DOI] [PubMed] [Google Scholar]
- Zhang Q., Murcia N. S., Chittenden L. R., Richards W. G., Michaud E. J., Woychik R. P., Yoder B. K. (2003). Loss of the Tg737 protein results in skeletal patterning defects. Dev. Dyn. 227, 78-90 [DOI] [PubMed] [Google Scholar]
- Zhao M., Ko S. Y., Liu J. H., Chen D., Zhang J., Wang B., Harris S. E., Oyajobi B. O., Mundy G. R. (2009). Inhibition of microtubule assembly in osteoblasts stimulates bone morphogenetic protein 2 expression and bone formation through transcription factor Gli2. Mol. Cell. Biol. 29, 1291-1305 [DOI] [PMC free article] [PubMed] [Google Scholar]