

Abstract
TLR4 agonists can be used as adjuvants to trigger innate immune responses of antigen-presenting cells (APCs) such as dendritic cells (DCs) to enhance vaccine-specific immunity. Adjuvant effects of TLR4 agonists are mediated by downstream signaling controlled by both MyD88 and TRIF adapter proteins. In this study, we investigated the adjuvanting capacity of glucopyranosyl lipid A (GLA), a chemically synthesized TLR4 agonist, to boost antigen-specific immunity elicited by DC-directed lentiviral vectors (DC-LV). We found that stimulation by this agonist in vitro can activate DCs in a TLR4-dependent manner. The agonist can significantly boost DC-LV-induced humoral and cellular immune responses, resulting in better antitumor reactions in response to tumor challenges. We observed that the adjuvant-mediated enhancement of cytotoxic CD8+ T cell responses is CD4+ T cell-dependent and determined that in vitro the agonist stimulation involves the participation of both MyD88 and TRIF pathways to activate DCs. In vivo immunization study however revealed that adjuvant effects depend more on the MyD88 signaling as TRIF-/- mice but not MyD88-/- mice were able to maintain the enhanced CD8+ T cell responses upon DC-LV immunization. Thus, our study supports the use of this TLR4 agonist as a potent adjuvant candidate for boosting DC-LV immunization.
Keywords: adjuvant, TLR4 agonist, dendritic cells, MyD88, TRIF, lentiviral vector
1. Introduction
There has been a growing interest in utilizing lentiviral vectors (LVs) as vaccine carriers to elicit antigen-specific humoral and cellular immune responses [1-4]. LVs present several desirable features of a virus-based vaccine vector [5]: they are able to transduce both dividing and non-dividing cells [6], capable of carrying large transgenes (up to 8 kb), and low in pre-existing anti-vector immunity [7], and they are currently being evaluated in human gene therapy trials for a wide range of human diseases [8]. Many studies have demonstrated the promise of LVs to generate vaccine-specific immunity targeting a broad range of infectious diseases and cancer [1, 9]. Although various routes of vaccine administration have been investigated and compared [10-12], subcutaneous injection remains the most potent and practical means for LVs to stimulate transgene-specific immune responses. Recent reports have convincingly shown that a subcutaneous injection of LVs can result in genetic modification of skin-derived dendritic cells (DCs) to have prolonged antigen expression and presentation [13-15]. Their subsequent migration to skin-draining lymph nodes and priming of the repertoire T cells are the major mechanism of action for the resulting immune responses to the delivered antigens. Because of the essential role of DCs in LV-mediated immunization, considerable effort has been devoted to developing LVs capable of targeting DCs to improve vaccine efficacy and safety [1, 16-22]. We have reported a targeting transduction system, in which the human immunodeficiency virus-1 (HIV-1)-based LV is enveloped with a mutant Sindbis virus glycoprotein (SVGmu) that, when injected subcutaneously into mice, can target DCs through its selective recognition of the attachment receptor DC-SIGN, a protein predominantly expressed on the DC surface [23]. Immunization with this vector system resulted in durable immune responses to several delivered immunogens and required only a modest dose of vector administration [23-26].
Our previous in vitro study showed a slight maturation of bone marrow-derived DCs (BMDCs) upon exposure to this DC-directed LV (DC-LV) system [23], presumably due to the interaction between SVGmu and DC-SIGN, and the transduction-mediated DC activation via Toll-like receptors [27-29]. We postulated that DC-stimulating molecular adjuvants such as agonists for TLR family proteins, when co-administered with DC-LV, could further improve the vaccine efficacy. The mammalian TLRs are a group of pattern recognition receptors expressed by innate immune cells and can be stimulated by structural motifs known as pathogen-associated molecular patterns (PAMPs) contained by bacteria, viruses, and fungi [30-32]. These stimulations can trigger downstream signal transduction pathways such as nuclear factor (NF)-κB and interferon regulatory factor (IRF), which will activate antigen-presenting cells (APCs) and promote inflammatory responses [31, 33, 34].
Among various known TLRs, TLR4 is the only one capable of inducing two distinct signaling pathways [32, 35]: 1) the MyD88-dependent pathway to activate NF-κB signaling and be responsible for induction of proinflammatory cytokines; 2) the TRIF-dependent pathway to mediate the activation of Type I interferons. Studies have shown that the ability to induce both pathways is essential for maximizing the immunostimulatory potentials of DCs [36]. The most widely known TLR4 agonist is lipopolysaccharide (LPS) that presents in the outer membrane of Gram-negative bacteria. Monophosphoryl lipid A (MPL) is a derivative of LPS exacted from Salmonella minnesota R595 [37] and exhibits only ~0.1% of the inflammatory toxicity of LPS [38, 39]. When used as an adjuvant, MPL enhances immunogen-specific immune responses by promoting the development of Th1 CD4+ T cells [40]. MPL has been approved as a component of adjuvant formulation for vaccines against human papilloma virus (HPV) and hepatitis B virus (HBV) [40]. Recently a synthetic TLR4 agonist, glucopyranosyl lipid A (GLA), has emerged as a more pure and chemically defined molecular adjuvant, in contrast to the heterogeneous mixture of MPL extracted from bacteria [41]. GLA has been demonstrated to be potent for assisting the generation of Th1-biased immune responses in experimental vaccines against tuberculosis [42], leishmaniasis [43], influenza [44], and malaria [45, 46]. It is currently being evaluated as an adjuvant in phase I clinical trials of an influenza virus vaccine [47].
In this report, we explore this TLR4 agonist as an adjuvant for immunization delivered by a DC-LV encoding the chicken ovalbumin (OVA) antigen. We show that GLA can activate BMDCs in vitro and significantly improve the immune responses in vivo by increasing the populations of both antigen-specific CD8+ and CD4+ T cells and improving the titers of various antibody isotypes specific for OVA. These enhancements resulted in improved protection against the growth of tumors yielding better survival rates in both prophylactic and therapeutic tumor challenge models. Moreover, we also found that the elevated CD8+ T cell responses provided by GLA are CD4+ T cell-dependent. Although the in vitro activation of DCs by GLA was observed to be mediated by both MyD88- and TRIF-dependent pathways, our DC-LV immunization assays showed that GLA is a more MyD88-biased agonist of TLR4 for augmenting vaccine-specific immunity.
2. Materials and methods
2.1. Mice and reagents
6–8 week old female C57BL/6 mice were purchased from the Charles River Laboratories. The strain of B6.B10ScN-Tlr4lps-del/JthJ (designated as TLR4-/-) and C57BL/6J-Ticam1Lps2/J (designated as TRIF-/-) mice were purchased from the Jackson Laboratory and maintained in the animal facilities of the California Institute of Technology (Caltech) and the University of Southern California (USC). B6.129/SvJ-MyD88tm1AKI (designated as MyD88-/-) mice were a gift from Prof. S. Akira (Osaka University, Osaka, Japan) and maintained at Caltech and USC. All animal procedures were performed in accordance with the guidelines set by the National Institutes of Health, Caltech, and USC on the Care and Use of Animals. The aqueous formulated GLA (GLA-AF, used for in vitro experiments) and the oil-in-water stable emulsion formulated GLA (GLA-SE, used for in vivo immunization) [41] were prepared at the Infectious Disease Research Institute (IDRI, Seattle, WA, USA).
2.2. Lentiviral vector construct and production
The lentiviral backbone plasmid FUW-TfROVA was constructed by insertion of the cDNA consisting of the first 118 amino acids of the membrane-anchoring domain of murine transferrin receptor) fused downstream with the truncated chicken ovalbumin (OVA, amino acids 139-386) into FUW [48]; FUW is a HIV-1-derived lentiviral plasmid composed of an internal human ubiqutin-C promoter to drive transgene expression and woodchuck responsive element to improve stability of the RNA transcript [49]. We employed a previously reported procedure of transient transfection of 293T cells to produce the DC-LV-OVA vector [23]. Briefly, 293T cells cultured in a 15-cm tissue culture plate (BD Biosciences, San Jose, CA, USA) were transfected via a standard calcium phosphate precipitation method with the following plasmids: the lentiviral backbone plasmid FUW-TfROVA (37.5 μg), the plasmid encoding the mutant Sindbis virus glycoprotein (SVGmu) (18.75 μg), and the packaging plasmids (pMDLg/pRRE and pRSV-Rev, 18.75 μg for each). The viral supernatants were harvested twice at 48 and 72 h post-transfection, pooled, and filtered through a 0.45-mm filter (Corning, Lowell, MA, USA). The concentrated viral pellets were obtained after ultracentrifugation of the viral supernatants at 50,000 ×g for 90 min, and were then resuspended in an appropriate volume of HBSS for in vivo injection.
2.3. BMDC generation and activation
We employed a previously described procedure to generate bone marrow-derived DCs (BMDCs) with various genetic backgrounds [23]. Briefly, bone marrow from the femurs and tibias of mice was grown in RPMI 1640 with 10% heat-inactivated FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.05 mM 2-ME, and 20 ng/ml GM-CSF (J558L supernatant) after the red blood cells were lysed. Cultures were initiated by placing 107 bone marrow cells in 10 ml of medium onto 100-mm petri dishes (Falcon 1029 plates; BD Labware, Franklin Lakes, NJ). On day 3, another 10 ml of J558L-conditioned medium was added. On day 6, suspension cells were collected. BMDCs were seeded at a density of 0.5 million/ml in 24-well plates (BD Labware), treated with GLA-AF (1 μg/ml) or left untreated, supernatants were collected 24 h later for cytokine measurements. Cells were also collected for antibody staining and flow cytometric analysis.
2.4. Flow cytometric analysis of surface markers of BMDCs
Single cell suspensions were incubated with anti-mouse CD16/CD32 Fc blocking antibody and then stained with fluorophore-conjugated monoclonal antibodies against specific BMDC surface markers, including CD80, CD86, H2-Kb, and I-Ab. All antibodies were purchased from Biolegend (San Diego, CA). Stained cells were assayed using BD LSRII flow cytometer (BD Biosciences) and acquired data was analyzed using FlowJo software (Tree Star, Ashland, OR).
2.5. Supernatant ELISA assay
ELISA was used to detect cytokine/chemokine levels after BMDC activation. Specific combination of capture and detection antibodies was purchased either from R&D Systems (Minneapolis, MN) for assaying IL-6, IL-12p70, IL-15, RANTES/CCL5, IP-10/CXCL10, and IL-1β, or from eBioscience (San Diego, CA) for assaying TNF-α. We followed the manufacturer's recommended protocols to conduct these ELISA assays.
2.6. Immunization procedure
For immunization with DC-LV, mice were injected with replication-defective DC-LV-OVA (5×106 Transduction Units (TU)) at the rear footpad. For immunization with adjuvant, GLA-SE (20 μg) was administered at the base of tail when DC-LV-OVA was injected. For the experiment to deplete CD4+ T cells, the monoclonal depletion antibody GK1.5 (200 μg, BioXCell, West Lebanon, NH) was intraperitoneally injected at days 0, 2, 5, and 8 post-immunization. The vaccinated mice were analyzed for their immune responses 2 wk post-immunization.
2.7. Intracellular cytokine staining (ICCS)
Splenocytes from immunized or control mice were pooled and incubated with the OVA257-264 peptide (SIINFEKL) (1μg/ml) in the presence of costimulatory anti-CD28 antibody (2 μg/ml, BD Biosciences) for 2 h at 37 °C in a 96-well round-bottom plate in RPMI medium supplemented with 10% FBS (Sigma), 10 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine. Brefeldin A (BFA, Sigma, St. Louis, MO) was added (10 μg/ml) to wells to inhibit cytokine exporting for another 4 h. Surface staining was performed by incubating restimulated cells with anti-mouse CD16/CD32 Fc blocking antibody, followed by anti-mouse CD8 and anti-mouse CD4 antibodies. Cells were then permeabilized in 100 μl Cytofix/Cytoperm solution (BD Biosciences) at 4 °C for 10 min, washed with Perm/Wash buffer (BD Biosciences), and followed by intracellular staining with PE-conjugated anti-mouse IFN-γ at 4 °C for 15 min. The flow cytometry analysis was carried out using the FACSort instrument from BD Biosciences. For the multi-parameter ICCS analysis, cells were stained with the following surface monoclonal antibodies (Biolegend): anti-CD4-PerCP, anti-CD8-APC-Cy7, anti-CD44-Alexa488, and with the following intracellular monoclonal antibodies (BD Biosciences): anti-IFN-γ-APC, and anti-TNF-α-PE-Cy7. The ICCS data were acquired on a BD LSR II flow cytometer (BD Biosciences).
2.8. IL-2 ELISPOT assay
ELISPOT assays were performed for detecting IL-2 using a kit from Millipore (Billerica, MA) according to the manufacturer's instruction. Briefly, anti-mouse IL-2 antibody (10 μg/ml in PBS) was used as the capture antibody and plated with 100 μl/well on 96-well MultiScreen-IP plates overnight at 4 °C. The plate was decanted and blocked with the RPMI medium containing 10% FBS at 37 °C for 2 h. Splenocytes from mice were plated at 5 × 105 cells/well in 100 μl complete medium in company with the CD4 epitope OVA323-339 peptide (ISQAVHAAHAEINEAGR) (10 μg/ml). After 18 h incubation at 37 °C, cells were lysed and plates were detected by 1 μg/ml biotinylated anti-IL-2 antibody (BD Biosciences) for 2 h at room temperature. Plates were further washed and incubated with the 1,000-fold-diluted streptavidin-alkaline phosphate conjugate for 45 min at room temperature. After a final extensive washing, spots were identified by adding BCIP/NBTplus substrate (Millipore), and the number of IL-2 producing cells was quantified by an ELISPOT reader.
2.9. Statistics
All the statistics were calculated by either Origin Pro 7.0 or GraphPad Prism 5 software. Error Bars in all the figures represent SD except the tumor growth curve in therapeutic tumor challenge model, SEM was used. One-way ANOVA followed by a Bonferroni's multiple comparison test was used to determine significance of difference while animal survival curves were analyzed by log-rank (Mantel-Cox) test and the value of P < 0.05 was considered to be statistically significant.
3. Results
3.1. Activation of dendritic cells by the TLR4 agonist GLA in vitro
Pathogens binding to TLR4 can initiate downstream signal transduction and induce NF-κB activity, which is critical for DC activation and maturation [50]. To evaluate the activation status of DCs, we examined the expression of major histocompatibility complex (MHC) and costimulatory molecules on the surface of BMDCs after GLA stimulation. We used an aqueous formation of GLA (GLA-AF) for in vitro studies [41, 44]. As shown in the left panel of Fig. 1A, MHC I (H2-Kb) and MHC II (I-Ab) molecules were both elevated after GLA treatment, which is likely to enhance DCs’ antigen presentation capability. Expression of both CD80 and CD86 was enhanced, which should allow DCs to provide stronger costimulatory signals for T cell stimulation. Furthermore, the surface expression both of ICAM-1 and CD40 was also greatly increased by GLA treatment (data not shown). To examine whether activation of DCs by GLA is TLR4-dependent, we conducted a similar experiment using BMDCs lacking TLR4 expression (TLR4-/- BMDCs). All of these surface activation markers were unaltered following GLA stimulation of TLR4-/- BMDCs (Fig. 1A, right panel), confirming that GLA activation of DCs is strictly TLR4-dependent.
Fig. 1.
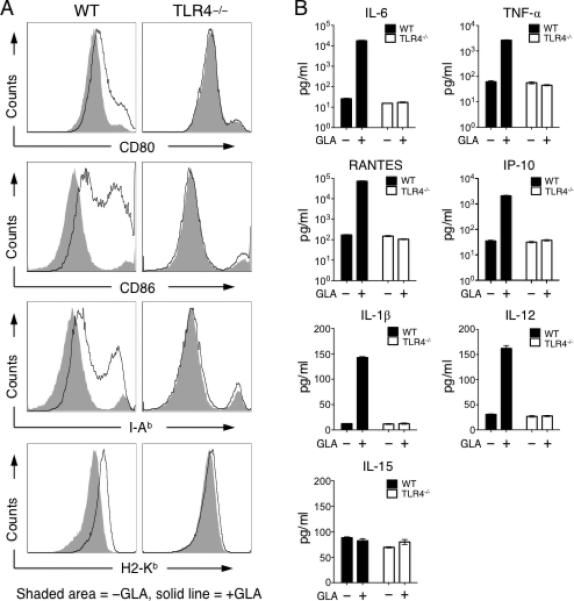
GLA-mediated activation of mouse BMDCs in vitro. A. FACS analysis of surface antigen presentation and costimulatory molecules of CD11c+ BMDCs from wild-type mouse (left) and TLR4-/- mouse (right) treated with aqueous GLA (solid line) or aqueous formulation lacking GLA (shaded area) for 24 h. Representative data from triplicate cultures is shown. B. Secretion of cytokines and chemokines of BMDC cultures from wild-type (black bar) and TLR4-/- mice (white bar) treated with (+) or without (□) GLA. Mean secretion +/- SD of triplicate culture is shown.
We then examined cytokine and chemokine secretion from BMDCs after GLA treatment. GLA stimulation significantly enhanced production and secretion of interleukin (IL)-6, TNF-α, RANTES, IP-10, modestly enhanced the amount of IL-1β and IL-12p70, but had little effect on IL-15 production (Fig. 1B). Proinflammatory cytokines IL-6, TNF-α and IL-12 are important for promoting the proliferation of CD8+ and CD4+CD25□ T cells, while limiting CD4+CD25+ regulatory T cell (Treg) proliferation [51-53]. RANTES is known to be involved in proper T cell function and proliferation [54]. IL-1β can drive proliferation of CD4+CD25+Foxp3□ effector and memory T cells, while inhibiting CD4+CD25+Foxp3+ Treg function [55]. IL-1β is also involved in CD4+ T cell-dependent antibody production and promotes the function of antigen-specific T helper cells [56]. The chemokine IP-10 is critical for effector T cell trafficking [57] and is involved in promoting T cell-based anti-tumor immunity [58]. Such elevated secretion of cytokines and chemokines was not observed in GLA-treated TLR4-/- BMDCs, confirming the essential role of TLR4 for GLA to activate DCs.
3.2. Adjuvant effect of the TLR4 agonist GLA on DC-LV-induced T cell immunity
We next tested the ability of GLA as an adjuvant to improve OVA-specific CD8+ and CD4+ T cell responses induced by subcutaneous immunization of DC-LV encoding the model OVA antigen (DC-LV-OVA). OVA was fused with the transferrin receptor transmembrane sequence for achieving balanced CD4 and CD8 responses [48]. Oil-in-water stable emulsion formulation of GLA (GLA-SE) was used for all our in vivo studies because such formulation resulted in more robust adjuvant responses as compared to aqueous GLA [40, 44]. We observed a higher level (approximately 2-3 times higher) of OVA-specific CD8+ T cells capable of producing IFN-γ and/or TNF-α from GLA-treated group than that of the untreated control group (Fig. 2A and B). An ELISPOT assay was employed to measure CD4+ T cell response. We found that GLA-treated mice on average had approximately 2-3 times more IL-2 producing cells than the control mice (Fig. 2B). When the same immunization protocol was applied to TLR4-/- mice, we observed no enhancement of OVA-specific CD8+ and CD4+ upon administration of GLA (Fig. 2C and D). This in vivo immunization study is in good agreement with our in vitro DC activation study, showing that GLA can bolster the response to a DC-LV-based genetic vaccine in a TLR4-dependent manner. In addition, in the absence of GLA, we noticed similar levels of OVA-specific CD8+ and CD4+ T cell responses induced by DC-LV immunization for wild-type and TLR4-/- mice, suggesting that DC-LV-based immunization generates a fraction of its vaccine-specific immune response in a manner that is independent of TLR4.
Fig. 2.
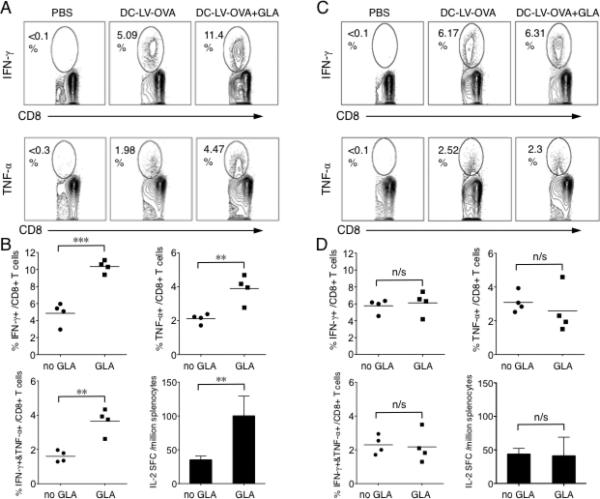
Adjuvant effects of GLA on boosting DC-LV-based vaccine-specific T cell immune responses in vivo. Wild-type (A, B) or TLR4-/- (C, D) mice were immunized at rear footpad with PBS, DC-LV-OVA, DC-LV-OVA plus GLA-SE (base of tail). 14 d later, splenocytes were collected and OVA-specific CD8+ T cells were analyzed by intracellular staining for IFN-γ or TNF-α expression after stimulation with OVA257-264 peptide for 6 h (A, C). The FACS data shown is one representative data of four analyzed mice. (B, D) Statistical data showing the percentage of IFN-γ+, TNF-α+, or IFN-γ+TNF-α+ cells within the CD8+ T cell population. Splenocytes were also pooled for an ELISPOT assay to analyze IL-2 secretion following stimulation with OVA323-339 peptide for 18 h. (***: P < 0.001; **: P < 0.01; *: P < 0.05 and n/s: not statistically significant; One-way ANOVA followed by a Bonferroni's multiple comparison test. Mean + SD is shown.)
3.3. Adjuvant effect of the TLR4 agonist GLA on DC-LV-induced antibody responses
Because we observed that GLA could improve DC-LV-elicited T cell responses, we investigated whether GLA could also facilitate vaccine-induced antibody responses. Mice were immunized with DC-LV-OVA plus and minus GLA adjuvant treatment. Two weeks post-immunization, sera were collected and OVA-specific titers of different antibody isotypes (IgG1, IgG2a, IgG2b, IgG3, IgA and IgM) were measured using an ELISA assay. GLA could broadly and greatly enhance antibody responses induced by DC-LV immunization (Fig. 3). The generation of elevated titers of both IgG1 (Fig. 3A) and IgG2a (Fig. 3B) suggests that GLA is likely able to bolster both Th1 and Th2 immune responses. Interestingly, we observed that GLA could enhance IgA antibody responses as well (Fig. 3E), which may be useful for vaccine applications that require mucosal immunity [59]. Notably, although GLA improved IgM antibody titer (Fig. 3F), the enhancment of IgM titer was not as significant as that of IgG. This might indicate a role for GLA in facilitating more efficient isotype switching from IgM to IgG.
Fig. 3.
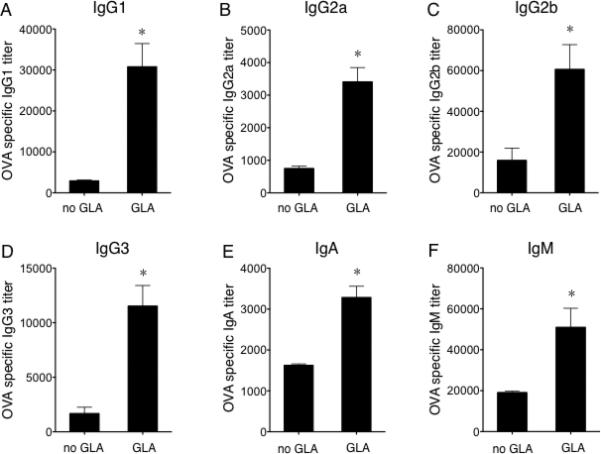
Adjuvant effects of GLA on boosting DC-LV-based vaccine-specific antibody immune responses in vivo. ELISA of OVA-specific IgG1 (A), IgG2a (B), IgG2b (C), IgG3 (D), IgA (E), IgM (F) antibody titers in sera of mice 14 d after immunization with DC-LV-OVA (left) or DC-LV-OVA plus GLA-SE (right). Results are shown as mean titer + SD of four mice per group. (*: P < 0.05; One-way ANOVA followed by a Bonferroni's multiple comparison test.)
3.4. Adjuvant effect of the TLR4 agonist GLA on the anti-tumor immunity delivered by DC-LV
The promising results of GLA as an adjuvant for enhancing vaccine-specific immune responses encouraged us to test whether GLA combined with a vector vaccine could generate better antitumor responses in mice. We employed the EG7 tumor cell line, which stably expresses chicken OVA, as a tumor model for this investigation [23]. In the tumor prophylactic model, mice were inoculated with EG7 tumor cells 14 days post-immunization. Aggressive tumor growth was seen in non-vaccinated control mice. Using the tumor size of 2000 mm3 as a surrogate endpoint of survival, none these mice could survive for more than 20 days. For the group immunized with DC-LV-OVA alone, although all mice developed tumors, tumor growth was slower and mice survived longer as compared to the non-vaccinated group. In contrast, for mice vaccinated with DC-LV-OVA and treated with GLA, only half of the mice developed tumors and those tumors grew much more slowly than did those in the vaccine-only group (Fig. 4A), resulting in a longer overall survival for these tumor-bearing mice (Fig. 4A, lower right). We also evaluated GLA in a therapeutic tumor model setting. Mice were challenged with a lethal dose of EG7 cells with immunization and GLA treatment administration 3 days later (Fig. 4B). As compared to the vaccine-only group, significantly slower tumor growth was observed for mice that received both DC-LV-OVA and GLA treatment and median survival time was 8 days longer (Fig. 4B, lower panel).
Fig. 4.
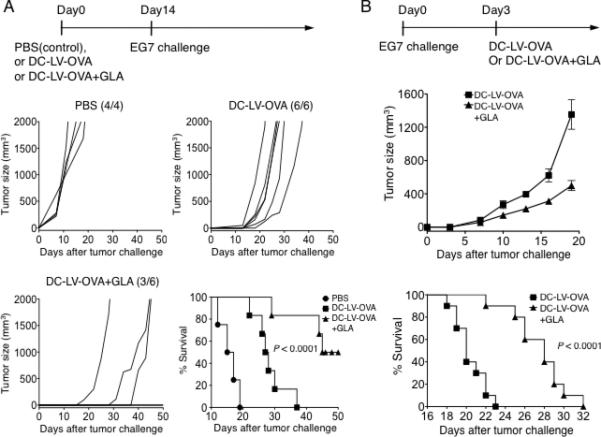
Adjuvant effects of GLA on DC-LV immunization to enhance antitumor immune responses. (A, upper) Schematic showing the immunization and tumor challenge strategy in the prophylactic model. (A, middle and lower left) Mice were vaccinated with PBS (four mice), DC-LV-OVA (six mice), or DC-LV-OVA plus GLA-SE (six mice) on day 0, followed by EG7 tumor challenge on day 14. Tumor growth was quantified as tumor volume (mm3) and plotted as a function of days after inoculation of EG7 cells. Three of the DC-LV-OVA plus GLA-SE mice never developed tumors. (A, lower right) Kaplan-Meier survival plot of mice vaccinated with PBS (●), DC-LV-OVA (■), or DC-LV-OVA plus GLA-SE (▲), P < 0.0001. The tumor size of 2000 mm3 was used as a surrogate endpoint of survival. (B, upper) Schematic showing the tumor inoculation on day 0 and immunization strategy on day 3 in the therapeutic model. (B, middle) Tumor growth was plotted as mean volume +/- SEM (n=10) as a function of days after EG7 tumor challenge. (B, lower) Kaplan-Meier survival plot of mice vaccinated with DC-LV-OVA (■) or DC-LV-OVA plus GLA-SE (▲), P < 0.0001.
3.5. Role of CD4+ T cells in GLA-augmented DC-LV immunization
It is well documented that CD4+ T helper cells play a pivotal role in orchestrating the generation of both effector and memory CD8+ cytotoxic T cells [60, 61], although CD8+ T cell responses can also be produced in a CD4+ T cell-independent manner [62-64]. To investigate the cellular mechanism underlying the enhanced vaccine responses delivered by GLA, we conducted experiments to deplete CD4+ T cells following the immunization. Four groups of mice were immunized with DC-LV-OVA, within which two groups also received GLA adjuvant at the same time. Depletion of CD4+ T cells by the GK1.5 antibody was carried out in one vaccinated group and in a group vaccinated along with GLA treatment. Flow cytometric analysis of the resulting splenocytes showed that the majority of CD4+ T cells (21.5% vs. 0.33%, Fig. 5A) were eliminated by the antibody treatment and these mice maintained such a depletion condition throughout the experiment. The intracellular staining of CD8+ T cells harvested from immunized mice for IFN-γ (Fig. 5B) showed higher (~19% vs. ~7%) OVA-specific CD8+ T cells in mice with the GLA-adjuvanted vaccination. On the contrary, with the CD4+ T cell depletion, the immune response dropped to about 2% and GLA treatment failed to improve the CD8+ T cell response in the CD4+ T cell-deficient condition (Fig. 5A and B). This data demonstrates that CD4+ T cells are necessary for the adjuvanting effect of GLA on lentiviral vector immunization. We also observed that the DC-LV-OVA-induced CD8+ T cell response was partially dependent on the presence of CD4+ T cells (Fig. 5A, DC-LV-OVA vs. DC-LV-OVA/GK1.5). To investigate whether such dependence is unique for DC-LV, we directly compared immunization by vectors enveloped with either SVGmu (DC-LV) or the glycoprotein derived from vesicular stomatitis virus (VSVG), which provides a broader tropism in both normal and CD4+ T cell-deficient conditions. Consistent with our previous results [23], when immunized with an identical dose, the SVGmu-enveloped vector (DC-LV) elicited a markedly higher CD8+ T cell response than that induced by the VSVG-enveloped vector (Fig. 5C). Depletion of CD4+ T cells lowered CD8+ T cell immunity in both situations, although the drop was more significant for SVGmu-enveloped vector than VSVG-enveloped vector (Fig. 5C).
Fig. 5.
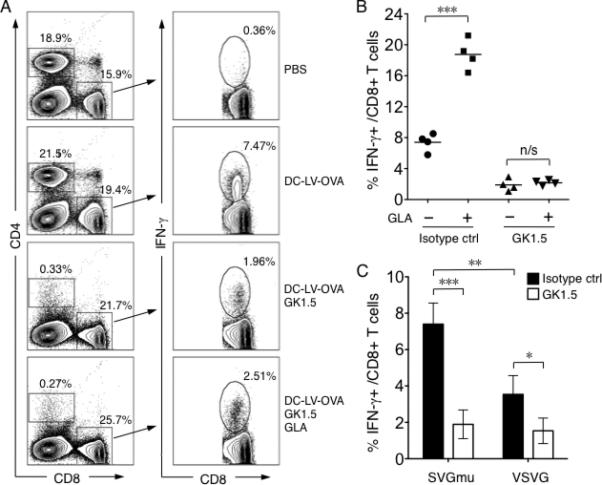
The role of CD4+ helper T cells in GLA-enhanced CD8+ T cell responses. A. FACS analysis of intracellular staining of IFN-γ from splenocytes of mice immunized with PBS, DC-LV-OVA, DC-LV-OVA plus GK1.5 antibody-mediated depletion of CD4+ T cells, or DC-LV-OVA plus GLA-SE and GK1.5. One representative data from a group of four mice is shown. Percentage shown in the left panel is the percentage of CD4+ T cells and CD8+ T cells. B. Statistical data showing OVA-specific CD8+ T cells by intracellular staining of IFN-γ following stimulation with OVA257-264 peptide. C. OVA-specific CD8+ T cell percentage by intracellular staining of IFN-γ after OVA257-264 peptide stimulation from mice immunized for 14 d with the same transduction units (TUs) of either SVGmu- (left) or VSVG- (right) enveloped LV-OVA. Black bar: isotype control antibody; white bar: GK1.5 antibody. (***: P < 0.001; **: P < 0.01; *: P < 0.05 and n/s: not statistically significant; One-way ANOVA followed by a Bonferroni's multiple comparison test. Mean +/- SD is shown.)
3.6. Study of the role of MyD88 and TRIF pathways in GLA-mediated activation of DCs in vitro
It has been reported that Monophosphoryl Lipid A (MPL), another TLR4 agonist, activates macrophage cells in a TRIF-biased rather than MyD88-biased manner when compared with its parent molecule LPS [65]. Because GLA is a relatively novel and unstudied synthetic molecule, elucidating the downstream signaling pathway involved for activating DCs by GLA can be very useful for understanding the mechanism of its adjuvant effect and exploring its potential applications in vaccine research. Among various Toll-like receptors, TLR4 is the only receptor that utilizes both TRIF- and MyD88-dependent pathways for signaling [32, 35]. To examine which pathway GLA/TLR4 depends on to trigger DC activation, BMDCs derived from wild-type, MyD88-/- and TRIF-/- mice were generated and treated with GLA for 1 day individually; no GLA treatment was included as controls. Treated BMDCs were subjected to analysis of their surface expression of costimulatory molecules (CD40, CD80, CD86, OX40), MHC class I molecule H2-Kb, and MHC class II molecule I-Ab. As shown in Fig. 6A, expression of these surface markers on both MyD88-/- and TRIF-/- BMDCs were generally lower (except CD80 which has similar levels in all three situations) than those expressed on wild-type cells after GLA treatment, indicating that MyD88 and TRIF both participated in the downstream signaling in response to GLA stimulation. To further this study, we collected supernatants from these BMDCs and assayed the secretion profile of a selective set of cytokines and chemokines (Fig. 6B). Surprisingly, lack of MyD88 expression almost completely abolished the ability of GLA-treated DCs to produce cytokines TNF-α, IL-6, and IL-1β, while they were able to maintain a modest production of chemokines RANTES and IP-10. On the other hand, as compared to MyD88-null cells, TRIF-/- BMDCs better retained the ability to secrete TNF-α, IL-6, and IL-1β in response to GLA treatment, although their levels of secretion were lower than that of the wild-type cells. This suggests that the production of these cytokines is less dependent on the TRIF-mediated pathway. In contrast, chemokines RANTES and IP-10 tend to depend more on TRIF expression because TRIF-/- BMDCs produced less of them compared to MyD88-/- cells upon GLA stimulation. Interestingly, we observed that DCs have an equal, partial dependence on MyD88 and TRIF for the secretion of IL-12 in response to GLA stimulation.
Fig. 6.
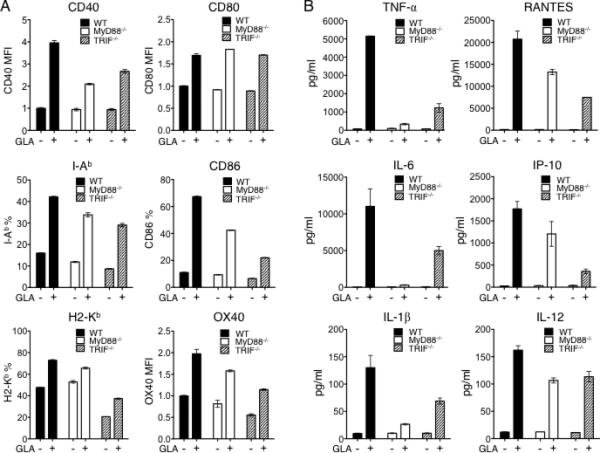
Involvement of MyD88 and TRIF signaling pathways in GLA-mediated activation of BMDCs in vitro. A. FACS analysis of surface activation markers of CD11c+ BMDCs from wild-type mouse (black), MyD88-/- mouse (white), or TRIF-/- mouse (dashed line) treated with (+) or without (□) aqueous GLA. Data is shown based on either percentage or normalized mean fluorescence intensity (MFI). B. Secretion of cytokines and chemokines of BMDCs derived from wild-type (black), MyD88-/- (white), or TRIF-/- mouse (dashed line) stimulated with (+) or without (□) GLA. The mean secretion +/- SD of triplicate culture is shown.
3.7. Study of the role of MyD88 and TRIF pathways in GLA-mediated activation of DCs in vivo
To examine the in vivo role of GLA adjuvant in augmenting DC-LV-based vaccination, we compared immune responses among vaccinated MyD88-/-, TRIF-/-, and wild-type mice. As seen previously, intracellular cytokine staining of splenocytes restimulated with the OVA257-264 peptide showed enhanced CD8+ T cell immunity in wild-type mice treated with GLA (Fig. 7A). Significantly lower antigen-specific CD8+ T cell immune responses were observed in MyD88-/- mice and GLA treatment did not improve the magnitude of the response (Fig. 7A and B). In sharp contrast, TRIF-/- mice could mount the same level of CD8+ T cell response as that of wild-type animals and a similarly enhanced level of immune responses was obtained upon GLA treatment. These results indicate that the MyD88 pathway, rather than the TRIF pathway, is essential for GLA-adjuvanted CD8+ T cell responses in vivo. We also assessed the OVA-specific CD4+ T cell immunity by an IL-2 ELISPOT assay for these treated mouse groups and found a similar trend to what was observed for CD8+ T cell responses (Fig. 7B, lower right), suggesting that GLA-enhanced CD4+ T cell immune response in vivo is also largely dependent on the MyD88 pathway. Another notable observation from this study is the important role of MyD88 in generating vaccine-specific immunity induced by DC-LV immunization. Comparing to the wild-type mice, the MyD88-/- mice had a markedly diminished CD8+ and CD4+ T cell responses after immunization, while TRIF-deficient mice could retain the similar level of the response magnitude (Fig. 7B). All of these data suggest that MyD88 rather than TRIF plays an important signaling transduction role for DC-LV vaccine to generate immune responses to immunogens in vivo.
Fig. 7.
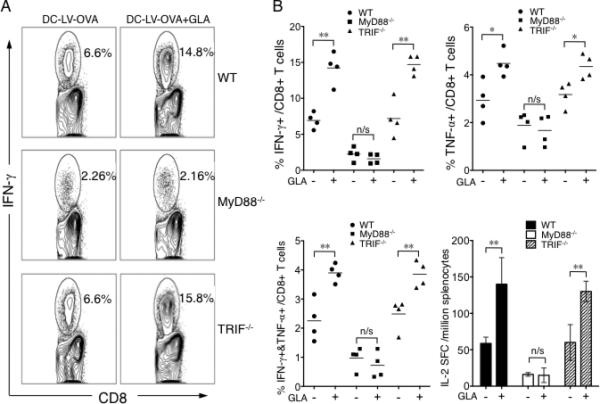
Involvement of MyD88 and TRIF signaling pathways in GLA-adjuvanted DC-LV immunization in vivo. A. Wild-type (upper), MyD88-/- (middle), and TRIF-/- (lower) mice were immunized with DC-LV-OVA, or DC-LV-OVA plus GLA-SE. 2 wk later, OVA-specific CD8+ T cells were analyzed by intracellular staining of IFN-γ following OVA257-264 peptide stimulation. The FACS data shown is representative of four mice tested. B. Statistical data showing OVA-specific CD8+ T cells analyzed by IFN-γ+ (upper left), TNF-α+ (upper right), or IFN-γ+TNF-α+ (lower left) populations for groups of mice described above with (+) or without (-) GLA treatment. (B, lower right) Splenocytes were also pooled for an ELISA assay of IL-2 production after stimulation with OVA323-339 peptide. (**: P < 0.01; *: P < 0.05 and n/s: not statistically significant; One-way ANOVA followed by a Bonferroni's multiple comparison test. Mean +/-SD is shown.)
4. Discussion
Antigen genes delivered to DCs by replication-deficient LVs generate both antigen-specific T cell and B cell responses in mice and rhesus macaques [1, 66]. Skin-derived DCs are the major cell targets for LV-based immunization [13-15]. However, several studies have shown that LVs are weak stimulators for activation of DCs by themselves [67], and further activation is needed [68, 69]. Activation of individual TLRs or combined TLRs could be incorporated in order to achieve stronger T cell responses [70-74]. We have developed a DC-targeted LV vector system where LVs are enveloped with an engineered Sindbis virus glycoprotein. This DC-LV system can target DCs through interaction with DC-SIGN and subcutaneous immunization with this vector has induced strong antigen-specific immunity [23]. Our in vitro study revealed that DC-SIGN-interacting DC-LV only modestly induces maturation of DCs [23]. Therefore, it is possible that further activation of DCs would improve the immune responses induced by DC-LV. Here we demonstrate that the synthetic TLR4 agonist GLA can be used as adjuvant to enhance DC-LV-mounted immune responses.
Some studies reported that LV-mediated transduction efficiency towards DCs could be reduced with prior to or simultaneous activation of DCs [75]. Other studies showed that transduction of LVs did not hamper the subsequent activation of DCs by TLRs [76]. The possible explanation is that upon maturation, internalization potency of DCs is down-regulated [77, 78], which may cause the reduced transduction. In light of these observations, we decided to employ a protocol to deliver LVs and GLA at different locations (footpad vs. base of tail). After administration, GLA could be transported to lymph nodes through lymphatic circulation and encounters DCs that have been modified by LVs. In addition, GLA could activate DCs that have not been transduced by LVs; these DCs could subsequently migrate to the lymph nodes to not only secrete inflammatory cytokines but also cross-prime antigens derived from LV-modified DCs through cross antigen presentation, which could eventually also enhance antigen-specific immunity.
Three signals are required for efficient stimulation of T cells by DCs: 1) an antigen-specific signal, involving the interaction of the MHC/peptide complex with T cell receptor (TCR); 2) a surface costimultory protein on DCs interacting with a receptor on T cells; and 3) a cytokine signal passed from DC to T cell [79, 80]. Our in vitro activation study, presented in Fig. 1, indicated that molecular signatures involved in all these three signals could be enhanced after BMDCs were treated by GLA, including the upregulation of MHC molecules, elevated expression of costimulatory molecules CD80 and CD86, and improved production of proinflammatory cytokines (IL-6, TNF-α, IL-12, and IL-1β) and chemokines (RANTES/CCL5 and IP-10/CXCL10); a comparable potency was observed upon LPS stimulation (data not shown). This is consistent with prior studies, in which either murine and human monocyte/macrophage cell lines [44] or murine BMDCs [41] secreted TNF-α, IL-6, and IP-10 in response to aqueous GLA stimulation. GLA-mediated DC maturation is TLR4-dependent, as the TLR4-/- BMDCs failed to respond to GLA. Our use of genetically deficient cells to directly demonstrate the TLR4 dependence of GLA complements previous use of anti-TLR4 antibody to successfully block GLA-mediated activation [44]. To further improve DCs potency, combination strategy of TLRs with silencing of inhibitory pathways [51, 80] can also be taken into consideration.
Both CD8+ cytotoxic and CD4+ T helper cells act coordinately to either kill tumor cells or clear infectious pathogens. In our study, we found that by co-administration of GLA along with DC-LV immunization, numbers of both CD8+ and CD4+ T cells specific for OVA antigen were greatly increased in mice. We confirmed that the enhancement is strictly TLR4-dependent, as no elevated response to GLA was seen in TLR4-deficient mice. In addition to T cell immunity, the antibody response is also important to clear infectious viruses and initiate Fc-mediate tumor cell killing by natural killer (NK) cells [81]. We found that GLA improved the titers of various antibody isotypes specific for OVA, including that of both IgG1 and IgG2a, indicating that GLA can boost both the Th1 and Th2 responses. We observed an enhanced IgA antibody titer, suggesting that GLA can be used as an adjuvant to facilitate the application of DC-LV vaccination for inducing mucosal immunity.
CD4+ T cells provide essential help to CD8+ T cell during the priming stage to generate effector and memory CD8 T cell responses [60, 61, 82]. On the other hand, it was reported that helper-independent cytotoxic T cell priming can be promoted by upregulating CD40L expression on DCs, and only TLR3 and TLR9, but not TLR2 and TLR4, have the capacity to stimulate CD40L expression [83]. This suggests that GLA, as a TLR4 agonist, may promote CD8+ cytotoxic T cell response in a CD4+ T helper cell-dependent manner. There are several hypotheses and models regarding how CD4+ T cells provide help to CD8+ T cells through APCs. One of them requires that activated CD4+ T cells and CD8+ T cells interact with the same APC [84, 85]. Another model is that CD4+ T cells can acquire the MHC-peptide complexes and costimulatory molecules from APCs, and then these CD4+ T cells carrying acquired antigen presentation components can efficiently stimulate CD8+ T cells [86]. We conducted a simple CD4+ T cells depletion experiment to study their role in GLA promotion of the CD8+ T cell response. Our results show that without help from CD4+ T cells, the CD8+ T cell response cannot be augmented by GLA. Either model could explain the involvement of CD4+ T cells with the adjuvant effect of GLA possibly dependent on CD4+ T cells to indirectly pass signals to CD8+ T cells. Interestingly, we also found that GLA failed to improve immune responses induced by DC-LV encoding the unmodified OVA sequence (without transferrin receptor membrane-binding segment) (Fig. S1). Considering that a much lower OVA-specific CD4+ T cell response is elicited with such an antigen configuration, it seems that the presence of immunogen-specific CD4+ T cells is vital for GLA to augment DC-LV-based vaccination. We also observed that CD4+ T cells play a critical role in generating primary CD8+ T cell responses induced by either DC-LV or VSVG-enveloped LVs, and depletion of CD4+ T cells markedly reduced vaccine-specific CD8+ T cell responses in either case. This is in good agreement with a recent report showing the important role of CD4+ T cells in LV-mediated immunization [15].
Activation of TLR4 triggers two different signaling pathways controlled respectively by MyD88/MAL, which is critical for regulating proinflammatory cytokines production, and TRIF/TRAM, which is responsible for mediating the induction of Type I interferons [32, 35]. Many of these signaling studies have been conducted in macrophage cells. One recent study compared MPL with LPS in mouse macrophages and showed that MPL is a more TRIF-biased TLR4 agonist and preferentially induces TRIF-mediated signals for macrophage activation [65]. However, their subsequent studies in BMDCs demonstrated that both MyD88 and TRIF pathways are involved in the MPL-mediated activation of DCs [87]. Shen et al. found that induction of both MyD88 and TRIF signaling pathways are critical for maximizing the capacity of DCs for T cells priming [36]. We found that GLA activates BMDCs in vitro in both MyD88- and TRIF-dependent pathways. This represents the first study to use genetically deficient DCs (MyD88-/- and TRIF-/-) to investigate signaling activation of GLA in DCs, and the data corroborates the result from a mouse inflammation microarray study that showed both MyD88- and TRIF-inducible genes responding to GLA stimulation [41]. In contrast, in our in vivo data we observed a more MyD88-biased signaling role for GLA to elicit adjuvant responses to DC-LV immunization. It is unknown whether the MyD88-biased role is unique for the interplay between GLA and DC-LV or it is more general and can be seen in other formats of vaccine delivery; more experiments are required to address these questions. One possible explanation is found in the behavior of the cytokines IL-6 and TNF-α, which appears to be key cytokines for priming CD8+ T cell responses in vivo [51]. Our in vitro study showed that MyD88-/- BMDCs almost completely lacked secretion of TNF-α and IL-6, while TRIF-/- BMDCs could still respond to GLA treatment and secrete a significant level of these two cytokines. An inability of MyD88-/- BMDCs to produce these cytokines in response to GLA could partially explain the strong dependence on the MyD88-dependent pathway for GLA augmentation of vaccine-specific immunity in vivo.
5. Conclusion
We have determined that GLA can activate DCs in vitro and augment humoral and cellular immunity elicited by DC-LV immunization in a TRL4-dependent manner. CD4+ helper T cells are indispensible for this adjuvant effect of GLA. Our in vitro assays confirm that both MyD88 and TRIF are able to participate in downstream signaling under GLA-mediated activation of DCs, but our in vivo immunization of genetically deficient mice by DL-LV suggests that MyD88 plays the greater role in mediating potent adjuvant effects of GLA to improve antigen-specific immune responses.
Supplementary Material
TLR4-dependent activation
Better T and B cell responses
MyD88-biased pathway
Acknowledgements
We thank Paul Bryson and April Tai for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health (R01AI68978 and P01CA132681), a grant from the Bill and Melinda Gates Foundation, a translational acceleration grant from the Joint Center for Translational Medicine and a grant from the California HIV/AIDS Research Program.
Abbreviations used in this article
- LV
lentiviral vector
- DC
dendritic cells
- GLA
glucopyranosyl lipid A
- MPL
monophosphoryl lipid A
- OVA
ovalbumin
- TLR
Toll-like receptor
- MHC
major histocompatibility complex
- APC
antigen-presenting cells
- BMDC
bone marrow-derived dendritic cell
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hu B, Tai A, Wang P. Immunization delivered by lentiviral vectors for cancer and infectious diseases. Immunol Rev. 2011;239(1):45–61. doi: 10.1111/j.1600-065X.2010.00967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He Y, Munn D, Falo LD., Jr Recombinant lentivector as a genetic immunization vehicle for antitumor immunity. Expert Rev Vaccines. 2007;6(6):913–24. doi: 10.1586/14760584.6.6.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He Y, Falo LD., Jr Lentivirus as a potent and mechanistically distinct vector for genetic immunization. Curr Opin Mol Ther. 2007;9(5):439–46. [PMC free article] [PubMed] [Google Scholar]
- 4.Pincha M, Sundarasetty BS, Stripecke R. Lentiviral vectors for immunization: an inflammatory field. Expert Rev Vaccines. 2010;9(3):309–21. doi: 10.1586/erv.10.9. [DOI] [PubMed] [Google Scholar]
- 5.Draper SJ, Heeney JL. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol. 2010;8(1):62–73. doi: 10.1038/nrmicro2240. [DOI] [PubMed] [Google Scholar]
- 6.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272(5259):263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 7.Kootstra NA, Verma IM. Gene therapy with viral vectors. Annu Rev Pharmacol Toxicol. 2003;43:413–39. doi: 10.1146/annurev.pharmtox.43.100901.140257. [DOI] [PubMed] [Google Scholar]
- 8.Escors D, Breckpot K. Lentiviral vectors in gene therapy: their current status and future potential. Arch Immunol Ther Exp. 2010;58(2):107–19. doi: 10.1007/s00005-010-0063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breckpot K, Aerts JL, Thielemans K. Lentiviral vectors for cancer immunotherapy: transforming infectious particles into therapeutics. Gene Ther. 2007;14(11):847–62. doi: 10.1038/sj.gt.3302947. [DOI] [PubMed] [Google Scholar]
- 10.Sigel MB, Sinha YN, VanderLaan WP. Production of antibodies by inoculation into lymph nodes. Methods Enzymol. 1983;93:3–12. doi: 10.1016/s0076-6879(83)93031-8. [DOI] [PubMed] [Google Scholar]
- 11.Maloy KJ, Erdmann I, Basch V, Sierro S, Kramps TA, Zinkernagel RM, et al. Intralymphatic immunization enhances DNA vaccination. Proc Natl Acad Sci U S A. 2001;98(6):3299–303. doi: 10.1073/pnas.051630798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senti G, Johansen P, Kundig TM. Intralymphatic immunotherapy. Curr Opin Allergy Clin Immunol. 2009;9(6):537–43. doi: 10.1097/ACI.0b013e3283310ff7. [DOI] [PubMed] [Google Scholar]
- 13.He Y, Zhang J, Donahue C, Falo LD., Jr Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity. 2006;24(5):643–56. doi: 10.1016/j.immuni.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furmanov K, Elnekave M, Lehmann D, Clausen BE, Kotton DN, Hovav AH. The role of skin-derived dendritic cells in CD8+ T cell priming following immunization with lentivectors. J Immunol. 2010;184(9):4889–97. doi: 10.4049/jimmunol.0903062. [DOI] [PubMed] [Google Scholar]
- 15.Goold HD, Escors D, Conlan TJ, Chakraverty R, Bennett CL. Conventional dendritic cells are required for the activation of helper-dependent CD8 T cell responses to a model antigen after cutaneous vaccination with lentiviral vectors. J Immunol. 2011;186(8):4565–72. doi: 10.4049/jimmunol.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopes L, Dewannieux M, Gileadi U, Bailey R, Ikeda Y, Whittaker C, et al. Immunization with a lentivector that targets tumor antigen expression to dendritic cells induces potent CD8+ and CD4+ T-cell responses. J Virol. 2008;82(1):86–95. doi: 10.1128/JVI.01289-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimura T, Koya RC, Anselmi L, Sternini C, Wang HJ, Comin-Anduix B, et al. Lentiviral vectors with CMV or MHCII promoters administered in vivo: immune reactivity versus persistence of expression. Mol Ther. 2007;15(7):1390–9. doi: 10.1038/sj.mt.6300180. [DOI] [PubMed] [Google Scholar]
- 18.Dresch C, Edelmann SL, Marconi P, Brocker T. Lentiviral-mediated transcriptional targeting of dendritic cells for induction of T cell tolerance in vivo. J Immunol. 2008;181(7):4495–506. doi: 10.4049/jimmunol.181.7.4495. [DOI] [PubMed] [Google Scholar]
- 19.Ageichik A, Buchholz CJ, Collins MK. Lentiviral Vectors Targeted to MHC II Are Effective in Immunization. Hum Gene Ther. 2011 doi: 10.1089/hum.2010.184. [DOI] [PubMed] [Google Scholar]
- 20.Ageichik A, Collins MK, Dewannieux M. Lentivector targeting to dendritic cells. Mol Ther. 2008;16(6):1008–9. doi: 10.1038/mt.2008.95. [DOI] [PubMed] [Google Scholar]
- 21.Goyvaerts C, De Groeve K, Dingemans J, Van Lint S, Robays L, Heirman C, et al. Development of the Nanobody display technology to target lentiviral vectors to antigen-presenting cells. Gene Ther. 2012 doi: 10.1038/gt.2011.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verhoeyen E, Cosset FL. Engineering the surface glycoproteins of lentiviral vectors for targeted gene transfer. Cold Spring Harbor Protoc. 2009;2009(8):pdb top59. doi: 10.1101/pdb.top59. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Yang H, Rideout K, Cho T, Joo KI, Ziegler L, et al. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol. 2008;26(3):326–34. doi: 10.1038/nbt1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H, Hu B, Xiao L, Wang P. Dendritic cell-directed lentivector vaccine induces antigen-specific immune responses against murine melanoma. Cancer Gene Ther. 2011;18(5):370–80. doi: 10.1038/cgt.2011.13. [DOI] [PubMed] [Google Scholar]
- 25.Dai B, Yang L, Yang H, Hu B, Baltimore D, Wang P. HIV-1 Gag-specific immunity induced by a lentivector-based vaccine directed to dendritic cells. Proc Natl Acad Sci U S A. 2009;106(48):20382–7. doi: 10.1073/pnas.0911742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu B, Dai B, Wang P. Vaccines delivered by integration-deficient lentiviral vectors targeting dendritic cells induces strong antigen-specific immunity. Vaccine. 2010;28(41):6675–83. doi: 10.1016/j.vaccine.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breckpot K, Escors D, Arce F, Lopes L, Karwacz K, Van Lint S, et al. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol. 2010;84(11):5627–36. doi: 10.1128/JVI.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breckpot K, Emeagi P, Dullaers M, Michiels A, Heirman C, Thielemans K. Activation of immature monocyte-derived dendritic cells after transduction with high doses of lentiviral vectors. Hum Gene Ther. 2007;18(6):536–46. doi: 10.1089/hum.2007.006. [DOI] [PubMed] [Google Scholar]
- 29.Tan PH, Beutelspacher SC, Xue SA, Wang YH, Mitchell P, McAlister JC, et al. Modulation of human dendritic-cell function following transduction with viral vectors: implications for gene therapy. Blood. 2005;105(10):3824–32. doi: 10.1182/blood-2004-10-3880. [DOI] [PubMed] [Google Scholar]
- 30.Janeway CA, Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 31.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 33.Lee HK, Iwasaki A. Innate control of adaptive immunity: dendritic cells and beyond. Semin Immunol. 2007;19(1):48–55. doi: 10.1016/j.smim.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327(5963):291–5. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–51. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Shen H, Tesar BM, Walker WE, Goldstein DR. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J Immunol. 2008;181(3):1849–58. doi: 10.4049/jimmunol.181.3.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baldridge JR, Crane RT. Monophosphoryl lipid A (MPL) formulations for the next generation of vaccines. Methods. 1999;19(1):103–7. doi: 10.1006/meth.1999.0834. [DOI] [PubMed] [Google Scholar]
- 38.Qureshi N, Kaltashov I, Walker K, Doroshenko V, Cotter RJ, Takayama K, et al. Structure of the monophosphoryl lipid A moiety obtained from the lipopolysaccharide of Chlamydia trachomatis. J Biol Chem. 1997;272(16):10594–600. doi: 10.1074/jbc.272.16.10594. [DOI] [PubMed] [Google Scholar]
- 39.Evans JT, Cluff CW, Johnson DA, Lacy MJ, Persing DH, Baldridge JR. Enhancement of antigen-specific immunity via the TLR4 ligands MPL adjuvant and Ribi.529. Expert Rev Vaccines. 2003;2(2):219–29. doi: 10.1586/14760584.2.2.219. [DOI] [PubMed] [Google Scholar]
- 40.Reed SG, Bertholet S, Coler RN, Friede M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009;30(1):23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 41.Coler RN, Bertholet S, Moutaftsi M, Guderian JA, Windish HP, Baldwin SL, et al. Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS One. 2011;6(1):e16333. doi: 10.1371/journal.pone.0016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldwin SL, Bertholet S, Kahn M, Zharkikh I, Ireton GC, Vedvick TS, et al. Intradermal immunization improves protective efficacy of a novel TB vaccine candidate. Vaccine. 2009;27(23):3063–71. doi: 10.1016/j.vaccine.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertholet S, Goto Y, Carter L, Bhatia A, Howard RF, Carter D, et al. Optimized subunit vaccine protects against experimental leishmaniasis. Vaccine. 2009;27(50):7036–45. doi: 10.1016/j.vaccine.2009.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldwin SL, Shaverdian N, Goto Y, Duthie MS, Raman VS, Evers T, et al. Enhanced humoral and Type 1 cellular immune responses with Fluzone adjuvanted with a synthetic TLR4 agonist formulated in an emulsion. Vaccine. 2009;27(43):5956–63. doi: 10.1016/j.vaccine.2009.07.081. [DOI] [PubMed] [Google Scholar]
- 45.Lousada-Dietrich S, Jogdand PS, Jepsen S, Pinto VV, Ditlev SB, Christiansen M, et al. A synthetic TLR4 agonist formulated in an emulsion enhances humoral and Type 1 cellular immune responses against GMZ2--a GLURP-MSP3 fusion protein malaria vaccine candidate. Vaccine. 2011;29(17):3284–92. doi: 10.1016/j.vaccine.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 46.Lumsden JM, Pichyangkul S, Srichairatanakul U, Yongvanitchit K, Limsalakpetch A, Nurmukhambetova S, et al. Evaluation of the safety and immunogenicity in rhesus monkeys of a recombinant malaria vaccine for Plasmodium vivax with a synthetic TLR4 agonist formulated in an emulsion. Infect Immun. 2011 doi: 10.1128/IAI.05257-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coler RN, Baldwin SL, Shaverdian N, Bertholet S, Reed SJ, Raman VS, et al. A synthetic adjuvant to enhance and expand immune responses to influenza vaccines. PLoS One. 2010;5(10):e13677. doi: 10.1371/journal.pone.0013677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rowe HM, Lopes L, Ikeda Y, Bailey R, Barde I, Zenke M, et al. Immunization with a lentiviral vector stimulates both CD4 and CD8 T cell responses to an ovalbumin transgene. Mol Ther. 2006;13(2):310–9. doi: 10.1016/j.ymthe.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 49.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295(5556):868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 50.Andreakos E, Williams RO, Wales J, Foxwell BM, Feldmann M. Activation of NF-kappaB by the intracellular expression of NF-kappaB-inducing kinase acts as a powerful vaccine adjuvant. Proc Natl Acad Sci U S A. 2006;103(39):14459–64. doi: 10.1073/pnas.0603493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14(3):258–65. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. 2004;173(8):5008–20. doi: 10.4049/jimmunol.173.8.5008. [DOI] [PubMed] [Google Scholar]
- 53.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164(1):183–90. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 54.Makino Y, Cook DN, Smithies O, Hwang OY, Neilson EG, Turka LA, et al. Impaired T cell function in RANTES-deficient mice. Clin Immunol. 2002;102(3):302–9. doi: 10.1006/clim.2001.5178. [DOI] [PubMed] [Google Scholar]
- 55.O'Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, et al. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J Immunol. 2006;176(12):7278–87. doi: 10.4049/jimmunol.176.12.7278. [DOI] [PubMed] [Google Scholar]
- 56.Nakae S, Asano M, Horai R, Iwakura Y. Interleukin-1 beta, but not interleukin-1 alpha, is required for T-cell-dependent antibody production. Immunology. 2001;104(4):402–9. doi: 10.1046/j.1365-2567.2001.01337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168(7):3195–204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 58.Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, et al. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med. 1995;182(1):155–62. doi: 10.1084/jem.182.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fagarasan S, Honjo T. Intestinal IgA synthesis: regulation of front-line body defences. Nat Rev Immunol. 2003;3(1):63–72. doi: 10.1038/nri982. [DOI] [PubMed] [Google Scholar]
- 60.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300(5617):337–9. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 61.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300(5617):339–42. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434(7029):88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 63.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421(6925):852–6. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 64.Smith C, Martinez M, Cooper L, Rist M, Zhong J, Khanna R. Generating functional CD8+ T cell memory response under transient CD4+ T cell deficiency: implications for vaccination of immunocompromised individuals. Eur J Immunol. 2008;38(7):1857–66. doi: 10.1002/eji.200737933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316(5831):1628–32. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 66.Beignon AS, Mollier K, Liard C, Coutant F, Munier S, Riviere J, et al. Lentiviral vector-based prime/boost vaccination against AIDS: pilot study shows protection against Simian immunodeficiency virus SIVmac251 challenge in macaques. J Virol. 2009;83(21):10963–74. doi: 10.1128/JVI.01284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Escors D, Lopes L, Lin R, Hiscott J, Akira S, Davis RJ, et al. Targeting dendritic cell signaling to regulate the response to immunization. Blood. 2008;111(6):3050–61. doi: 10.1182/blood-2007-11-122408. [DOI] [PubMed] [Google Scholar]
- 68.Breckpot K, Escors D. Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification. Endocr Metab Immune Disord Drug Targets. 2009;9(4):328–43. doi: 10.2174/187153009789839156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arce F, Kochan G, Breckpot K, Stephenson H, Escors D. Selective Activation of Intracellular Signalling Pathways In Dendritic Cells For Cancer Immunotherapy. Anticancer Agents Med Chem. 2011 doi: 10.2174/187152012798764679. [DOI] [PubMed] [Google Scholar]
- 70.Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol. 2004;5(5):508–15. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- 71.Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11(2):138–45. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 72.Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, et al. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. 2005;201(9):1435–46. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6(8):769–76. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Warger T, Osterloh P, Rechtsteiner G, Fassbender M, Heib V, Schmid B, et al. Synergistic activation of dendritic cells by combined Toll-like receptor ligation induces superior CTL responses in vivo. Blood. 2006;108(2):544–50. doi: 10.1182/blood-2005-10-4015. [DOI] [PubMed] [Google Scholar]
- 75.Breckpot K, Dullaers M, Bonehill A, van Meirvenne S, Heirman C, de Greef C, et al. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J Gene Med. 2003;5(8):654–67. doi: 10.1002/jgm.400. [DOI] [PubMed] [Google Scholar]
- 76.Esslinger C, Chapatte L, Finke D, Miconnet I, Guillaume P, Levy F, et al. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J Clin Invest. 2003;111(11):1673–81. doi: 10.1172/JCI17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reis e Sousa C, Stahl PD, Austyn JM. Phagocytosis of antigens by Langerhans cells in vitro. J Exp Med. 1993;178(2):509–19. doi: 10.1084/jem.178.2.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, Trombetta S, et al. Developmental control of endocytosis in dendritic cells by Cdc42. Cell. 2000;102(3):325–34. doi: 10.1016/s0092-8674(00)00038-6. [DOI] [PubMed] [Google Scholar]
- 79.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–67. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 80.Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. 2011;3(10):581–92. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 82.Dullaers M, Van Meirvenne S, Heirman C, Straetman L, Bonehill A, Aerts JL, et al. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther. 2006;13(7):630–40. doi: 10.1038/sj.gt.3302697. [DOI] [PubMed] [Google Scholar]
- 83.Johnson S, Zhan Y, Sutherland RM, Mount AM, Bedoui S, Brady JL, et al. Selected Toll-like receptor ligands and viruses promote helper-independent cytotoxic T cell priming by upregulating CD40L on dendritic cells. Immunity. 2009;30(2):218–27. doi: 10.1016/j.immuni.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med. 1997;186(1):65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393(6684):474–8. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 86.Xiang J, Huang H, Liu Y. A new dynamic model of CD8+ T effector cell responses via CD4+ T helper-antigen-presenting cells. J Immunol. 2005;174(12):7497–505. doi: 10.4049/jimmunol.174.12.7497. [DOI] [PubMed] [Google Scholar]
- 87.Cekic C, Casella CR, Eaves CA, Matsuzawa A, Ichijo H, Mitchell TC. Selective activation of the p38 MAPK pathway by synthetic monophosphoryl lipid A. J Biol Chem. 2009;284(46):31982–91. doi: 10.1074/jbc.M109.046383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.