

Abstract
Electrical and structural remodeling during the progression of cardiovascular disease is associated with adverse outcomes subjecting affected patients to overt heart failure (HF) and/or sudden death. Dysfunction in integral membrane protein trafficking has long been linked with maladaptive electrical remodeling. However, little is known regarding the molecular identity or function of these intracellular targeting pathways in the heart. Eps15 homology domain-containing (EHD) gene products (EHD1-4) are polypeptides linked with endosomal trafficking, membrane protein recycling, and lipid homeostasis in a wide variety of cell types. EHD3 was recently established as a critical mediator of membrane protein trafficking in the heart. Here, we investigate the potential link between EHD3 function and heart disease. Using four different HF models including ischemic rat heart, pressure overloaded mouse heart, chronic pacing-induced canine heart, and non-ischemic failing human myocardium we provide the first evidence that EHD3 levels are consistently increased in HF. Notably, the expression of the Na/Ca exchanger (NCX1), targeted by EHD3 in heart is similarly elevated in HF. Finally, we identify a molecular pathway for EHD3 regulation in heart failure downstream of reactive oxygen species and angiotensin II signaling. Together, our new data identify EHD3 as a previously unrecognized component of the cardiac remodeling pathway.
Keywords: EHD proteins, heart failure, cardiac remodeling, regulation, animal models
1.0 Introduction
Heart disease is a leading cause of death worldwide. Despite therapeutic advances, heart failure (HF) remains responsible for substantial morbidity and mortality [1] with most deaths occurring as a result of cardiac arrhythmia. Following insult or injury, the heart undergoes both electrical and structural remodeling at the level of the single cell in an attempt to maintain contractility and cardiac output. However, in the setting of severe and/or chronic injury, the remodeling process ultimately may become maladaptive leading to further progression of disease.
Alterations to the expression and function of integral membrane proteins have been identified in HF [2–5]. Despite research linking dysfunction in membrane protein trafficking with human heart disease, little is known regarding the molecular identity of the intracellular targeting pathways underlying membrane protein remodeling. Eps15 homology domain (EHD) proteins (1–4) are key conductors of endosome-based protein trafficking and membrane protein recycling in a wide variety of species and cell types [6–9].
We recently demonstrated that EHD proteins are expressed in cardiac muscle and play key roles in membrane protein targeting and regulation [10]. In fact, one EHD gene product, EHD3, appears critical for cardiac myocyte membrane protein targeting and cell excitability. Specifically, consistent with the established role of EHD3 in endosomal trafficking in other cell types [7], EHD3 plays an indispensable role in the proper trafficking of the Na/Ca exchanger (NCX) to the myocyte cell membrane. In fact, knock-down of EHD3 in ventricular myocytes results in accumulation of NCX1 in intracellular peri-nuclear compartments, and loss of functional INCX. Currently, the link between EHD proteins and cardiac pathology is unknown. Based on this new role for EHD3 in heart, and the established roles of EHD proteins in membrane protein targeting and regulation in other cell types, we hypothesized that EHD3 levels would be altered in heart failure- where both membrane electrical and structural alterations are hallmarks of the disease.
Here we report the first link between EHD3 and HF. EHD3 levels are consistently increased across a host of animal models of heart disease including ischemic HF in rat, pressure overload-induced HF in mouse, and a canine model of HF induced by chronic tachypacing. Importantly, EHD3 levels are significantly altered in human HF. We also demonstrate a simultaneous increase of NCX1 expression in heart failure, providing evidence for a potential role of these proteins in membrane remodeling following myocardial insult. We provide the first evidence for a mechanism that modulates EHD3 levels in heart failure, supporting an unexpected new link between oxidative stress, angiotensin II (AngII), and EHD3 levels. Collectively, our data provide the first link between EHD3 proteins and human cardiovascular disease, providing a potential new pathway for future therapies to regulate membrane remodeling in disease.
2.0 Methods
For detailed Methods see On-line Supplement.
Primary cardiomyocyte cultures
Neonatal (post-natal day 1) hearts from wild-type and mutant mice euthanized via complete cervical dislocation (in accordance with NIH guidelines) were extracted and enzymatically-dispersed with trypsin and collagenase, plated on fibronectin-coated glass plates, and maintained in defined media as described [11].
Immunoblotting of heart proteins
Immunoblotting of cardiac tissue and cell lysates was performed as described [12]. Whole heart homogenates were centrifuged at 1,000 ×g to remove nuclei. Supernatant was used for protein quantification and immunoblotting. Normalized band densities were measured with Adobe Photoshop 8.0.
Immunofluorescence
Sections of rat hearts and adult mouse ventricular myocytes were washed with phosphate-buffered saline (PBS, pH 7.4) and fixed in 4% paraformaldehyde (37 °C) or ice cold ethanol, respectively. After blocking (see On-line Supplement) cells were incubated in primary antibody overnight at 4 °C (anti-EHD3, 1:50, Sigma Aldrich), washed and incubated with secondary antibody and co-labeled with Topro 3AM (1:2000, Invitrogen) to visualize nuclei. Images were collected on Zeiss 510 Meta confocal microscope. Cropping and linear contrast adjustments were made in in Adobe Photoshop. Figures represent typical staining patterns.
Histology
Rat hearts were harvested and fixed in 4% PFA. Tissues were sectioned and stained with Masson’s trichrome.
Immunoblots
Hearts or heart pieces were frozen in dry ice and ground into a fine powder. The powder was resuspended in lysis buffer and homogenyzed. EHD3 expression in ventricular lysates was determined by SDS-PAGE and immunoblotting using anti-EHD3, and anti-actin or anti-GAPDH (Santa Cruz Biotechnology) antibodies [10]. When applicable, myocytes were treated with ROS scavenger, N-(2-Mercaptopropionyl) glycine (2 mM, Sigma Aldrich), for 10 minutes. Normalized band densities were measured with Adobe Photoshop 8.0.
Human tissue samples
Left ventricular tissue was obtained from explanted hearts of patients undergoing heart transplantation through The Cooperative Human Tissue Network: Midwestern Division at The Ohio State University. Approval for use of human subjects was obtained from the Institutional Review Board of The Ohio State University. Left ventricular tissue from healthy donor hearts not suitable for transplantation was obtained through the Iowa Donors Network and the National Disease Research Interchange. The investigation conforms to the principles outlined in the Declaration of Helsinki. Age and sex were the only identifying information acquired from tissue providers.
Induction of myocardial infarction (MI) or sham operation
Adult male Sprague–Dawley rats (Harlan Sprague Dawley Inc.) weighing 350 to 375 g were anesthetized (ketamine 90 mg/kg + xylazine 10 mg/kg, IP) and underwent coronary ligation to induce HF (n=7) or sham (n=5), as described [13]. Adequate anesthesia was monitor via pain response to hind limb toe pinch. Rats underwent echocardiography under sedation (ketamine 25 mg/kg, IP) to assess left ventricular (LV) function and the extent of ischemic injury. Measurements of ischemic zone as a percentage of LV circumference, LV ejection fraction (EF), and LV end-diastolic volume (EDV) were made as described previously [14]. Large myocardial infarction was defined by an ischemic zone >39%. Six weeks later, rats underwent repeat echocardiography under sedation to ensure chronic HF. Hearts were excised from deeply anesthetized rats (0.2 mL/10g, 2% Avertin, I.P.) via rapid thoracotomy.
Mouse heart failure model with proximal aortic banding
Adult male B6 mice were subjected to pressure overload by TAC surgery as previously described [15]. Briefly, mice were anesthetized (ketamine, 100 mg/kg I.P. plus xylazine, 5 mg/kg), intubated, and placed on a respirator (120 breaths min−1, 0.1 mL tidal volume). Anesthesia was monitored by repeated hind limb response to pinch. Aorta was exposed via a midline sternotomy. A 6.0 Prolene suture was placed around the aorta distal to the brachiocephalic artery. The suture was tightened around a blunted 27-gauge needle placed next to the aorta (n=7). The needle was removed, and the chest was closed. Echocardiography was performed at regular intervals for two months to assess cardiac function. Banded mice had significantly decreased EF (68±5% vs. 49±6%, p<0.05) and significantly increased heart to body weight ratio (0.57±0.03 vs. 1.12±0.09, p<0.05) when compared with Sham mice. Age-matched littermates were use as sham operated controls (n=7). Hearts were harvested from sacrificed mice (2% Avertin, 20 μL/g, I.P.) via rapid thoracotomy. Adequate anesthesia was monitored via pain response to hind limb toe pinch.
Canine nonischemic dilated cardiomyopathy induced by chronic tachypacing
HF via chronic tachypacing was induced as previously described [16]. All dogs were initially verified to have normal cardiac function by examination by a veterinarian. Adult hound-type dogs of either sex (2–3 years of age) received pre-anesthetics of butorphanol 0.2 – 0.4 mg/kg IM, and morphine (0.5 – 2 mg/kg IV) followed by anesthesia induction with isoflurane; anesthesia was maintained with isoflurane (1.5 – 2.5 %). An RV pacemaker lead was implanted in the RV apex with fluoroscopic guidance followed by implantation of a modified Prevail 8086 pacemaker (Medtronic, Inc., Minneapolis, MN, USA). Following recovery from the pacemaker implant, the RV was paced at 180 bpm for 2 weeks; 200 bpm for the next 6 weeks, followed by 180 bpm for the duration of the protocol. Sequential echocardiograms were performed at baseline and during brief periods of sinus rhythm during butorphanol sedation (0.5 mg/kg, im) to confirm the presence of reproducible dilated cardiomyopathy. After 1 or 4 months of tachypacing, animals were administered butorphanol (0.2–0.4 mg/kg IM) followed by pentobarbital (25 mg/kg IV) or isoflurane (5% for induction, 2 % for anesthesia). After achieving a deep level of anesthesia, the heart was rapidly removed. Cardiac tissues were snap frozen in liquid nitrogen and stored at −80°C until used. Control dogs were sacrificed in parallel.
All animal studies were performed in accordance with the American Physiological Society Guiding Principles for Research Involving Animals and Human Beings, and approved by the University of Iowa or The Ohio State University Institutional Animal Care and Use Committee. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Statistics
When appropriate, data were analyzed using a two-tailed Students t-test, and values less than p<0.05 were considered significant. Values are expressed as the mean ± standard error.
3.0 Results
3.1 EHD3 levels are increased in a chronic rat heart failure model
We recently identified EHD3 as a new cardiac protein with roles in myocyte membrane protein targeting [10]. As a first step to analyze EHD3 protein regulation in cardiovascular disease, we examined EHD3 levels in a well-validated rat model of ischemic HF [17]. This model allowed for the assessment of EHD response to acute myocardial injury induced by infarct (MI). Remodeling post-MI begins nearly immediately in the border zone of the MI, and persists for weeks to months in the entire myocardium until cardiac function is normalized. Various neurohormonal signaling cascades are triggered as the result of the subsequently impaired cardiac output, many of which have been linked to myocyte apoptosis and hypertrophic remodeling [18].
Five weeks post-coronary ligation, overall heart size was increased with a thickening of the right ventricular free wall and septal wall, accompanied by a pronounced thinning of the left ventricular free wall within the infarct zone (Figure 1A & B) and depressed cardiac function. Average EDV was 1225±65 and 397±28 mm3 in infarcted and control rats, respectively (p<0.05). EF averaged 28.9±3.6% in infarcted rats versus 71.8±5.3% in Sham rats (p<0.05). Notably, EHD3 levels were significantly elevated in protein lysates from failing left ventricle (Figure 1C–D, 2.7-fold increased, n=7, p<0.05) compared to control. We also observed increased EHD3 levels in right ventricle free wall in HF lysates (Figure 1E–F, ~1.8-fold increased, n=5, p<0.05). Thus, EHD3 protein levels are increased in both remodeled ventricles in HF by immunoblot. Notably, and consistent with prior findings in a model of three to five week post-coronary artery ligation in canine [10], we observed no significant changes in ankyrin-B expression (previously identified EHD3 partner) in chronic rat HF left or right ventricle samples (Supplemental Figure 1). These data suggest that ankyrin-B and EHD3 levels are differentially regulated at this later stage of remodeling.
Figure 1. EHD3 expression in rat model of ischemic HF.
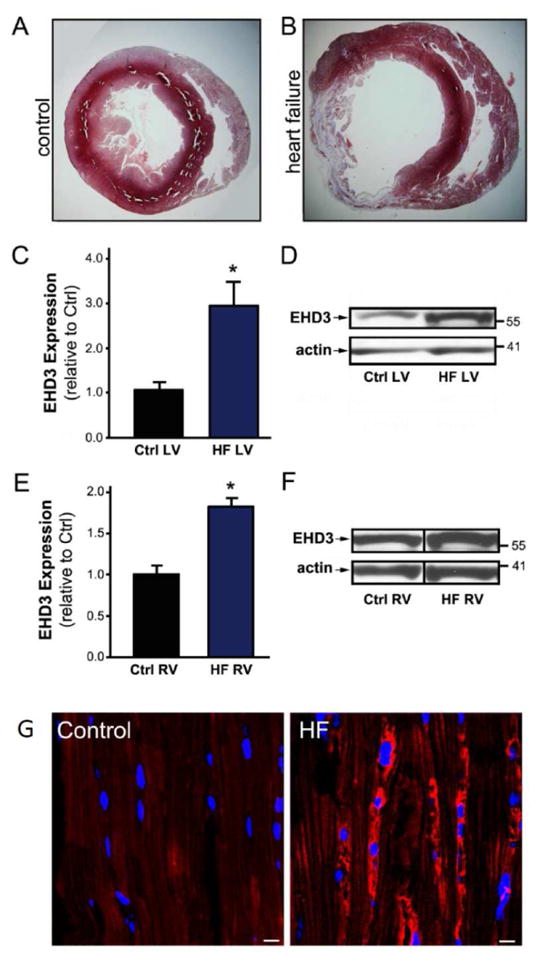
A & B) Tissue cross sections of control (A) and infarcted rat hearts (B). C) EHD3 expression was significantly increased in LV HF vs. control (n=7). D) Representative western blot of LV tissue. E) EHD3 expression was significantly increased in RV HF vs. control (n=5). F) Representative western blot of RV tissue. G) EHD3 expression visualized by immunofluorescence in control (left) and failing rat hearts (right). EHD3 stained in red, nuclei in blue. EHD3 levels are significantly increased in HF tissue samples. Scale bar = 20 μm.
Elevations in EHD3 protein levels by immunoblot were paralleled by increased EHD3 immunostaining in failing rat hearts. We not only observed increased EHD3 immunolabeling, but also peri-nuclear accumulation of EHD3 in left ventricle sections of rats with HF using EHD3-specific antibodies (Figure 1G). Together, our data provide the first evidence that EHD3 expression is increased in HF, consistent with their known role in endosome-based membrane protein targeting in cell models [6, 8].
3.2 EHD3 levels are elevated in a pressure overload mouse model of heart failure
We next evaluated EHD3 levels in a validated mouse model of non-ischemic HF induced by trans-aortic constriction (TAC) [19, 20]. This model allows assessment of EHD function in a nonischemic model of heart failure, for comparison to the ischemic rat model. Pressure-overload induced by TAC has been shown to trigger signaling pathways activated by mechanical stretch leading to activation of growth factors including angiotensin II (AngII) [21, 22]. Consistent with the rat ischemic HF model, EHD3 levels were significantly increased (1.75-fold, n=6 TAC; n=5 Sham; p<0.05) in the left ventricle of banded mice versus hearts from sham mice (Figure 2; see Methods). These data support a role for the EHD protein family in cardiac remodeling during hemodynamic overload. Our collective data in two different models of rodent heart failure reveal elevated EHD3 levels, suggesting a fundamental role for EHD3 in the remodeling process.
Figure 2. EHD3 expression is increased in mouse heart failure model.
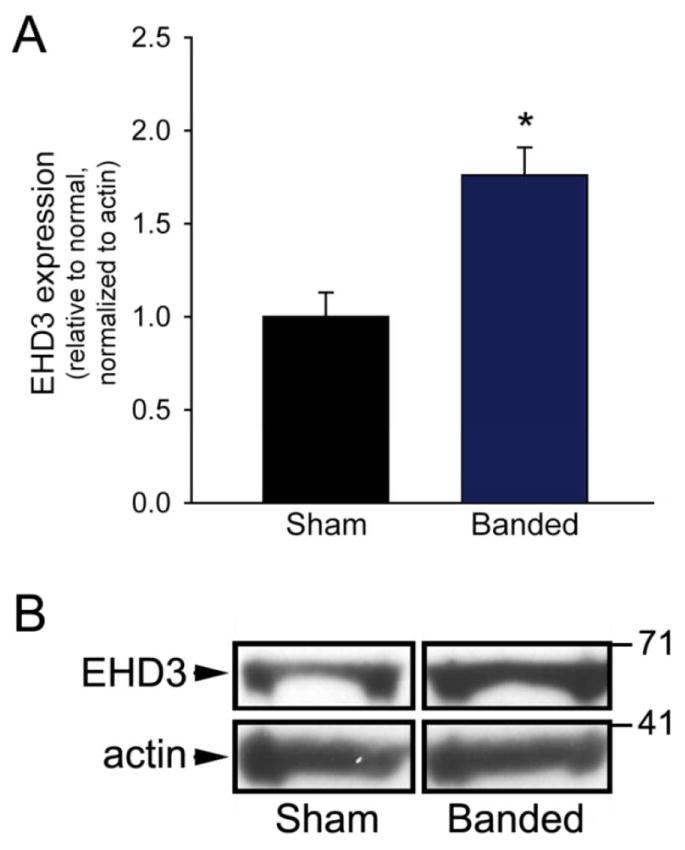
A) EHD3 expression is significantly increased in hearts (LV) of mice 8 weeks after TAC compared to Sham operated littermates. B) Representative immunoblot of LV tissue.
3.3 EHD3 levels are elevated in large animal model of heart failure
Rodent models of HF have been effectively used to identify and analyze specific protein pathways in cardiac pathology. However, the translation of rodent studies for understanding human disease may often be hampered by differences in the physiology of humans versus rodents (e.g., differences in heart rate, heart size, and metabolism). Therefore, we next used a large animal (canine) model of HF to evaluate the regulation of EHD3. This model provides a number of unique characteristics. First, cardiomyopathy induced by chronic tachypacing is non-ischemic in nature and does not require structural or exogenous hormonal insult to the heart. Second, the remodeling of the heart takes place over a longer term than the other models used here (up to 4 months). Therefore, assessing EHD function within this model provides insights into the EHD response to non-ischemic stress and how EHD function may change with long-term stimulus.
HF in canines was induced by rapid tachypacing for either 1 or 4 months, and cardiac function was assessed by echocardiogram (cardiomegaly, mitral regurgitation, left ventricular fractional shortening of less than 12%). EHD3 levels were compared by immunoblot from right atria, right ventricle, and left ventricle from control dogs, or after 1 or 4 months of pacing. Interestingly, similar to findings in rodent models of HF, we observed dramatic increases in EHD3 levels in all chambers (Figure 3, n=8 ctrl, n=4; 1 & 4 month, p<0.05). The alterations in EHD3 were equally prominent at 1 and 4 months of rapid pacing compared to control groups. These data suggest an important role of EHD3 in key remodeling events that occur during the primary phases of the development of heart disease as well as prolonged stress. In summary, data from several very different animal models of heart failure displayed increased levels of EHD3 protein expression, suggesting a fundamental and conserved role of EHD proteins in the response of the heart to stress.
Figure 3. EHD3 levels are elevated in canine HF model.
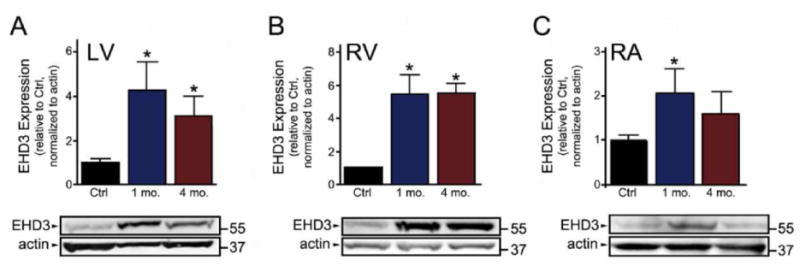
EHD3 expression in LV, RV and RA (A, B, and C, respectively) isolated from a canine model of tachypaced-induced HF. EHD3 expression was significantly increased in all chambers following 1 month of pacing, with significant increases maintained in the LV and RV after 4 months (n=8 for control; n=4 for 1 and 4 month, p<0.05).
3.4 EHD3 levels are increased in human heart failure
As a final step to assess the regulation of EHD3 in heart disease, we measured protein levels in left ventricle tissue from failing human hearts. Left ventricular free wall tissue was obtained from de-identified donors with history of non-ischemic HF. Samples from patients with documented HF showed significant increases in EHD3 protein levels versus control hearts (Figure 4, n=6 & 7 respectively, p<0.05). In summary, EHDs are differentially regulated in animal models of mammalian HF of widely variable etiologies, and in human heart failure, indicating that this response to cardiac stress is likely evolutionarily conserved and may be part of an even more universal mechanism regulating protein trafficking in heart at baseline and in disease.
Figure 4. EHD3 levels are elevated in human HF.
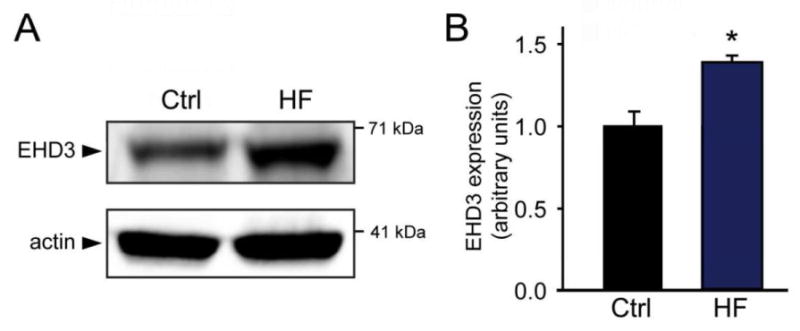
EHD3 expression in LV isolated from non-ischemic human HF tissue. A) Representative immunoblots of human HF samples. B) EHD3 expression is significantly increased in human HF versus non-failing controls (n=7, ctrl; 6 HF, respectively, p<0.05).
3.5 NCX1 expression levels are increased in HF
We previously demonstrated that NCX membrane targeting is dependent on EHD3 expression [10]. Therefore, we next tested the relationship of NCX expression with the increased expression of EHD3 in HF tissue examined. In all forms of HF examined, NCX1 protein expression was significantly increased (Figure 5). These data are consistent with the role of EHD3 for NCX membrane targeting in myocytes at baseline and in NCX remodeling in disease.
Figure 5. Increased NCX expression in HF models.
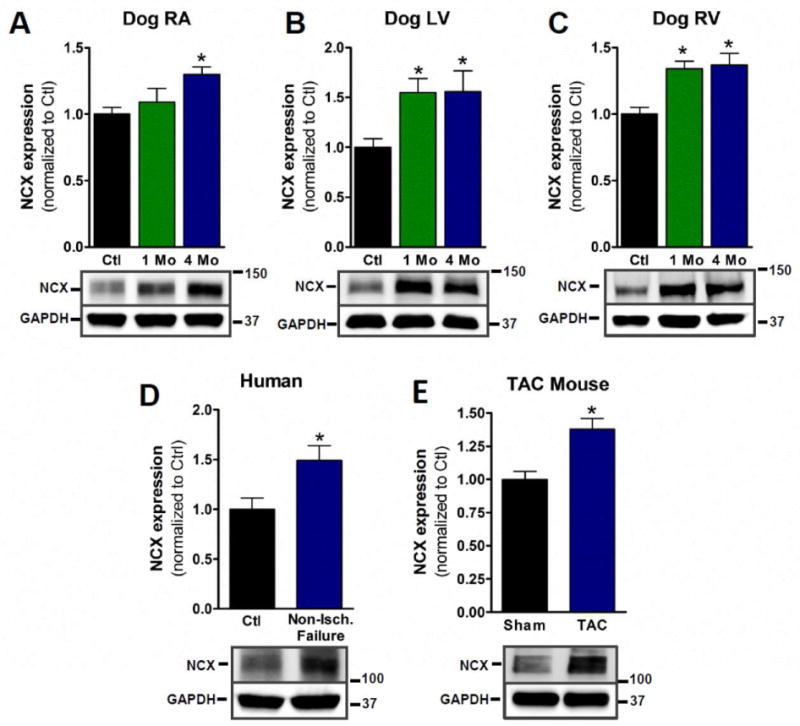
A–C) NCX is significantly increased in all chambers of dog HF model examined (n=4, 5 & 4, respectively, p <0.05). D) NCX expression in non-ischemic human HF (n=8 for control, 6 for non-ischemic, p<0.05). E) NCX expression in TAC mouse model (n=5, p<0.05).
3.6 EHD3 expression is regulated by reactive oxygen species (ROS)
Multiple molecular pathways play critical roles in the development and progression of HF including neuro-hormonal stimulation, inflammatory pathways, and apoptosis [23]. Reactive oxygen species (ROS) are oxygen-based second messenger molecules implicated in a host of human cardiac physiology and pathophysiological processes including cardiac hypertrophy and apoptosis. As a first step in assessing the pathways that may trigger elevated EHD3 expression in HF, we exposed cardiomyocytes to oxidative stress. Specifically, we analyzed EHD3 expression in mouse neonatal cardiomyocytes following 30, 60 or 90 minutes of treatment with hydrogen peroxide (75 μM) using a well-established protocol [24]. Notably, we observed an initial decrease in EHD3 expression following 30 and 60 minutes of treatment. However, EHD3 levels were nearly 1.4 fold higher than control levels following 90 minutes hydrogen peroxide treatment (Figure 6A–B, n=3, p<0.05). This increase was reversed in myocytes pretreated with ROS scavenger, 2-MPG (2 mM, Figure 6C). Similar results were observed in adult mouse myocytes (Figure 6D). As previously reported, EHD3 is localized to the perinuclear region in untreated myocytes with a small peripheral distribution also visible, consistent with its role in endosomal trafficking [10]. After treatment with H2O2, a striking increase in EHD3 was observed in both the perinuclear and cytosolic compartments as visualized by immunofluorescence (Supplemental Figure 2). These data suggest that EHD3 levels are dynamically regulated by ROS.
Figure 6. EHD3 expression is mediated via ROS downstream of angiotensin II.
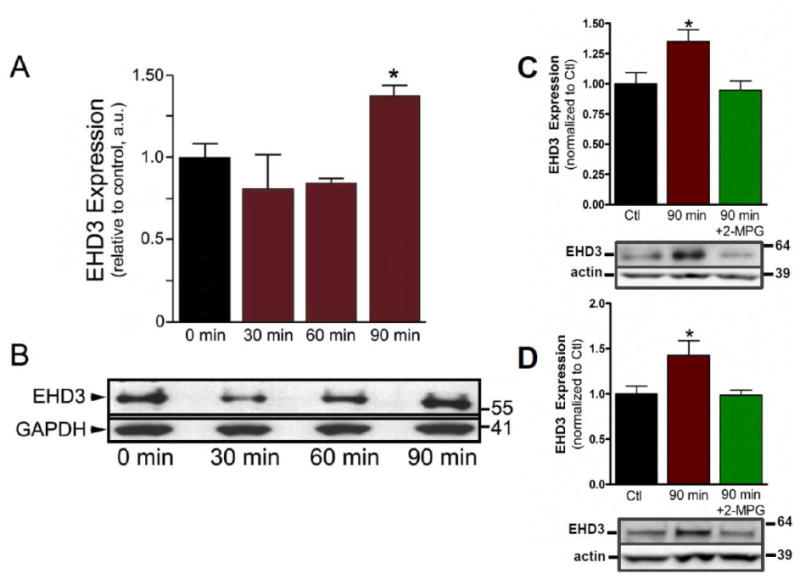
Each experiment represents all myocytes isolated from two hearts. A) EHD3 expression is significantly increased in neonatal cardiomyocytes after treatment with H2O2 (75 μM) for 90 min. (n=3). B) Representative western blot of experiments in A. C) H2O2-depenent increase in EHD3 expression is reversed by ROS scavenger, 2-MPG, with representative western blot (n=6, p<0.05). D) EHD3 expression in adult cardiomyocytes is increased after H2O2 treatment and in presence of ROS-scavenger with representative western blot (n=5, p<0.05).
3.7 ROS production downstream of AngII via NADPH oxidase regulates EHD3 expression in cardiomyocytes
Oxidative stress produces a number of global changes to myocyte cell physiology. Recent studies implicate nicotinamide-adenine dinucleotide phosphate (NADPH) oxidases (Nox) as a major source for cardiac ROS production [24, 25]. Five Nox isoforms have been described, but only Nox2 and Nox4 are expressed in cardiomyocytes [25, 26]. AngII causes increased ROS levels via an NADPH mechanism [27]. To determine whether EHD3 responds to increased ROS downstream of AngII, we treated mouse cardiomyocytes with AngII (75nM) for one, three, or 24 hours. Similar to treatment with hydrogen peroxide, we observed that with AngII treatment the EHD3 levels initially decreased, but then the expression increased with time (Figure 7, blue bars, n=4).
Figure 7. EHD3 expression depends on NADPH oxidase activity.
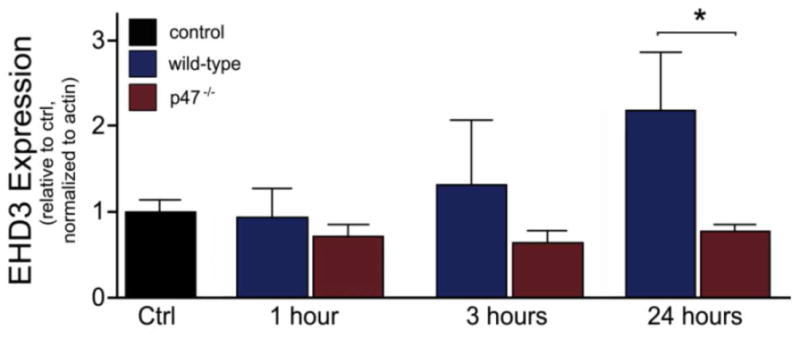
EHD3 expression is blunted in AngII-stimulated neonatal myocytes (75 nM) isolated from p47−/− hearts versus WT (n=4 WT, n=5, 3, 3, 6 for p47−/− treatments, respectively).
We performed parallel experiments in cardiomyocytes lacking p47phox (p47−/−), a critical subunit of NADPH oxidase. This mouse strain is unable to activate NADPH oxidase and produce superoxide which is the main source of ROS due to AngII-dependent stimulation [27]. Notably, in contrast to wild-type cardiomyocytes, p47-deficient cardiomyocytes showed no difference in EHD3 expression even following 24 hours of AngII-treatment compared to control. EHD3 expression was significantly attenuated in p47−/− hearts versus WT after 24 h (Figure 7, red bars, n=5, p<0.05). While there are likely other factors that regulate EHD3 expression in response to cardiac insult, our data demonstrate that EHD3 protein levels are regulated by ROS downstream of AngII and provide the first example of a common pathophysiological pathway underlying EHD3 activation.
4.0 Discussion
Here, we identify EHD3 as dynamically regulated in the failing heart. Notably, EHD3 expression is increased in both large and small models of HF with distinct etiologies. These models provide the first glimpse into how EHD3 responds in the various, progressive stages of HF. Critically, the increase in EHD3 expression in animal models was observed in human heart failure samples. These data suggest that the dynamic regulation of EHD3 in response to heart disease and subsequent cardiac remodelling is a conserved, universal response, and this response is likely initiated early in and maintained throughout the progression of cardiac disease. Furthermore, we show that EHD3 expression is dynamically regulated downstream of the AngII signaling cascade and is dependent upon the formation of ROS in cardiomyocytes.
EHD proteins share a common evolutionary past with the orthologous protein, RME-1, found in lower metazoans such as C. elegans and D. melanogaster [8]. In these species a single gene encodes RME-1. EHD1-4 are ubiquitously expressed in human tissue, and are encoded by four separate genes located on different chromosomes [28, 29]. EHD1-4 show high homology with RME-1 (67%), and the cellular functions of this proteins superfamily is conserved between species. In C. elegans lacking functional RME-1, endosomal recycling of integral membrane proteins was impaired but was rescued by the expression of any human EHD subtype. Furthermore, knockdown experiments in HeLa cells show that EHD1 and EHD2 mediate the exit of proteins from the endocytic recycling compartment (ERC), while EHD4 facilitates the transfer of proteins from the early endosome to the ERC [7]. A similar role for EHD3 was also described [30]. Recently, it was reported that EHD1-4 all bind to lipid tubules by forming dimers which oligomerize and tubulate charged liposomes [6, 31]. Thus, as proposed by others, EHD3 in heart may serve roles reminiscent of dynamin, acting to tubulate membranes and participate in budding vesicles from endosomal membrane-bound compartments [9, 32]. In other mammalian cell types, EHD proteins regulate membrane targeting of the transferrin receptor, MHC1 and 2 receptors, glucose transporter 4, and β1 integrin [6, 33–35]. Other binding partners of EHD include ARF6 [36], ankyrin-B [10] and various Rab effector proteins, confirming the role of EHD proteins in endosomal vesicular trafficking. Together, this evidence supports the hypothesis that EHD proteins serve as mediators of endocytic trafficking, and that EHDs will likely play similar roles in the heart. EHD1-4 are known to be expressed in the heart [10]. We are just beginning to understand the function of EHD3 in the heart, while specific roles for EHD1, EHD2 and EHD4 are completely unknown and require further investigation. Elevations in EHD3 levels in HF are likely produced through multiple molecular pathways. In fact, our data demonstrate that elevated EHD3 levels in heart failure are accompanied by transcriptional changes in EHD3 mRNA (Supplemental Figure 3).
Immunofluorescence studies in intact myocytes reveal increases in both perinuclear and cytosolic expression of EHD3 during HF and following stimulation with H2O2 (Supplemental Figure 2). While all published data on EHD3 have largely focused on anterograde/retrograde plasma membrane trafficking there is no data to believe that this is an exclusive role. In fact, cytoplasmic EHD3 localization may confer new roles for this protein family in trafficking to intracellular membranous compartments such as mitochondria or the sarcoplasmic reticulum[37]. In fact, both mitochondrial and SR proteins are known to be differentially regulated during HF, making these organelles compelling targets for further investigations into EHD-dependent trafficking [38].
We have previously reported that EHD3 is intimately involved in the proper trafficking of the Na/Ca exchanger (NCX) in cardiomyocytes [10]. NCX is the primary mode of Ca2+ efflux in the myocyte, however it also contributes to Ca2+ influx when it is in the “reverse mode” and intracellular sodium concentration is elevated. In nearly all reports, NCX expression is increased in HF (including human) [39–41]. The same was observed in human, dog and mouse HF models used here. This increased expression is likely a compensatory mechanism to restore normal contractility during heart failure [40, 42]. While a direct causal relationship is difficult to prove, we speculate that EHD3 is upregulated in HF in response to the increased demand put upon EHD3-dependent trafficking pathways, in this particular case the demand for increased NCX trafficking. Therefore, we speculate that increased EHD3 expression observed in human and animal models of HF is part of a larger, conserved response to restore normal heart function (by increasing NCX expression) during remodeling.
Increased ROS is commonly associated with HF. Changes in NADPH oxidase expression, localization, and activity have all been linked to increased ROS production in HF with etiologies ranging from MI, idiopathic dilated cardiomyopathy, to pressure overload. In particular, both NADPH isoforms expressed in the heart, Nox2 and Nox4, play a substantive role in oxidative stress during HF [43, 44]. The NADPH subunit, p22phox, was also shown to be increased in HF and critical to mediating ROS production[45]. Futher compounding this NADPH-dependent oxidative stress, a decrease in antioxidant enzyme activity has also been demonstrated in the failing heart [46]. Oxidative stress, however, is not exclusive to increased NADPH activity in HF. The functional uncoupling of nitric oxide synthase resulting in the production of superoxide has also been implicated as a source of oxidative stress during HF [47].
Activation of the renin-angiotensin-aldosterone system is one of the pathological processes associated with HF. Further, this system has been shown to be activated in all the animal models used here. AngII-induced hypertrophy and aldosterone stimulation are associated with increased ROS production in cardiac tissue via NADPH oxidase stimulation [25]. Critically, Byrne et al. previously demonstrated that Nox2 was essential for AngII-induced cardiac hypertrophy, while Nox4 plays no role in this response [48]. Nox2 requires the p47phox subunit for its activation and production of ROS, Nox4 does not [49]. Compared to control, EHD3 expression levels do not change in myocytes isolated from p47−/− mice when stimulated with AngII. These data suggest that the observed AngII-dependent increase in EHD3 depends on Nox2 activation, and the known increase in Nox2 expression in HF may play a central role in regulating this response [43, 45]. While, admittedly, our in vitro studies implement a brief timecourse of AngII treatement that do not directly address hypertrophy, our results strongly suggest that EHD3 expression is regulated within the AngII signalling cascade, and at a minimum may be involved in the early phase of the cellular remodeling process. To our knowledge, these are the first data demonstrating regulation of any EHD protein expression downstream in a neuro-hormonal signaling cascade in excitable tissue.
In summary, combined with recent findings [10], our new findings linking altered regulation in heart failure provide additional support for the role of EHD proteins in membrane protein regulation in health and disease. While our findings link activation with elevated ROS and AngII, we predict that other stimuli will also likely regulate EHD3 levels in attempts to restore membrane protein homeostasis. Future experiments in animal models lacking EHD proteins will be critical for defining the specific cell biological roles of these fascinating proteins in vivo in health and disease.
Supplementary Material
Highlights.
EHD3 is identified as a new protein associated with cardiac remodeling and disease.
EHD3 is differentially expressed in human HF and three animal models of HF.
Regulation of EHD3 is downstream AngII signaling and depends on ROS formation.
Acknowledgments
Funding
This work was supported in part by a grant from the Saving Tiny Hearts Society (PJM). This work was also supported by the National Institutes of Health [HL084583, HL083422 to PJM]; [HL079031, HL62494, HL70250 to MEA]; [CA105489, CA87986, CA99163, and CA116552 to HB]; [HL089836 to CAC]; [HL073986 to RBF]; [HL096805 to TJH]; Pew Scholars Trust (PJM), American Heart Association Established Investigator Award (PJM), Gilead Sciences Research Scholars Program (TJH), and Fondation Leducq Award to the Alliance for Calmodulin Kinase Signaling in Heart Disease (PJM, MEA, TJH). Pacemakers and leads were donated by Medtronic, Inc. (Minneapolis, MN, USA).
We would like to thank Shunguang Wei and Yang Yu for their important work in generating the infracted rat model.
Footnotes
Disclosures
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McMurray JJ, Pfeffer MA. Heart failure. Lancet. 2005;365:1877–89. doi: 10.1016/S0140-6736(05)66621-4. [DOI] [PubMed] [Google Scholar]
- 2.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–83. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Kaprielian R, Wickenden AD, Kassiri Z, Parker TG, Liu PP, Backx PH. Relationship between K+ channel down-regulation and [Ca2+]i in rat ventricular myocytes following myocardial infarction. J Physiol. 1999;517 ( Pt 1):229–45. doi: 10.1111/j.1469-7793.1999.0229z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Nolan B, Kutschke W, Hill JA. Na+-Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. J Biol Chem. 2001;276:17706–11. doi: 10.1074/jbc.M100544200. [DOI] [PubMed] [Google Scholar]
- 5.Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, et al. Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–53. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- 6.Caplan S, Naslavsky N, Hartnell LM, Lodge R, Polishchuk RS, Donaldson JG, et al. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. Embo J. 2002;21:2557–67. doi: 10.1093/emboj/21.11.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.George M, Ying G, Rainey MA, Solomon A, Parikh PT, Gao Q, et al. Shared as well as distinct roles of EHD proteins revealed by biochemical and functional comparisons in mammalian cells and C. elegans. BMC cell biology. 2007;8:3. doi: 10.1186/1471-2121-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grant B, Zhang Y, Paupard MC, Lin SX, Hall DH, Hirsh D. Evidence that RME-1, a conserved C. elegans EH-domain protein, functions in endocytic recycling. Nat Cell Biol. 2001;3:573–9. doi: 10.1038/35078549. [DOI] [PubMed] [Google Scholar]
- 9.Grant BD, Caplan S. Mechanisms of EHD/RME-1 protein function in endocytic transport. Traffic. 2008;9:2043–52. doi: 10.1111/j.1600-0854.2008.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gudmundsson H, Hund TJ, Wright PJ, Kline CF, Snyder JS, Qian L, et al. EH domain proteins regulate cardiac membrane protein targeting. Circ Res. 2010;107:84–95. doi: 10.1161/CIRCRESAHA.110.216713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunha SR, Mohler PJ. Obscurin Targets Ankyrin-B and Protein Phosphatase 2A to the Cardiac M-line. J Biol Chem. 2008;283:31968–80. doi: 10.1074/jbc.M806050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francis J, Weiss RM, Wei SG, Johnson AK, Felder RB. Progression of heart failure after myocardial infarction in the rat. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1734–45. doi: 10.1152/ajpregu.2001.281.5.R1734. [DOI] [PubMed] [Google Scholar]
- 14.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–31. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishijima Y, Feldman DS, Bonagura JD, Ozkanlar Y, Jenkins PJ, Lacombe VA, et al. Canine nonischemic left ventricular dysfunction: a model of chronic human cardiomyopathy. J Card Fail. 2005;11:638–44. doi: 10.1016/j.cardfail.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Francis J, Weiss RM, Johnson AK, Felder RB. Central mineralocorticoid receptor blockade decreases plasma TNF-alpha after coronary artery ligation in rats. Am J Physiol Regul Integr Comp Physiol. 2003;284:R328–35. doi: 10.1152/ajpregu.00376.2002. [DOI] [PubMed] [Google Scholar]
- 18.Tilley DG, Rockman HA. Role of beta-adrenergic receptor signaling and desensitization in heart failure: new concepts and prospects for treatment. Expert Rev Cardiovasc Ther. 2006;4:417–32. doi: 10.1586/14779072.4.3.417. [DOI] [PubMed] [Google Scholar]
- 19.Xiao CY, Chen M, Zsengeller Z, Li H, Kiss L, Kollai M, et al. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. The Journal of pharmacology and experimental therapeutics. 2005;312:891–8. doi: 10.1124/jpet.104.077164. [DOI] [PubMed] [Google Scholar]
- 20.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, et al. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci U S A. 1991;88:8277–81. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–71. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 22.Malhotra R, Sadoshima J, Brosius FC, 3rd, Izumo S. Mechanical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes In vitro. Circ Res. 1999;85:137–46. doi: 10.1161/01.res.85.2.137. [DOI] [PubMed] [Google Scholar]
- 23.Hilfiker-Kleiner DLU, Drexler H. Molecular Mechanisms in Heart Failure: Focuse on Cardiac Hypertrophy, Inflammation, Angiogensis, and Apoptosis. JACC. 2006;48(Supplement A) [Google Scholar]
- 24.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–84. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 26.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–6. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki H, Frank GD, Utsunomiya H, Higuchi S, Eguchi S. Current understanding of the mechanism and role of ROS in angiotensin II signal transduction. Curr Pharm Biotechnol. 2006;7:81–6. doi: 10.2174/138920106776597667. [DOI] [PubMed] [Google Scholar]
- 28.Pohl U, Smith JS, Tachibana I, Ueki K, Lee HK, Ramaswamy S, et al. EHD2, EHD3, and EHD4 encode novel members of a highly conserved family of EH domain-containing proteins. Genomics. 2000;63:255–62. doi: 10.1006/geno.1999.6087. [DOI] [PubMed] [Google Scholar]
- 29.Mintz L, Galperin E, Pasmanik-Chor M, Tulzinsky S, Bromberg Y, Kozak CA, et al. EHD1--an EH-domain-containing protein with a specific expression pattern. Genomics. 1999;59:66–76. doi: 10.1006/geno.1999.5800. [DOI] [PubMed] [Google Scholar]
- 30.Naslavsky N, Rahajeng J, Sharma M, Jovic M, Caplan S. Interactions between EHD proteins and Rab11-FIP2: a role for EHD3 in early endosomal transport. Mol Biol Cell. 2006;17:163–77. doi: 10.1091/mbc.E05-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blume JJ, Halbach A, Behrendt D, Paulsson M, Plomann M. EHD proteins are associated with tubular and vesicular compartments and interact with specific phospholipids. Exp Cell Res. 2007;313:219–31. doi: 10.1016/j.yexcr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Daumke O, Lundmark R, Vallis Y, Martens S, Butler PJ, McMahon HT. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007;449:923–7. doi: 10.1038/nature06173. [DOI] [PubMed] [Google Scholar]
- 33.Guilherme A, Soriano NA, Furcinitti PS, Czech MP. Role of EHD1 and EHBP1 in perinuclear sorting and insulin-regulated GLUT4 recycling in 3T3-L1 adipocytes. J Biol Chem. 2004;279:40062–75. doi: 10.1074/jbc.M401918200. [DOI] [PubMed] [Google Scholar]
- 34.Jovic M, Naslavsky N, Rapaport D, Horowitz M, Caplan S. EHD1 regulates beta1 integrin endosomal transport: effects on focal adhesions, cell spreading and migration. J Cell Sci. 2007;120:802–14. doi: 10.1242/jcs.03383. [DOI] [PubMed] [Google Scholar]
- 35.Walseng E, Bakke O, Roche PA. Major histocompatibility complex class II-peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J Biol Chem. 2008;283:14717–27. doi: 10.1074/jbc.M801070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma M, Naslavsky N, Caplan S. A role for EHD4 in the regulation of early endosomal transport. Traffic. 2008;9:995–1018. doi: 10.1111/j.1600-0854.2008.00732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poupon V, Begue B, Gagnon J, Dautry-Varsat A, Cerf-Bensussan N, Benmerah A. Molecular cloning and characterization of MT-ACT48, a novel mitochondrial acyl-CoA thioesterase. J Biol Chem. 1999;274:19188–94. doi: 10.1074/jbc.274.27.19188. [DOI] [PubMed] [Google Scholar]
- 38.Hollander JM, Baseler WA, Dabkowski ER. Proteomic remodeling of mitochondria in heart failure. Congest Heart Fail. 2011;17:262–8. doi: 10.1111/j.1751-7133.2011.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sipido KR, Volders PG, Vos MA, Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc Res. 2002;53:782–805. doi: 10.1016/s0008-6363(01)00470-9. [DOI] [PubMed] [Google Scholar]
- 40.Sipido KR, Volders PG, de Groot SH, Verdonck F, Van de Werf F, Wellens HJ, et al. Enhanced Ca(2+) release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000;102:2137–44. doi: 10.1161/01.cir.102.17.2137. [DOI] [PubMed] [Google Scholar]
- 41.Sipido KR, Volders PG, Schoenmakers M, De Groot SH, Verdonck F, Vos MA. Role of the Na/Ca exchanger in arrhythmias in compensated hypertrophy. AnnNYAcadSci. 2002;976:438–45. doi: 10.1111/j.1749-6632.2002.tb04773.x. [DOI] [PubMed] [Google Scholar]
- 42.Weisser-Thomas J, Kubo H, Hefner CA, Gaughan JP, McGowan BS, Ross R, et al. The Na+/Ca2+ exchanger/SR Ca2+ ATPase transport capacity regulates the contractility of normal and hypertrophied feline ventricular myocytes. J Card Fail. 2005;11:380–7. doi: 10.1016/j.cardfail.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Krijnen PA, Meischl C, Hack CE, Meijer CJ, Visser CA, Roos D, et al. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J Clin Pathol. 2003;56:194–9. doi: 10.1136/jcp.56.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–70. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukui T, Yoshiyama M, Hanatani A, Omura T, Yoshikawa J, Abe Y. Expression of p22-phox and gp91-phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem Biophys Res Commun. 2001;281:1200–6. doi: 10.1006/bbrc.2001.4493. [DOI] [PubMed] [Google Scholar]
- 46.Hill MF, Singal PK. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol. 1996;148:291–300. [PMC free article] [PubMed] [Google Scholar]
- 47.Nishijima Y, Sridhar A, Bonilla I, Velayutham M, Khan M, Terentyeva R, et al. Tetrahydrobiopterin depletion and NOS2 uncoupling contribute to heart failure-induced alterations in atrial electrophysiology. Cardiovasc Res. 2011;91:71–9. doi: 10.1093/cvr/cvr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, et al. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93:802–5. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- 49.Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 2006;21:269–80. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.