

Abstract
Respiratory systems are constantly being challenged by pathogens. Lung epithelial cells serve as a first line of defense against microbial pathogens by detecting pathogen-associated molecular patterns (PAMPs) and activating downstream signaling pathways, leading to a plethora of biological responses required for shaping both the innate and adaptive arms of the immune response. Acute-phase proteins (APPs), such as type 1 plasminogen activator inhibitor (PAI-1), play important roles in immune/inflammatory responses. PAI-1, a key regulator for fibrinolysis and coagulation, acts as an APP during acute phase response (APR) such as acute tissue injury (ALI), inflammation, and sepsis. However, the role of PAI-1 in the pathogenesis of these diseases still remains unclear, especially in bacterial pneumonia. In this study, we showed that PAI-1 expression is upregulated following nontypeable Haemophilus influenzae (NTHi) infection. PAI-1 knockout (KO) mice failed to generate early immune responses against NTHi. Failure of generating early immune responses in PAI-1 KO mice resulted in reduced bacterial clearance and prolonged disease process, which in turn led to enhanced inflammation at late stage of infection. Moreover, we also found that NTHi induces PAI-1 via activation of TLR2-MyD88-MKK3-p38 MAPK signaling pathway. These data suggest that PAI-1 plays critical role in earl host defense response against NTHi infection. Our study thus reveals a novel role of PAI-1 in infection caused by NTHi, one of the most common gram-negative bacterial pathogens in respiratory systems.
Keywords: Nontypeable Haemophilus influenzae, type 1 plasminogen activator inhibitor, inflammation, pneumonia, innate immune response
Introduction
Respiratory systems are constantly being challenged by pathogens. Pneumonia, a lung infection, is one of the most common and potentially serious infections, causing two million deaths annually among young children in low-income countries. It is also the most common cause of death from infection in the USA [1]. Lung epithelial cells are the most abundant cell type in lung, which are lining the airway surface and thus serve as a first defense against microbial pathogens. Epithelial cells detect microbial pathogens via pattern recognition receptors (PRRs) such as toll-like receptors (TLRs). Recognition of pathogen-associated molecular patterns (PAMPs) by PRRs activates downstream signaling pathways, leading to a plethora of biological responses required for shaping both the innate and adaptive arms of the immune response [2,3,4]. Thus, early activation of PRRs by PAMPs and their downstream signaling pathways, such as Mitogen-activated protein kinases (MAPKs) and Nuclear Factor-kappa B (NF-κB), have been found to be critical for host defense against microbial infections as the failure in this early defense response due to the genetic defects in PRRs or their key downstream signaling molecules resulted in uncontrolled bacterial growth and detrimental inflammation [5,6]. Therefore, immediate and adequate activation of PRRs, their signaling pathways and subsequent immune responses is required for antimicrobial protection.
The immediate host defense response, also known as an acute-phase response (APR), was originally thought to be nonspecific physiological and biochemical responses to the most forms of tissue damage, infection, inflammation, and malignant neoplasia. Expressions of acute-phase proteins (APPs), such as proteinase inhibitors, coagulation factors, complements, and transport proteins, are rapidly upregulated following infectious and/or non-infectious injuries. APPs, thus, have been known as biological marker proteins for the presence of inflammation and/or injuries [7,8]. A growing body of study, however, indicates that APPs are not only inflammation markers but also important regulators for immune/inflammatory responses including recruitment of inflammatory cells into the local tissues, opsonization of microbial pathogens, and prevention of microbial dissemination by trapping pathogens. However, their precise role in immune/inflammatory responses is not yet fully understood.
Type 1 plasminogen activator inhibitor (PAI-1) is a key regulator for fibrinolysis and coagulation by counteracting with plasminogen activators (PAs), such as urokinase (uPA) and tissue-type PA (tPA) [9]. Beside its original role in fibrinolysis and coagulation, PAI-1 acts as an APP during APR such as acute tissue injury (ALI), inflammation, and sepsis [10,11,12,13,14]. Especially in lung tissues, the involvement of PAI-1 in disease processes of ALI and pneumonia has been suggested, but the role of PAI-1 in these diseases still remains unclear. We previously reported that PAI-1 expression is upregulated following Streptococcus pneumoniae (S.p.) infection and plays a critical for protecting against S.p. cytolytic toxin pneumolysin-induced ALI [15]. Hence, deficiency of PAI-1 in mice results in enhanced alveolar hemorrhage and mortality during severe S.p. infection. However, the role of PAI-1 in infection with nontypeable Haemophilus influenzae (NTHi), the most frequently isolated gram-negative bacterial pathogen in respiratory infection, is totally unknown.
In this study, we found that PAI-1 expression is upregulated following NTHi infection both in vitro epithelial cells and in vivo lung tissues of mice. PAI-1 knockout (KO) mice exhibited low expression of cytokines and chemokines and reduced polymorphonuclear (PMN) leukocytes recruitment into airway. Failure of early immune responses in PAI-1 KO mice indeed resulted in reduced bacterial clearance and enhanced disease progress, thereby resulting in enhanced inflammation at late stage of infection. In addition, we also found that NTHi induces PAI-1 via activation of TLR2-MyD88-MKK3-p38 MAPK signaling pathway. Together, these data suggest that PAI-1 plays critical role in earl host defense response during NTHi infection.
Materials and Methods
Reagents
SB203580, PD98059 and SP600125 were purchased from Calbiochem (La Jolla, CA, USA). Total PAI-1 antigen ELISA kit and active PAI-1 antigen ELISA kit were purchased from Molecular Innovations (Novi, MI, USA).
Bacterial strains and culture condition
NTHi strain 12, a clinical isolate, was used in this study [16]. Cells were treated with NTHi at a concentration of 150 multiplicity of infection (MOI). For in vivo animal experiments, mid-log phase of NTHi obtained from 6 hours after incubation were prepared at the concentration of 1 × 108 colony forming unit (CFU) per ml in saline by centrifugation followed by washing with sterile saline.
Cell Culture
Human lung epithelial A549 and cervix epithelial HeLa cells were maintained as described previously [15,17]. All media were supplemented with 10% fetal bovine serum, penicillin (100 U/ml) and streptomycin (0.1 mg/ml). All cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C.
Plasmids, Transfection and Luciferase Assay
The PAI-1 Luc report construct (PAI-1 Luc) and dominant-negative mutant (DN) forms of TLR2, TLR4, MyD88, p38α, and p38β2 were described previously [18,19]. Transient transfection of cells was carried out in triplicate with TransIT-LT1 reagent (Mirus, Medison, WI) following manufacturer's instruction. For experiments with inhibitors, the transfected cells were pretreated with chemical inhibitors including SB203580 (10 μM), PD98059 (10 μM), and SP600125 (10 μM) for 1 hour prior to NTHi treatment. Luciferase activity was conducted as described previously [15,17].
Real-time Quantitative PCR (Q-PCR) Analysis
Q-PCR analysis of human PAI-1 and mouse PAI-1, TNF-α, IL-1β, MIP-2, and KC was conducted as follows. Total RNA was isolated using TRIzol reagent (Invitrogen) following the manufacturer's instructions. The reverse transcription reaction was performed using TaqMan reverse transcription reagents (Applied Biosystems). PCR amplification was performed with SYBR Green Universal Master Mix (Applied Biosystems). Reactions were amplified and quantified by using an ABI 7500 sequence detector and the manufacturer's software (Applied Biosystems). Relative quantity of human and mouse mRNA was obtained by using the comparative threshold cycle (Ct) Method and was normalized using human and mouse GAPDH as an endogenous control. Primers for human PAI-1 and mouse PAI-1, TNF-α, IL-1β, MIP-2, and KC were described previously [15,17,20,21].
Animal Experiments
C57BL/6 mice were purchased from National Cancer Institute, NIH. TLR2 KO, MyD88 KO, MKK3 KO, and PAI-1 KO mice were descried previously [15,17,20,21]. Eight weeks old male mice were used in these experiments. For NTHi infection, animals were intratracheally (i.t.) inoculated with 5 × 106 CFU of NTHi in 50 μl of saline. Mice were then sacrificed by intraperitoneal (i.p.) injection of sodium pentobarbital (100 mg/kg in 100 μl of saline) at the time indicated in the figures, and blood was collected from abdominal vena cava. Bronchoalveolar lavage (BAL) was conducted as described previously to collect BAL fluid (BALF) and polymorphonuclear (PMN) leukocytes [17,21]. Total RNA was isolated from the lung tissues using TRIzol reagent (Invitrogen) following manufacturers instruction. Reverse transcription and Q-PCR analysis were conducted as described above. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at University of Rochester.
Statistical Analysis
Statistical analysis was performed with student t-test. P values of less than 0.05 were considered statistically significant.
Results and Discussion
Expression of PAI-1 is upregulated during NTHi infection both in epithelial cells in vitro and in lung of mice in vivo
Plasminogen activator inhibitors (PAIs) are a subgroup of the serine protease inhibitor (serpin) superfamily including PAI-1, PAI-2, PAI-3, and protease nexin 1 (PN-1) [22,23]. Among them, PAI-1 is the most extensively studied serpin member because of its pleiotropic functions in biological processes such as wound healing, tumor angiogenesis, bone remodeling, asthma, rheumatoid arthritis, fibrosis, sepsis, and acute lung injury (ALI) [13]. It has been reported that PAI-1 expression is up-regulated during bacterial pneumonia with Klebsiella pneumoniae (K.p.), Pseudomonas aeruginosa (P.a.), and Streptococcus pneumoniae (S.p.), and lung epithelial cells indeed were found to express PAI-1 following intratracheal (i.t.) inoculation of lipopolysaccharide (LPS) in mice [15,24,25,26,27]. However, the role of PAI-1 in infection with NTHi, the most common gram-negative bacterial pathogen in respiratory systems, is unknown. To determine the role of PAI-1 in NTHi infection, we first investigated if PAI-1 expression is regulated during NTHi infection in epithelial cells and also in lung tissues of mice. Following NTHi treatment, NTHi up-regulated PAI-1 expression at mRNA level (Fig. 1A) and also at transcriptional level in a dose-dependent manner (Fig. 1B). Next, we determined if PAI-1 expression is indeed upregulated in vivo in lung tissues of mice following NTHi infection. Consistent with our in vitro finding, mRNA expression of PAI-1 was greatly upregulated in the lung tissues of mice following NTHi infection (Fig. 1C), and enhanced expression level of PAI-1 antigen was also detected in the bronchoalveolar lavage fluid (BALF) of mice infected with NTHi (Fig. 1D).
Figure 1. Expression of PAI-1 is upregulated during NTHi infection both in epithelial cells in vitro and in lung tissues of mice in vivo.
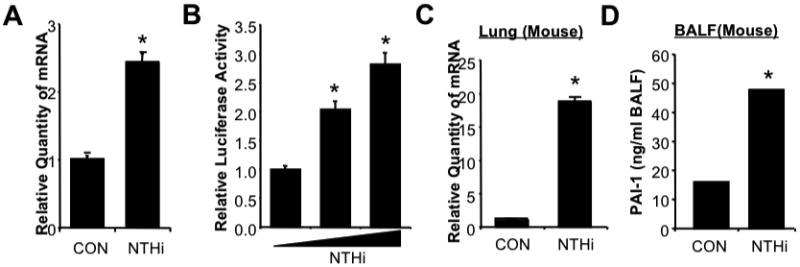
A. NTHi induces PAI-1 mRNA expression in airway epithelial A549 cells. Cells were treated with NTHi or PBS as control for 5 hours, and mRNA expression of PAI-1 was measured by using real time quantitative RT-PCR (Q-PCR) analysis. B. NTHi induces PAI-1 transcription in epithelial cells. Cells were transfected with PAI-1-Luc construct and treated with NTHi or PBS 48 hours after transfection. PAI-1 transcription was measured 5 hours after NTHi treatment by using luciferase assay. C. NTHi induces PAI-1 mRNA expression in lung tissues of wild-type (WT) mice. C57BL/6 mice were intratracheally (i.t.) inoculated with NTHi or saline as a control, and mRNA expression of PAI-1 was measured from the lung tissues of mice inoculated with NTHi or saline by using Q-PCR analysis. D. NTHi induces PAI-1 protein expression in lungs of WT mice. C57BL/6 mice were i.t. Inoculated with NTHi or saline as a control, and expression of total PAI-1 protein was measured from the bronchoalveolar lavage fluid (BALF) of mice inoculated with NTHi or saline by using total PAI-1 antigen ELISA assay kit. The values presented in A-D are Mean ± SD (n=3). *, p<0.05 compared with CON; CON: control.
Deficiency of PAI-1 impairs early host defense against NTHi infection, thereby resulting in uncontrolled inflammation at late stage of infection in PAI-1 KO mice
Since we found that PAI-1 expression is upregulated following NTHi infection, and we also previously reported that PAI-1 plays a critical role for early defense against bacterial infection with S.p. [15], we determined if PAI-1 plays a critical role on NTHi pneumonia by assessing early innate immune responses following NTHi infection. PAI-1 KO mice and their littermate wildtype (WT) control mice were i.t. inoculated with NTHi, and mRNA expression of pro-inflammatory cytokines and chemokines were measured 6 hours after infection. As shown in Fig. 2A, deficiency of PAI-1 in PAI-1 KO mice results in decreased expression of pro-inflammatory cytokines such as IL-1β and TNF-α We also found that mRNA expression of chemokines MIP-2 and KC was significantly inhibited in the lung tissues of PAI-1 KO mice, thereby resulting in decreased PMN recruitment into airway (Fig. 2B). Early innate immune responses against bacterial pathogens are critical for controlling microbial infections by inhibiting bacterial growth. We thus evaluated if reduced early innate immune responses against NTHi in PAI-1 KO mice affect pathogenesis process of NTHi in these mice. Twenty-four hours after NTHi infection, bacterial load in lung and body temperature of mice were measured in PAI-1 KO and their littermate WT control mice. Bacterial counts in the lung tissues were significantly enhanced in PAI-1 KO mice compared to that in WT mice (Fig. 2C). In addition, PAI-1 KO mice showed severe hypothermia, a physiological index of disease severity of pneumonia in mice [20], compared to WT mice infected with NTHi (Fig. 2D). Furthermore, failure for controlling bacterial outgrowth at early stage of infection in PAI-1 KO mice results in enhanced expression of pro-inflammatory cytokines IL-1β and TNF-α and chemokine MIP-2 (Fig. 2E) at 24 and 48 hours after infection. These data indicate that early upregulation of PAI-1 by NTHi is critical for early defense against NTHi infection by enhancing anti-microbial immune responses and thus serves to limit bacterial growth in lung.
Figure 2. Deficiency of PAI-1 impairs early defense against NTHi infection, thereby resulting in uncontrolled inflammation at late stage of infection in PAI-1 KO mice.
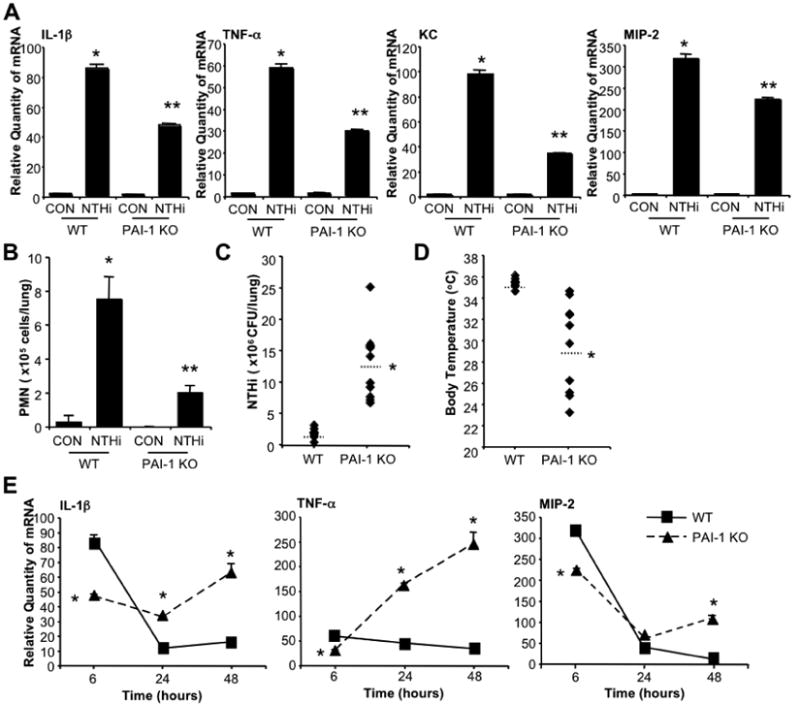
A. PAI-1 KO mice fail to induce early pro-inflammatory responses against NTHi infection. PAI-1 KO mice and their littermate WT control mice were i.t. inoculated with NTHi or saline as a control, and mRNA expression of pro-inflammatory cytokines IL-1β and TNF-α and chemokines KC and MIP-2 were measured from the lung tissues of mice 6 hours after NTHi inoculation. B. Polymorphonuclear (PMN) leukocyte migration was inhibited in PAI-1 KO mice following NTHi infection. PAI-1 KO and littermate WT mice were i.t. inoculated with NTHi or saline as a control, and PMN cells migrated into lung was measured from BALF of mice inoculated with NTHi or control. C & D. PAI-1 KO mice showed reduced bacterial clearance against NTHi infection. PAI-1 KO and littermate WT mice were i.t. inoculated with NTHi, and in vivo bacteria growth was measured from the lung tissues of mice 24 hours after NTHi inoculation (C), and body temperature was measured before sacrifice (D). E. Loss of early defense against NTHi infection in PAI-1 KO mice results in enhanced inflammation at late stage of infection. PAI-1 KO and littermate WT mice were i.t. inoculated with NTHi, and mRNA expression of pro-inflammatory cytokines IL-1β and TNF-α and chemokine MIP-2 was measured from the lung tissues of mice 6, 24, and 48 hours after NTHi infection. The values presented in A - D are Mean ± SD (n=3 in A & B, n=10 in C & D). *, p<0.05 compared with CON in WT; **, p<0.05 compared with NTHi in WT. The values presented in E are Mean ± SD (n=3); *, p<0.05 compared with NTHi in WT. CON: control; WT: wild-type; KO: knock-out.
NTHi induces PAI-1 upregulation via TLR2/MyD88 pathway
Having identified PAI-1 as a critical defense factor for NTHi infection, we next sought to determine the molecular mechanism by which PAI-1 expression is upregulated by NTHi. Because recognition of PAMPs by PRR such as TLRs plays an important role in initiating innate immune response in epithelial cells [2,3,4,5,6], we first determined if TLRs are responsible for mediating NTHi-induced PAI-1 expression. Overexpressing dominant-negative (DN) mutant construct of TLR2 and its cytoplasmic adaptor MyD88, but not TLR4, greatly inhibited NTHi-induced PAI-1 transcriptional activity (Fig. 3A). To further confirm their involvement in vivo, TLR2 KO, MyD88 KO, and their littermate WT control mice, were i.t. inoculated with NTHi, and mRNA expression of PAI-1 was measured 6 hours after NTHi infection in lung tissues of mice. Consistent with our in vitro findings, PAI-1 mRNA expression was significantly lower in TLR2 KO and MyD88 KO mice compared to WT mice (Fig. 3B & C), thereby indicating that PAI-1 induction by NTHi is dependent on TLR2/MyD88 pathway.
Figure 3. NTHi induces PAI-1 upregulation via TLR2/MyD88 pathway.
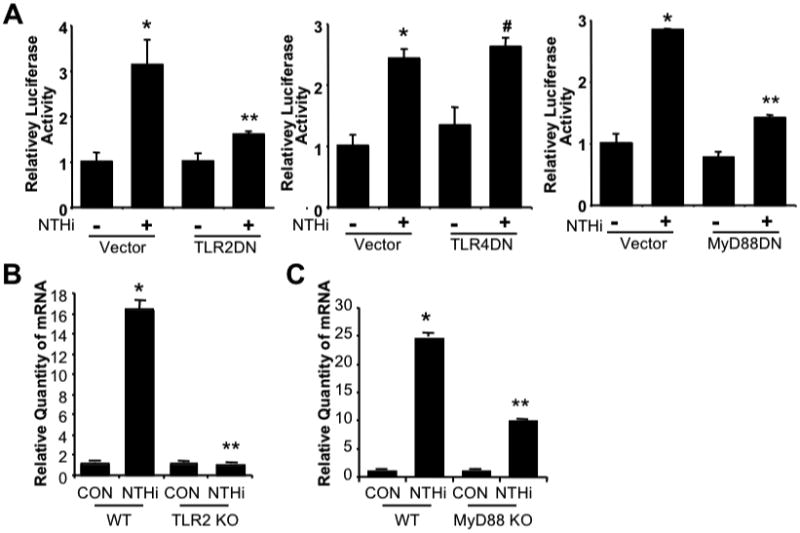
A. NTHi-induced PAI-1 expression is dependent on TLR2/MyD88, but not on TLR4 in vitro in epithelial cells. Cells were transfected with PAI-1-Luc construct with Vector, TLR2 DN, TLR4 DN, or MyD88 DN for 48 hours. Transfected cells were then treated with NTHi or PBS as a control, and PAI-1 transcription was measured 5 hours after NTHi or control treatment by luciferase assay. B & C. NTHi induces PAI-1 expression via TLR2 & MyD88 in vivo in mice. TLR2 KO, MyD88 KO, or their littermate WT control mice were i.t. inoculated with NTHi or saline as a control, and mRNA expression of PAI-1 was measured from the lung tissues of mice 6 hours after NTHi or control inoculation. The values presented in A-C are Mean ± SD (n=3). *, p<0.05 compared with CON; #,p>0.05 compared with NTHi in Vector (A) or WT (B & C). CON: control; WT: wild-type; KO: knock-out; DN; dominant-negative.
NTHi-induced PAI-1 expression is mediated by MKK3-p38 MAPK
Next, we determined which signaling pathway downstream of TLR2/MyD88 is involved in NTHi-induced PAI-1 expression. Because it has been reported that MAPK signaling pathways are involved in PAI-1 expression and NTHi potently activates MAPKs, especially for p38 MAPK [16,19,28], we determined if MAPKs are involved in NTHi-induced PAI-1 expression in lung. Epithelial cells were first pretreated with specific chemical inhibitors for MAPK p38, ERK, and JNK, and PAI-1 mRNA expression and transcriptional activity were then measured. Interestingly, SB203580, a specific p38 MAPK inhibitor, inhibited NTHi-induced PAI-1 mRNA expression and also transcriptional activity of PAI-1, whereas ERK inhibitor PD98059 or JNK inhibitor SP600125 exhibits no effect on NTHi-induced PAI-1 upregulation (Fig. 4A & B). These data suggest that p38 MAPK is likely involved in NTHi-induced PAI-1 expression in lung. We further determined if p38 MAPK is indeed critically involved in NTHi-induced PAI-1 expression by assessing the effect of p38αDN and p38β2 DN on NTHi-induced PAI-1 expression. Overexpressing p38α(AF) (p38αDN) and p38β2(AF) (p38β2 DN) greatly inhibited NTHi-induced PAI-1 mRNA expression (Fig. 4C) and also transcriptional activity of PAI-1 in epithelial cells in vitro (Fig. 4D). Moreover, NTHi-induced PAI-1 expression was significantly reduced in lung tissues of MKK3 KO mice compared to littermate WT in vivo. Together, these data indicate that NTHi-induced PAI-1 expression is mediated by MKK3-p38 MAPK signaling pathway both in vitro and in vivo.
Figure 4. NTHi-induced PAI-1 expression is mediated by MKK3-p38 MAPK.
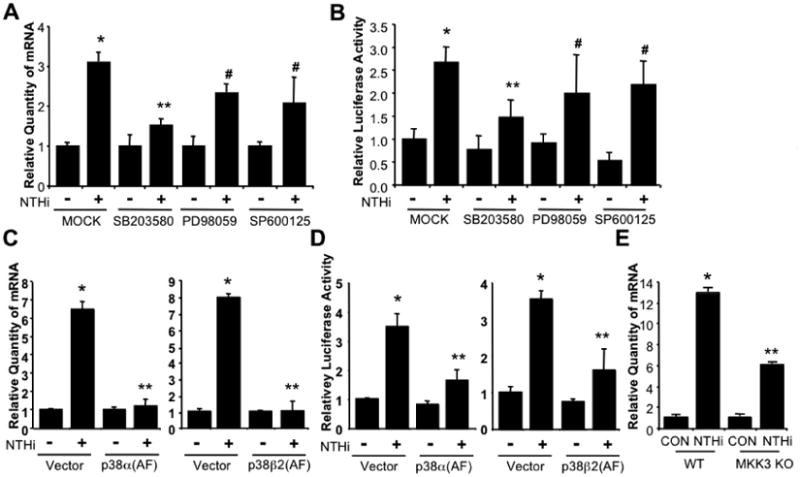
A & B. PAI-1 induction by NTHi is mediated by p38 MAPK but not ERK or JNK MAPKs in vitro in epithelial cells. Cells were pre-treated with specific chemical inhibitors for p38 (SB203580, 10 μM), ERK (PD98059, 10 μM), or JNK (SP600125, 10 μM) for 1 hour followed by NTHi or saline treatment. PAI-1 mRNA expression was measured by Q-PCR analysis 5 hours after NTHi treatment (A). Cells transfected with PAI-1-Luc were pre-treated with specific chemical inhibitors for p38 (SB203580, 10 μM), ERK (PD98059, 10 μM), or JNK (SP600125, 10 μM) for 1 hour followed by NTHi or saline treatment. PAI-1 transcriptional activity was measured by luciferase assay 5 hours after NTHi treatment (B). C & D. p38 MAPK mediates NTHi-induced PAI-1 expression in vitro in cells. Cells were transfected with control Vector, p38α(AF), or p38β2(AF) for 48 hours and then treated with NTHi or saline. mRNA expression of PAI-1 was measured 5 hours after NTHi treatment by Q-PCR analysis (C). Cells were co-transfected with PAI-1-Luc with or without p38α(AF) or p38β2(AF) for 48 hours and then treated with NTHi or saline. PAI-1 transcription was measured by luciferase assay 5 hours after NTHi treatment (D). E. NTHi induces PAI-1 expression in a MKK3 dependent manner. MKK3 KO and their littermate WT control mice were i.t. inoculated with NTHi, and mRNA expression of PAI-1 was measured from the lung tissues of mice 6 hours after NTHi or control inoculation by Q-PCR analysis. The values presented in A-E are Mean ± SD (n=3). *, p<0.05 compared with CON; #, p>0.05 compared with NTHi in MOCK (A & B), Vector (C & D), or WT (E). CON: control; WT: wild-type; KO: knock-out; DN; dominant-negative.
PAI-1 is a key regulator for fibrinolysis and coagulation by counteracting with PAs, uPA and tPA, and inhibiting fibrinolytic activity of PAs
Besides its original/classical role in homeostasis, pleiotropic functions of PAI-1 in biological processes such as wound healing, tumor angiogenesis, bone remodeling, asthma, rheumatoid arthritis, fibrosis, sepsis, and ALI have been reported [10,11,12,13,14]. Clinical reports showed that PAI-1 expression is upregulated in lung tissues of ALI patients, and higher PAI-1 level in these patients is closely associated with poor outcomes in clinic [29,30]. However, growing body of studies reported protective role of PAI-1 in respiratory infections with bacterial pathogens including K.p., P.a., and S.p. [15,25,26]. It is thus not clear if PAI-1 is detrimental or beneficial to the host during bacterial infection in lung. In this study, we found that NTHi induces PAI-1 expression in lung, and early induction of PAI-1 is critical for anti-microbial defense against NTHi by inducing expression of pro-inflammatory cytokines and chemokines, recruiting PMNs into airway, and inhibiting bacterial growth in lung. We thus believe that our study reveals the novel role of PAI-1 in NTHi infection, the most common gram-negative bacterial pathogens in respiratory systems.
PAI-1 expression is induced by NTHi in lung tissues of mice.
Early induction of PAI-1 is critical for anti-microbial defense against NTHi.
NTHi induces PAI-1 via activation of TLR2-MyD88-MKK3-p38 MAPK signaling pathway.
Acknowledgments
This work was supported by grants from National Institute of Health DC005843 (to JDL) and Yeungnam University Research Grants in 2010 (to CHW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hemila H, Louhiala P. Vitamin C for preventing and treating pneumonia. Cochrane database of systematic reviews. 2007:CD005532. doi: 10.1002/14651858.CD005532.pub2. [DOI] [PubMed] [Google Scholar]
- 2.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annual review of immunology. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Werling D, Jungi TW. TOLL-like receptors linking innate and adaptive immune response. Veterinary immunology and immunopathology. 2003;91:1–12. doi: 10.1016/s0165-2427(02)00228-3. [DOI] [PubMed] [Google Scholar]
- 5.Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. Journal of dental research. 2011;90:417–427. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Neill LA. Signal transduction pathways activated by the IL-1 receptor/toll-like receptor superfamily. Current topics in microbiology and immunology. 2002;270:47–61. [PubMed] [Google Scholar]
- 7.Baumann H, Gauldie J. The acute phase response. Immunology today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 8.Germolec DR, Frawley RP, Evans E. Markers of inflammation. Methods in molecular biology. 2010;598:53–73. doi: 10.1007/978-1-60761-401-2_5. [DOI] [PubMed] [Google Scholar]
- 9.Hermans PW, Hazelzet JA. Plasminogen activator inhibitor type 1 gene polymorphism and sepsis. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2005;41(7):S453–458. doi: 10.1086/431996. [DOI] [PubMed] [Google Scholar]
- 10.Dellas C, Loskutoff DJ. Historical analysis of PAI-1 from its discovery to its potential role in cell motility and disease. Thrombosis and haemostasis. 2005;93:631–640. doi: 10.1160/TH05-01-0033. [DOI] [PubMed] [Google Scholar]
- 11.Fay WP. Plasminogen activator inhibitor 1, fibrin, and the vascular response to injury. Trends in cardiovascular medicine. 2004;14:196–202. doi: 10.1016/j.tcm.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 12.Kucharewicz I, Kowal K, Buczko W, Bodzenta-Lukaszyk A. The plasmin system in airway remodeling. Thrombosis research. 2003;112:1–7. doi: 10.1016/j.thromres.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 13.Lijnen HR. Pleiotropic functions of plasminogen activator inhibitor-1. Journal of thrombosis and haemostasis: JTH. 2005;3:35–45. doi: 10.1111/j.1538-7836.2004.00827.x. [DOI] [PubMed] [Google Scholar]
- 14.Shetty S, Padijnayayveetil J, Tucker T, Stankowska D, Idell S. The fibrinolytic system and the regulation of lung epithelial cell proteolysis, signaling, and cellular viability, American journal of physiology. Lung cellular and molecular physiology. 2008;295:L967–975. doi: 10.1152/ajplung.90349.2008. [DOI] [PubMed] [Google Scholar]
- 15.Lim JH, Stirling B, Derry J, Koga T, Jono H, Woo CH, Xu H, Bourne P, Ha UH, Ishinaga H, Andalibi A, Feng XH, Zhu H, Huang Y, Zhang W, Weng X, Yan C, Yin Z, Briles DE, Davis RJ, Flavell RA, Li JD. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity. 2007;27:349–360. doi: 10.1016/j.immuni.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Wang B, Lim DJ, Han J, Kim YS, Basbaum CB, Li JD. Novel cytoplasmic proteins of nontypeable Haemophilus influenzae up-regulate human MUC5AC mucin transcription via a positive p38 mitogen-activated protein kinase pathway and a negative phosphoinositide 3-kinase-Akt pathway. The Journal of biological chemistry. 2002;277:949–957. doi: 10.1074/jbc.M107484200. [DOI] [PubMed] [Google Scholar]
- 17.Lim JH, Ha U, Sakai A, Woo CH, Kweon SM, Xu H, Li JD. Streptococcus pneumoniae synergizes with nontypeable Haemophilus influenzae to induce inflammation via upregulating TLR2. BMC immunology. 2008;9:40. doi: 10.1186/1471-2172-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jono H, Shuto T, Xu H, Kai H, Lim DJ, Gum JR, Jr, Kim YS, Yamaoka S, Feng XH, Li JD. Transforming growth factor-beta -Smad signaling pathway cooperates with NF-kappa B to mediate nontypeable Haemophilus influenzae-induced MUC2 mucin transcription. The Journal of biological chemistry. 2002;277:45547–45557. doi: 10.1074/jbc.M206883200. [DOI] [PubMed] [Google Scholar]
- 19.Shuto T, Imasato A, Jono H, Sakai A, Xu H, Watanabe T, Rixter DD, Kai H, Andalibi A, Linthicum F, Guan YL, Han J, Cato AC, Lim DJ, Akira S, Li JD. Glucocorticoids synergistically enhance nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via a negative cross-talk with p38 MAP kinase. The Journal of biological chemistry. 2002;277:17263–17270. doi: 10.1074/jbc.M112190200. [DOI] [PubMed] [Google Scholar]
- 20.Lim JH, Ha UH, Woo CH, Xu H, Li JD. CYLD is a crucial negative regulator of innate immune response in Escherichia coli pneumonia. Cellular microbiology. 2008;10:2247–2256. doi: 10.1111/j.1462-5822.2008.01204.x. [DOI] [PubMed] [Google Scholar]
- 21.Lim JH, Jono H, Koga T, Woo CH, Ishinaga H, Bourne P, Xu H, Ha UH, Li JD. Tumor suppressor CYLD acts as a negative regulator for non-typeable Haemophilus influenza-induced inflammation in the middle ear and lung of mice. PloS one. 2007;2:e1032. doi: 10.1371/journal.pone.0001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergmann S, Hammerschmidt S. Fibrinolysis and host response in bacterial infections. Thrombosis and haemostasis. 2007;98:512–520. [PubMed] [Google Scholar]
- 23.Janciauskiene S. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochimica et biophysica acta. 2001;1535:221–235. doi: 10.1016/s0925-4439(01)00025-4. [DOI] [PubMed] [Google Scholar]
- 24.Abraham E, Gyetko MR, Kuhn K, Arcaroli J, Strassheim D, Park JS, Shetty S, Idell S. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. Journal of immunology. 2003;170:5644–5651. doi: 10.4049/jimmunol.170.11.5644. [DOI] [PubMed] [Google Scholar]
- 25.Goolaerts A, Lafargue M, Song Y, Miyazawa B, Arjomandi M, Carles M, Roux J, Howard M, Parks DA, Iles KE, Pittet JF. PAI-1 is an essential component of the pulmonary host response during Pseudomonas aeruginosa pneumonia in mice. Thorax. 2011;66:788–796. doi: 10.1136/thx.2010.155788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renckens R, Roelofs JJ, Bonta PI, Florquin S, de Vries CJ, Levi M, Carmeliet P, van't Veer C, van der Poll T. Plasminogen activator inhibitor type 1 is protective during severe Gram-negative pneumonia. Blood. 2007;109:1593–1601. doi: 10.1182/blood-2006-05-025197. [DOI] [PubMed] [Google Scholar]
- 27.Shetty S, Bdeir K, Cines DB, Idell S. Induction of plasminogen activator inhibitor-1 by urokinase in lung epithelial cells. The Journal of biological chemistry. 2003;278:18124–18131. doi: 10.1074/jbc.M207445200. [DOI] [PubMed] [Google Scholar]
- 28.Jaulmes A, Sansilvestri-Morel P, Rolland-Valognes G, Bernhardt F, Gaertner R, Lockhart BP, Cordi A, Wierzbicki M, Rupin A, Verbeuren TJ. Nox4 mediates the expression of plasminogen activator inhibitor-1 via p38 MAPK pathway in cultured human endothelial cells. Thrombosis research. 2009;124:439–446. doi: 10.1016/j.thromres.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Prabhakaran P, Ware LB, White KE, Cross MT, Matthay MA, Olman MA. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury, American journal of physiology. Lung cellular and molecular physiology. 2003;285:L20–28. doi: 10.1152/ajplung.00312.2002. [DOI] [PubMed] [Google Scholar]
- 30.Tsangaris I, Tsantes A, Bonovas S, Lignos M, Kopterides P, Gialeraki A, Rapti E, Orfanos S, Dimopoulou I, Travlou A, Armaganidis A. The impact of the PAI-1 4G/5G polymorphism on the outcome of patients with ALI/ARDS. Thrombosis research. 2009;123:832–836. doi: 10.1016/j.thromres.2008.07.018. [DOI] [PubMed] [Google Scholar]