

SUMMARY
Constitutive Kras and NF-κB activation is identified as signature alterations in pancreatic ductal adenocarcinoma (PDAC). However, how NF-κB is activated in PDAC is not yet understood. Here, we report that pancreas-targeted IKK2/β inactivation inhibited NF-κB activation and PDAC development in KrasG12D and KrasG12D;Ink4a/ArfF/F mice, demonstrating a mechanistic link between IKK2/β and KrasG12D in PDAC inception. Our findings reveal that KrasG12D-activated AP-1 induces IL-1α, which, in turn, activates NF-κB and its target genes IL-1α and p62, to initiate IL-1α/p62 feedforward loops for inducing and sustaining NF-κB activity. Furthermore, IL-1α overexpression correlates with Kras mutation, NF-κB activity, and poor survival in PDAC patients. Therefore, our findings demonstrate the mechanism by which IKK2/β/NF-κB is activated by KrasG12D through dual feedforward loops of IL-1α/p62.
INTRODUCTION
The mutational activation of Kras is an early event in PDAC development and has been detected in 80%–95% of PDAC, and mutational inactivation of Ink4a/Arf tumor suppressor genes can be identified in approximately 50%–75% of PDAC (Hruban et al., 2000). Several experimental animal models were established to determine the functions of mutated Kras in induction of pancreatic intraepithelial neoplasia (PanIN) and PDAC (Aguirre et al., 2003; Bardeesy et al., 2006; Hingorani et al., 2003). However, the key signaling pathways that function downstream of Kras remained unidentified.
Several previous studies have shown a key role of the NF-κB signaling pathway in Ras-driven cancers using animal models of cancer. For example, knockout of IKK2/β inhibited H-Ras-driven melanoma (Yang et al., 2010), inhibition of NF-κB bypassed restraints on oncogenic Ras-stimulated growth in induction of invasive human epidermal neoplasia (Dajee et al., 2003), and loss of IKK2/β in hepatocyte enhances inflammation, tumor promotion, and progression (He et al., 2010; Maeda et al., 2005). We previously showed that RelA/p50NF-κB is constitutively activated in almost 70% of pancreatic cancer specimens and inhibition of NF-κB activity by a mutant IκBα inhibited PDAC cell tumorigenesis (Fujioka et al., 2003; Wang et al., 1999). Thus, these studies demonstrated that IKK2/β/NF-κB has a distinct function in different types of cells. However, it was unclear whether IKK2/β/NF-κB has either prooncogenic or tumor suppressive role in mutant Kras-induced PDAC in mouse models.
Accumulating evidence shows that various signals, including mutant Kras and cytokines activate NF-κB, which, in turn, integrates proinflammatory signals and promote tumorigenesis (Staudt, 2010). For instance, binding of interleukin-1alpha (IL-1α) to its receptor induces K63-linked polyubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) and activates transforming growth factor-β-activated kinase 1 (TAK1), which induces activation of IKK2/β, c-Jun N-terminal kinase, and p38 MAPK to activate NF-κB and AP-1 (Skaug et al., 2009; Wang et al., 2001). The turnover of signal-induced K63-polyubiquitination of TRAF6 is prevented by signaling adaptor p62 to prolong IKK2/β and NF-κB activation (Wooten et al., 2005), Furthermore, p62 is an important NF-κB mediator in lung tumorigenesis (Duran et al., 2008), However, the mechanisms that regulate p62 to control IKK2/β activation to the extent of stimulation were unknown. Another report showed that mutant Kras-driven transformation requires TBK1 kinase activation (Barbie et al., 2009). However, the work did not reveal the mechanisms for constitutive NF-κB activation by TBK1 in mutant Kras-driven tumor. Studies showed that mutant Kras-associated RAL guanine nucleotide exchange factors promote TBK1 kinase activation via an unknown mechanism, which, in turn, activates c-rel/p50NF-κB, (Chien et al., 2006), and that kinase-inactive TBK1 inhibits TANK-mediated alternative NF-κB activation pathway, but does not block canonical NF-κB activation induced by TNF-α or IL-1 (Pomerantz and Baltimore, 1999). Thus, the mechanisms through which oncogenic Kras activates the canonical NF-κB pathway in PDAC remained unknown.
In the present study, we investigated the role of IKK2/β/NF-κB activation and expression of IL-1α and p62 in KrasG12D-induced PDAC development and explored the underlying mechanisms by which IKK2/β/NF-κB is activated by KrasG12D.
RESULTS
Generation of Mutant Mouse Strains with Pancreas-Specific Expression of KrasG12D and Inactivation of IKK2/β with or without Concurrent Ink4a/Arf Deletion
To determine the function of constitutive NF-κB activity in PDAC development, we targeted IKK2/β deletion in the pancreas of the KrasG12D mice with and without Ink4a/Arf inactivation. The mouse strain with floxed IKK2/β alleles (IKK2/βF/F) (Li et al., 2003) is utilized in breeding with the mice that harbors a Pdx1-Cre transgene (Pdx1-Cre) (Gu et al., 2002) and a latent KrasG12D knockin allele (KrasLSL-G12D) (Hingorani et al., 2003), and those that carry the floxed Ink4a/ Arf (Ink4a/ArfF/F), KrasLSL-G12D, and Pdx1-Cre alleles (Aguirre et al., 2003). Generation of IKK2/βF/F genotypes in Pdx1-Cre;KrasLSL-G12D and Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F strains are schematically depicted in Figure 1A. Cre-mediated excision of the silencing cassette and subsequent recombination generated a single LoxP site was detected in the pancreas, but not in other organs such as liver in all of the mouse lines (Figure 1B). Consistent with expression of the mutant Kras allele at endogenous level, GTP-bound Ras protein was increased only in protein extracts from the pancreata of the mice carrying Pdx1-Cre;KrasLSL-G12D alleles (Figure 1C). The third coding exon-deleted IKK2/β alleles were detected in a mosaic pattern in the pancreata of all Pdx1-Cre;IKK2/βF/F mice (Figure 1D). As a result, NF-κB DNA binding activities were notably reduced in nuclear extracts from the pancreata of Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice, but not in those of Pdx1-Cre;KrasLSL-G12D mice (Figures 1E and 1F). Both p16Ink4a and p19Arf genes were specifically deleted in the pancreata of the mutant mice (Figure 1G). Thus, these results show that the Pdx1-Cre transgene concurrently deleted LoxP-containing alleles to target expression of KrasG12D and inactivation of IKK2/βF/F and/or Ink4a/ArfF/F alleles specifically in the pancreata of these mutant mice.
Figure 1. Generation of Mouse Strains with Pancreas-Specific KrasG12D Expression and Inactivation of IKK2/β with or without Parallel Deletion of Ink4a/Arf.
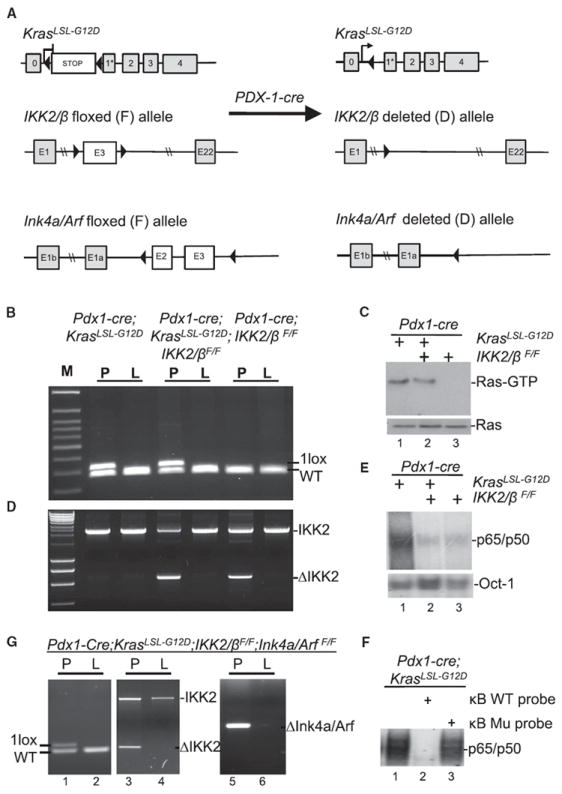
(A) Graphic representation of the targeted KrasLSL-G12D, IKK2/β, and Ink4a/Arf alleles before and after Cre-mediated excision and recombination.
(B) The presence of recombined KrasG12D allele in the pancreata (P) but not in the livers (L) of compound mutant mice was revealed by PCR.
(C) Ras-GTP and total Ras levels in whole pancreatic protein extracts of 3-month-old Pdx1-Cre;KrasLSL-G12D, Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F, and Pdx1-Cre; IKK2/βF/F mice. Elevated levels of Ras-GTP were observed only in compound mutant mice with Pdx1-Cre;KrasLSL-G12D alleles.
(D) The recombined IKK2/βF/F allele was detected only in the pancreata (P), not in the livers (L), of mice carrying Pdx1-Cre;IKK2/βF/F alleles.
(E) EMSA was performed to determine the levels of NF-κB DNA binding activity in the pancreata carrying Pdx1-Cre;KrasLSL-G12D, Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F, or Pdx1-Cre;IKK2/βF/F alleles. Nuclear extracts from mouse pancreata were used in this analysis with a κB probe. Oct-1 probe was used as a loading control.
(F) EMSA was performed with 32P-labeled κB probe in the presence and absence of both unlabeled wild-type (WT) and mutant (Mu) κB probes to determine the specificity of NF-κB DNA binding activity detected in the pancreata of Pdx1-Cre;KrasLSL-G12D mice as indicated.
(G) Pancreas-specific deletion of Ink4a/Arf alleles was confirmed in Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F;Ink4a/ArfF/F mice. The presence of the recombined KrasG12D allele and the exon 3-deleted IKK2/β allele in the pancreata (P) (lane 5) but not in the livers (L) (lane 6) of compound mutant mice was revealed by PCR.
IKK2/β Activity Required for Oncogenic Kras-Induced PanIN and PDAC
To examine the role of IKK2/β in PDAC development, we characterized a cohort of Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F and Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F;IKK2/βF/F mice and their control littermates. Our findings revealed that these mutant mice did not develop PDAC, unlike their control mice (Figures 2A and 2B). Chi-square analysis showed that the Pdx1-Cre;KrasLSL-G12D genotypes with IKK2/β are associated with the observed chronic pancreatis (CP), PanIN lesions, cystic ductal lesion (CDL), and PDAC (Figure 2B), suggesting IKK2/β plays a causal role in PDAC development. Consistent with these results, Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F;IKK2/βF/F mice remained free of PDAC for over 12 months, whereas the median survival of Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F mice was about 3.5 months, as previously described (Aguirre et al., 2003; Figure 2C).
Figure 2. Suppression of Oncogenic KrasG12D-Induced Histological Progression of PanIN and PDAC with or without Concurrent Deletion of Ink4a/Arf by Inactivation of IKK2/β.
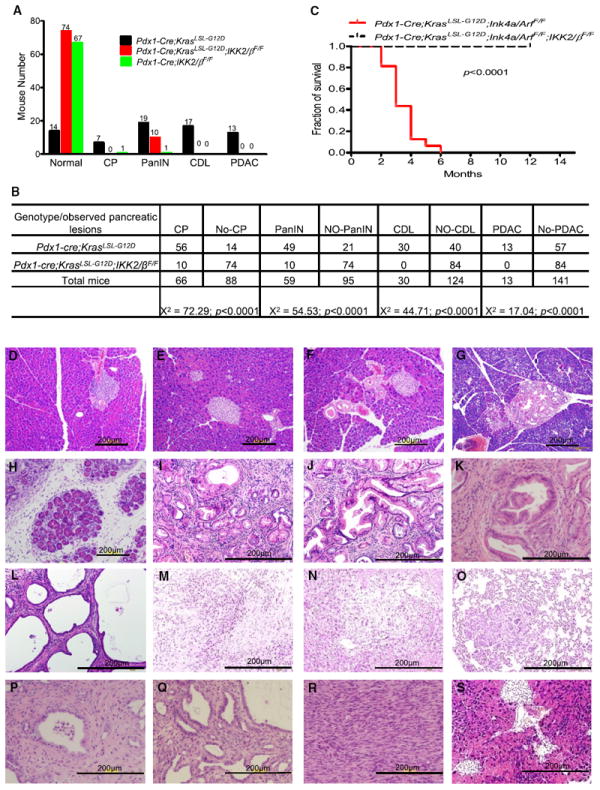
(A) Numbers of mutant mice that developed PDAC, cystic ductal lesions (CDL), PanIN, or chronic pancreatitis (CP), or remained healthy, in cohorts of Pdx1-Cre;KrasLSL-G12D, Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F, and Pdx1-Cre;IKK2/βF/F mice.
(B) Chi-square analysis of the association between Pdx1-Cre;KrasLSL-G12D and Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F, and the observed phenotypes in (A). Note: CP was found in PanIN, CDL, and PDAC; PanIN was coexisted with CDL and PDAC; and CDL was observed in PDAC.
(C) Kaplan-Meier PDAC-free survival curve for Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F (n = 16) and Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F;Ink4a/ArfF/F mice (n = 15). According to the approved animal protocol, mice that presented in a moribund state were killed for autopsy.
(D–S) Representative pancreatic histologic views. (D) Normal pancreas from a wild-type mouse. (E) Histologic appearance of normal pancreas from a Pdx1-Cre;IKK2/βF/F mouse. (F) Histologic appearance of normal pancreas from a Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mouse. (G) A rare PanIN-1 from Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mouse. (H–O) Pdx1-Cre;KrasLSL-G12D mice. (H) Chronic pancreatitis (I) PanIN-1. (J) PanIN-2. (K) PanIN-3. (L) Cystic ductal lesion. (M) PDAC. (N) PDAC liver metastasis. (O) PDAC lung metastasis. (P-R) Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F mice. (P) PanIN. (Q) Cystic ductal lesion. (R) PDAC. (S) Histologic appearance of normal pancreas from a Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F;IKK2/βF/F mouse. See also Figure S1.
The littermates of all the mutant mice were born with mendelian frequencies and weight similar to those of wild-type controls. Organs appeared to be formed normally and no phenotypic differences were observed before the age of 2 months in Pdx1-Cre;KrasLSL-G12D mice and 6 weeks in Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F mice. No pancreatic ductal lesions or other abnormalities were detected and the expression levels of glucagon, insulin, amylase, CK-19, and staining of duct-specific lectins, Dolichos biflorus agglutinin (DBA) in various IKK2/βF/F mice are consistent with those observed in the pancreas of wild-type mice (Figures 2E and 2F; Figure S1 available online). Altogether, the normal histology, marker gene expression, and life span suggest that knockout of IKK2/β in mouse pancreas did not result in abnormal pancreas development in acinar, ductal, and islet of the pancreas or apparent pathological phenotypes.
In Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice (Figures 2F and 2G), only 12% (10 of 84) of the mice developed stage 1 PanIN lesions, which consisted primarily of elongated mucinous ductal cells, and no PDAC was observed for over 12 months, whereas 19% (13 of 70) of Pdx1-Cre;KrasLSL-G12D mice had PDAC in 8–12 months (Figure 2M), 24% (17 of 70) had CDL during the same time period (Figure 2L), and 27% (19 of 70) had stage 1, 2, or 3 PanIN lesions (Figures 2I–2K) at about 3–6 months. Moreover, most CP was associated with all the PanIN and PDAC, and only 10% of Pdx1-Cre;KrasLSL-G12D mice (7 of 70) developed CP without PanIN or PDAC pathological lesions (Figures 2A and 2H). These findings suggest that inactivation of IKK2/β inhibited PDAC and CP might be a precursor lesion in Kras-induced PDAC.
Serial histological surveys of the pancreas of all the mice are shown in Figures 2D–2S. Pdx1-Cre;KrasLSL-G12D mice had PanIN at 3 months of age and had invasive tumors by 10 months of age, and 3 of 70 had liver or lung metastatic lesions by 8–12 months of age (Figures 2N and 2O). The progressive premalignant lesions with ductal histology and PDAC were observed (Figures 2M–2O and 2R). These PDAC histological lesions and incidences are consistent with those of the mice harboring KrasLSL-G12D or KrasLSL-G12D;Ink4a/ArfF/F in earlier studies (Aguirre et al., 2003; Tuveson et al., 2004). These results demonstrate that IKK2/β is required for oncogenic Kras-induced PanIN and PDAC, thus establishing a definitive role of IKK2/β in PDAC development in vivo.
Attenuation of Inflammatory and Proliferative Responses by Pancreas-Targeted Inactivation of IKK2/β
To determine whether chronic inflammation and proliferative responses were inhibited in the pancreas of Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice, immunohistochemical (IHC) analyses were performed. Levels of proliferation markers CyclinD1, Ki-67, and inflammatory marker COX-2 expression were substantially higher in PanIN and PDAC from Pdx1-Cre;KrasLSL-G12D mice, than in histologically normal pancreas from Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice (Figure 3A). This finding suggests that inactivation of IKK2/β interrupted mutant Kras-stimulated cell proliferation and inflammation.
Figure 3.
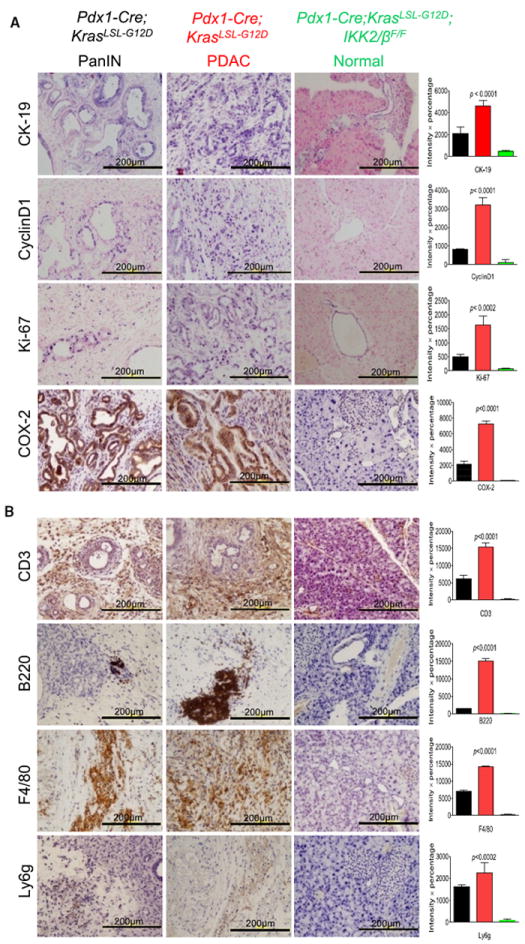
Comparison of the Cell Proliferation, Inflammation, and Immune Responses in the Pancreas between Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F and Pdx1-Cre;KrasLSL-G12D Mice
(A) Expression of CyclinD1, Ki-67, and COX-2 is elevated in PanIN and PDAC from Pdx1-Cre;KrasLSL-G12D mice, but not in the normal pancreas of Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice. Positive immunostaining for Cytokeratin-19 (CK-19) verifies the ductal phenotype of PanIN lesions, PDAC, and normal duct.
(B) Sections of formalin-fixed PanIN lesions and PDAC from 8-month old Pdx1-Cre;KrasLSL-G12D and normal duct tissue from Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice underwent IHC staining with anti-CD3 antibody as a T cell marker, anti-B220 as a B cell marker, anti-F4/80 as a macrophage marker, and anti-Ly6g as a neutrophil marker. Error bars represent ± standard deviation (SD) from the data of five mice for each of the two genotypes.
The observation of CP or proinflammatory responses in Pdx1-Cre;KrasLSL-G12D mice prompted us to examine whether these mutant mice developed lymphocytic infiltration around pancreatic lesions. IHC staining revealed that the infiltrating lymphocytes consisted of CD3-positive T cells, B220-positive B cells, F4/80-positive macrophages, and Ly6g-positive neutrophil (Figure 3B), whereas parallel staining did not reveal significant lymphocytic infiltration in the pancreata of Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice (Figure 3B). This phenotype of lymphocytic infiltration in the pancreata of Pdx1-Cre;KrasLSL-G12D mice has a close resemblance to those reported for various mouse strains with pancreatitis, such as IκBαm/m mice defective in IκBα-mediated negative regulation of NF-κB (Peng et al., 2010). So, the absence of hallmarks of cancer-related inflammation in Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice is due to inactivation of IKK2/β. This suggests chronic inflammation is a key factor in promoting PDAC development as observed in mouse models for other cancers (Karin, 2008), and one of the essential roles regulated by IKK2/β in Kras-induced PDAC inception.
Inhibition of Key Proinflammatory Cytokines in the Pancreas of Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F Mice
To further analyze Kras-induced inflammatory responses, gene expression was profiled and a number of known and previously unknown NF-κB regulated genes were identified by Gene Set Enrichment analyses (GSEA) using gene ontology and NF-κB target gene sets (Figures 4A and 4B; Figures S2A–S2E). Importantly, GSEA of significant gene upregulation in Pdx1-Cre;KrasLSL-G12D revealed that they are strongly associated to positive nodal status, high risk, higher tumor stage, and poor survival in PDAC patients by comparing with 102 PDAC cDNA microarray files (GSE21501) (Figure 4C). Two- and 5-fold enriched expression in Pdx1-Cre;Kras LSL-G12D;IKK2/βF/F is correlated to low risk (Figures S2A–S2E). These findings further indicate that the significant role of activated IKK2/β in PDAC development.
Figure 4. Gene Ontology and Gene Set Enrichment Analyses between Pancreata from Pdx1-Cre;KrasLSL-G12D and Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F Mice and Profiling Cytokine Expression.

(A) Enriched expression of NF-κB downstream target gene sets in Pdx1-Cre;KrasLSL-G12D;IKK2/βWT compared to Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F. The heat map represents top enriched genes in Pdx1-Cre;KrasLSL-G12D;IKK2/βWT. NES, normalized enrichment score; NOM p value, nominal p value; FDR, false discovery rate q value. (red, high expression; blue, low expression).
(B) GSEA analyses identify the enriched gene sets expressed either in Pdx1-Cre;KrasLSL-G12D;IKK2/βWT or Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F using 198 KEGG pathway gene sets. One gene set are enrich in Pdx1-Cre;KrasLSL-G12D;IKK2/βWT and twenty seven gene sets are enriched in Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F. Five NF-κB pathway-related gene sets are noteworthy enriched in Pdx1-Cre;KrasLSL-G12D; IKK2/βWT. Enriched gene sets were selected based on statistical significance (FDR q value < 0.25 and normalized p value < 0.05).
(C) GSEA analyses of significant gene upregulation in Pdx1-Cre;KrasLSL-G12D revealed that they are strongly correlated to positive nodal status, high risk, higher tumor stage, and poor survival in PDAC patients by using 102 PDAC cDNA microarray data (GSE21501). Two- and 5-fold enriched expression in Pdx1-Cre;KrasLSL-G12D; IKK2/βF/F is correlated to low risk. ns, not significant (FDR q value > 0.25 and/or normalized p value > 0.05).
(D) Analysis of differential cytokine gene expression between pancreatic cancer and normal pancreas (from 5- to 12-month-old Pdx1-Cre;KrasLSL-G12D mice and age-matched Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice) by real-time PCR arrays.
(E) Determination of IL-1α expression levels in pancreas, liver, and lung from Pdx1-Cre;KrasLSL-G12D, Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F, Pdx1-Cre;IKK2/βF/F, and wild-type (WT) mice.
(F) Evaluation of IL-1α and IL-1β expression levels in sera from Pdx1-Cre;KrasLSL-G12D, Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F, Pdx1-Cre;IKK2/βF/F, and wild-type (WT) mice. Error bars represent ±SD of the data from three mice in each genotype as indicated. See also Figure S2 and Tables S1–S3.
To further explore the role of IKK2/β in PDAC development, we compared additional gene expression profiles from Pdx1-Cre;KrasLSL-G12D and Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice using Affymetrix arrays. Our results showed that there is a little difference between the expression profiles of several IKK2/β/NF-κB-regulated genes, such as IL-1α, IL-1β, and c-jun in the histologically normal pancreas from Pdx1-Cre;KrasLSL-G12D and Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice, which might be due to the small fractions of Pdx1-targed cells (Figures S2F and S2G; Table S1). Furthermore, the analysis of the gene expression profiles among the normal pancreas, PanIN, and PDAC from Pdx1-Cre;KrasLSL-G12D mice revealed that these profiles are consistent with those involved in PDAC development as those identified between Pdx1-Cre;KrasLSL-G12D and Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice (Figures 4A–4C; Table S2), and are associated with signaling pathways and cellular functions of genes in tumorigenesis (Figures S2H and S2I). The progressive increases in the expression of several NF-κB regulated genes from low expression in the histological normal pancreas to high expression levels in PanIN and PDAC suggest the involvement of the NF-κB-regulated genes in Kras-induced PDAC development.
Among 20 of the differentially expressed genes with a greater than 10-fold increases in expression in the pancreatic tissues of Pdx1-Cre;KrasLSL-G12D mice (Figure 4A), most of them are cytokines, chemokines, and their receptors, suggesting a role of these molecules in Kras-induced PDAC in vivo. To substantiate the differences in cytokine gene expression between 5- to 12-month-old Pdx1-Cre;KrasLSL-G12D and age-matched Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice, we performed real-time PCR arrays with 84 cytokines and chemokines and validated 9 cytokines with highest expression levels, which includes IL-1α, IL-1β, and IL-1 receptor type 2 (Figure 4D; Table S3).
We next investigate the expression of IL-1α and IL-1β, since the expression of both cytokines is among the highest. The levels of IL-1α were significantly elevated in pancreatic tissues and IL-1β levels were slightly higher in serum of Pdx1-Cre;KrasLSL-G12D mice, but not in wild-type, Pdx1-Cre;IKK2/βF/F, and Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice (Figures 4E and 4F). These results, together with the lymphocytic infiltration, are consistent with chronic inflammation and the role of IKK2/β in cytokine expression, which may, in turn, promote a protumorigenic micro-environment. IL-1α is an NF-κB target gene and a strong NF-κB inducer (Mori and Prager, 1996; Osborn et al., 1989), but IL-1α induction by Kras is previously unknown, suggesting a possible mechanistic link between IL-1α expression induced by the KrasG12D and IKK2/β/NF-κB activation.
Downregulation of Expression of IKK2/β/NF-κB Upstream Signaling Molecules by Inactivation of IKK2/β
To elucidate the downstream pathways of mutant Kras that activate IKK2/β, we carried out IHC analysis of PanIN and PDAC to determine the status of inflammation related signaling pathways, including TAK1, p65/RelA, c-Fos, p62, IL-1α, and Rantes (Chemokine [C-C motif] ligand 5). Our results show that levels of c-Fos, TAK1, p62, activated-p65/RelA, IL-1α, and Rantes were substantially higher in PanIN and PDAC from Pdx1-Cre;KrasLSL-G12D mice than in histologically normal pancreata from Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice (Figure 5A). These results suggest that IKK2/β regulates the expression of TAK1, p62, c-Fos, IL-1α, and Rantes as well as NF-κB activation in the diseased pancreas of Pdx1-Cre;KrasLSL-G12D mice. To determine whether the Kras-induced signaling pathways are also activated in human PDAC, IHC staining was performed using six different PDAC patient specimens with their adjacent normal tissues as controls for a pilot study. The increased levels of IL-1α, TAK1, c-Fos, nuclear p65/RelA recognized by a specific antiactivated p65/RelA antibody (Zabel et al., 1993), p62, and phosphorylated AKT were observed in human PDAC (Figure 5B). Taken together, these results suggest that inactivation of IKK2/β interrupts oncogenic Kras-mediated pathways regulating expression of c-Fos, IL-1α, TAK1, and p62, and activation of AKT and NF-κB, but how these signaling molecules are regulated by oncogenic Kras through IKK2/β pathways remained largely unclear.
Figure 5. Analysis of Signaling Pathways in PanIN, PDAC, and Histologically Normal Pancreas from Compound Mutant Mice and Human PDAC Patients.
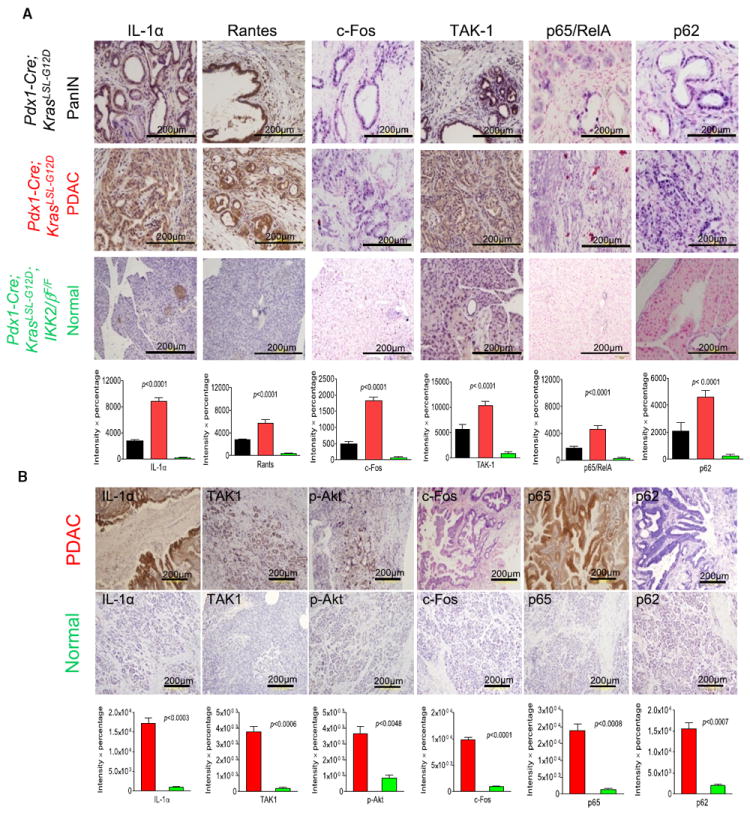
(A) Immunohistochemical analysis with anti-IL-1α, anti-Rantes (Chemokine [C-C motif] ligand 5), anti-c-Fos, anti-TAK1, anti-p62, and anti-p65 antibodies in sections of formalin-fixed PanIN lesions and PDAC from Pdx1-Cre;KrasLSL-G12D mice, and of normal duct tissues from Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice. Error bars represent ±SD from the data of five mice for each of the three genotypes.
(B) Immunohistochemical staining for IL-1α, TAK1, pAKT, c-Fos, p65, and p62 in sections of formalin-fixed human PDAC and adjacent histologically normal pancreatic tissues. Error bars represent ±SD from six human PDAC specimens.
IL-1α Overexpression Correlates with Kras Mutation and NF-κB Activation in Human PDAC Specimens and Poor Survival in PDAC Patients
To determine whether IL-1α overexpression is correlated with mutant Kras in human PDAC specimens, and is induced by KrasG12D in mouse PanIN and PDAC, we sequenced the Kras gene from paraffin sections of PDAC specimens from 14 patients who had not undergone neoadjuvant therapies, and from 9 PDAC patient specimens derived from the orthotopic xenograft mouse model implanted with freshly isolated patient pancreatic cancer for excluding normal human stromal cells (Kim et al., 2009). Ninety-three percent (13 of 14) Kras gene from PDAC paraffin sections has substitutional mutation at codon 12, from GGT (Gly) to GAT (Asp). 71% (5 of 7) Kras mutations identified in human PDAC tissue xenograft also have aspartic acid at codon 12, whereeas 29% (2 of 7) were changed to GTT (valine). The results of IL-1α expression, analyzed by chi-square test, showed that the presence of mutated Kras gene positively correlated with IL-1α overexpression in PDAC patient specimens (Figure 6A). Further study of IL-1α expression using human PDAC tissue microarray (TMA) demonstrated that IL-1α over-expression was found in most of the human PDAC tissues. Kaplan-Meier survival analysis indicated that high levels of IL-1α expression are associated with poor survival in PDAC patients (p = 0.016, log rank test) (Figure 6B).
Figure 6. Clinical Correlations among Mutant Kras, IL-α Overexpression, and NF-κB Activation in Human PDAC.
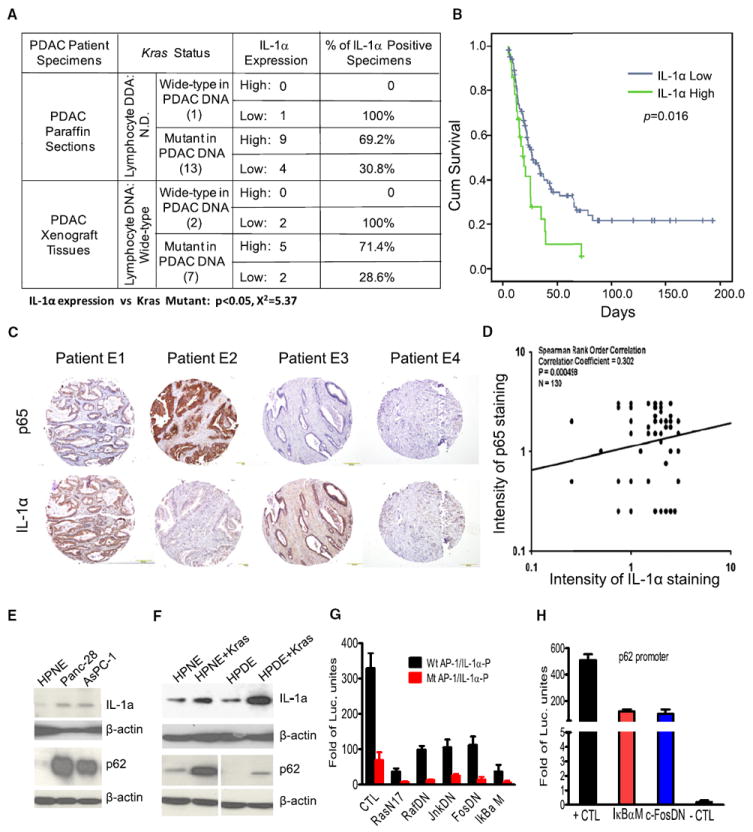
(A) The percentages of IL-1α positivity in PDAC tissues carrying a mutant Kras gene. Chi-square test was used to demonstrate the positive correlation between overexpression of IL-1α and the presence of a mutant Kras in PDAC tissues.
(B) Kaplan-Meier survival analysis of PDAC patients with and without high levels of IL-1α expression using TMA.
(C) Immunohistochemical staining for p65 and IL-1α in human PDAC tissue microarrays. Representative IHC stainings are shown, with four combinations of immunostaining patterns, E1: p65 high, IL-1α high; E2: p65 high, IL-1α low; E3: p65 low, IL-1α high; E4: p65 low, IL-1α low.
(D) Scores of activated NF-κB are plotted against those of IL-1α overexpression. Spearman’s rank order correlation was used to demonstrate the positive correlation between NF-κB activity and expression levels of IL-1α in TMA.
(E) Western blot analysis showing IL-1α and p62 overexpression in pancreatic cancer cell lines MDAPanc-28 and AsPc-1.
(F) Western blot analysis of IL-1α and p62 expression in hTERT-immortalized human pancreatic ductal HPNE and HPDE cell lines.
(G) Reporter gene assay for analyzing IL-1α promoter regulation by Kras, AP-1, and NF-κB pathways using mPDAC cells.
(H) Reporter gene assay for analyzing the regulation of p62 promoter by AP-1 and NF-κB pathways using mPDAC cells. Error bars represent ±SD from three independent experiments. See also Figure S3.
To determine the association between NF-κB activity and expression levels of IL-1α in human PDAC for validating the relevance of the observations in Pdx1-Cre;KrasLSL-G12D and Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mice, IHC staining for p65 and IL-1α in patient PDAC TMA was performed. 42 of 131 (32%) human PDAC samples showed very strong IL-1α staining and 47 (36%) had extremely intense staining for NF-κB activities (Figure 6C). Data analysis of these by Spearman’s rank order correlation showed the positive correlation between NF-κB activities and expression levels of IL-1α (Figure 6D). Altogether, the results suggest that the oncogenic Kras-mediated pathway induced IL-1α overexpression, which triggered NF-κB activation. To determine how early activated NF-κB can be detected during PDAC development, we analyzed the staining of p65/RelA in our pancreatic TMA, and the results show 49% (20/41) positive staining in PanIN stage (Figures S3A and S3B), suggest that NF-κB activation is one of the earliest molecular alterations observed in the development of pancreatic cancer.
To determine whether expression of IL-1α and p62 was induced in by KrasG12D, we examined the levels of IL-1α and p62 expression in human PDAC cell lines MDAPanc-28, AsPc-1, and in immortalized human pancreatic ductal cell lines HPDE and HPNE, with or without stable expression of mutant Kras. Expression of both IL-1α and p62 was elevated in MDAPanc-28 and AsPc-1 cells (Figure 6E) and in HPDE and HPNE cells expressing oncogenic Kras (Figure 6F). The expression of c-Fos and activation of Jun N-terminal kinase (JNK) was also induced in mutant Kras HPNE cells (Figure S3C).
To determine whether oncogenic Kras-induced AP-1 activity played a major role in the IL-1α and p62 expression, we performed IL-1α and p62 promoter analysis in mPDAC cells, an early passage mouse PDAC cell lines established from the PDAC of Pdx1-Cre;KrasLSL-G12D mice. Our reporter gene assays show that IL-1α promoter with mutated AP-1 sites had no responses, whereas activity of IL-1α promoter with wild-type AP-1 sites was strongly inhibited by expression of a mutant of ras (RasN17), c-fos (FosDN), and IκBα (IκBαM), as well as kinase-dead raf and jnk (Figure 6G). Similarly, p62 promoter was strongly inhibited by expression of FosDN and IκBαM (Figure 6H). The results showed that IL-1α and p62 promoters in mPDAC cells were regulated by Kras through AP-1 and NF-κB transcription factors.
Identifying Feedforward Mechanisms that Sustain Constitutive NF-κB Activation in Oncogenic KrasG12D-Induced PDAC
To determine whether IL-1α serves a mechanistic link between KrasG12D and NF-κB activation, we utilized two human PDAC cell lines and two early passage mouse PDAC cell lines, mPDAC-1 and mPDAC-2, which were established from PDAC of Pdx1-Cre;KrasLSL-G12D mice. These cells were treated with anti-IL-1α and anti-IL-1β neutralizing antibodies for 0, 4, and 8 hr and the nuclear extracts were analyzed. The results showed that anti-IL-1α, but not anti-IL-1β, neutralizing antibodies blocked constitutive NF-κB activation (Figures 7A and 7B). These findings suggest that secreted IL-1α from these cells activates NF-κB through autocrine mechanism.
Figure 7. Elucidation of Feedforward Signaling Pathways that Sustain Constitutive NF-κB Activation in Oncogenic KrasG12D-Induced PDAC.

(A) Nuclear extracts of human PDAC cell lines MDAPanc-28 and AsPc-1 and mouse PDAC cell lines mPDAC-1 and mPDAC-2 (derived from Pdx1-Cre;KrasLSL-G12D), treated with anti-IL-1α neutralizing antibody (2 μg/ml) for 0, 4, or 8 hr or with anti-IL-1β neutralizing antibody (2 μg/ml) for 0 or 8 hr as indicated, were analyzed by EMSA to determine NF-κB activity using a probe containing an NF-κB DNA binding site. Oct-1 DNA binding activities were determined as loading controls.
(B) EMSA was performed to determine the specificity of inducible RelA/p50 NF-κB DNA binding activity. Competition and supershift assays were performed using 20 μg of nuclear protein from mPDAC-2 cells as indicated.
(C) Western blot was performed to determine the expression of p62 in mPDAC cells (CTL), mPDAC cells (p62-shRNA) expressing p62shRNA, and mPDAC cells (SB-shRNA) expressing scrambled control shRNA with anti-p62 antibody. Relative protein loading was determined by the use of anti-β-actin antibody.
(D) NF-κB activities in the nuclear extracts of mPDAC cells (CTL), mPDAC cells (p62-shRNA) expressing p62shRNA, and mPDAC cells (SB-shRNA) expressing scrambled control shRNA stimulated with IL-1α for the times indicated were determined by EMSA. Oct-1 DNA binding activities were determined as loading controls in (A) and (D).
(E) NF-κB-dependent expression of p62 was determined by western blot analysis in protein extracts from MDAPanc-28 and AsPc-1 cells expressing a Flag-tagged IκBα mutant (Flag-IκBαM) and their control cells expressing a puromycin resistance vector (CTL/Puro). Flag-IκBαM expression was verified by using anti-Flag antibody. Relative protein loading was determined by the use of anti-β-actin antibody.
(F) IL-1α-regulated p62 expression was determined by real-time PCR in MDAPanc-28, AsPc-1, mPDAC-1, and mPDAC-2 treated with anti-IL-1α neutralizing antibody (2 μg/ml) for 8 hr or IL-1α (10 ng/ml) for 1 hr as indicated.
(G) IKK2/β-induced p62 expression was determined by western blot analysis using MDAPanc-28, AsPc-1, mPDAC-1, and mPDAC-2 cells expressing IKK2/β-shRNA (IKK2-shRNA) and their control cells expressing a puromycin resistance vector (CTL/Puro) and scrambled shRNA (CTL-shRNA). IKK2/β expression was verified by using anti-IKK2/β antibody. Relative protein loading was determined by the use of anti-β-actin antibody.
(H) IL-1α-regulated p62 expression was determined by western blot analysis in protein extracts of MDAPanc-28, AsPc-1, mPDAC-1, and mPDAC-2 cells treated with anti-IL-1α neutralizing antibody (2 μg/ml) for 8 hr or IL-1α (10 ng/ml) for 1 hr as indicated. Relative protein loading was determined by using anti-β-actin antibody.
(I) The sequence alignment between mouse and human p62 promoter regions was presented with AP-1 and NF-κB binding sites indicated.
(J) The activities of the AP-1 and κB binding sites in mouse p62 promoter were determined. mPDAC cells were stimulated with IL-1α for one hour and ChIP assays were performed with anti-c-Fos and anti-p65/NF-κB antibodies and IgG as negative control by using real-time PCR.
(K) Analysis of p62 promoter activities in mPDAC-1 cells. The luciferase reporter gene activities are presented with the schematic illustration of the different luciferase reporter constructs of the p62 promoter constructs with mutated AP-1 and κB binding sites and control plasmids as indicated. Luciferase activity was measured as described in Experimental Procedures. Error bars represent ±SD from three independent experiments. See also Figure S4.
Although p62 was induced by Kras to trigger IKK2/β activation in lung cancer cells (Duran et al., 2008), the mechanisms are still lacking. To determine whether and how p62 is involved in KrasG12D-induced IKK2/β/NF-κB activation, we stimulated p62-knockdown cells and control cells expressing a puromycin resistant vector and scrambled shRNA with IL-1α for 0, 0.5, 2, or 8 hr and then analyzed NF-κB activity by EMSA (Figures 7C and 7D). The results revealed that constitutive NF-κB activity was decreased in both mPDAC and AsPc-1 p62 knockdown cells (Figure 7D; Figures S4A and S4B). Interestingly, NF-κB activity was induced to the same peak level by IL-1α at 0.5 hr stimulation in the control and p62 knockdown cells, remained at the same level at the end of 2 hr stimulation, and was inhibited at the end of 8 hr of IL-1α stimulation for the p62 knockdown cells compared with their control cells (Figure 7D; Figures S4A and S4B). These findings suggest that p62 expression is dispensable for IL-1α-induced NF-κB activation at the early phase (Figure 7D, lanes 4–9; Figures S4A and S4B), but is required for IL-1α-induced long-term NF-κB activation (Figure 7D, lanes 10–12; Figures S4A and S4B). Since p62 expression was substantially increased in PanIN and PDAC from Pdx1-Cre;KrasLSL-G12D mice in comparison to those from histologically normal pancreata from Pdx1-Cre;KrasLSL-G12D;IKK2/β F/F mice (Figures 5A and 5B), it is possible that p62 is one of the previous unknown downstream target genes regulated by NF-κB. To test this possibility, we showed p62 expression was substantially inhibited in MDAPanc-28 and AsPc-1 cells by expressing IκBαM in comparison with their control cells (Figure 7E). Our results also show that both p62 mRNA and protein levels were increased by IL-1α stimulation and reduced by the anti-IL-1α neutralizing antibody and by silencing IKK2/β expression in MDAPanc-28, AsPc-1, mPDAC-1, and mPDAC-2 cells (Figures 7F–7H; Figure S4C), demonstrating that p62 expression is regulated by NF-κB activity. To further demonstrate the regulation of p62 expression by NF-κB, we identified two κB and three AP-1 binding sites in both 3.0 kb human and mouse p62 promoters, which have extensive DNA sequence similarity (Figure 7I). The results from ChIP assays revealed that NF-κB BS2 and AP-1 BS3 sites showed higher binding activity upon IL-1α stimulation in mPDAC-1 cells (Figure 7J). EMSA showed that NF-κB BS2 site displayed the most κB binding activity, consistent with the results of ChIP assay, whereas the three AP-1 binding sites showed similar binding activity in mPDAC-1 cells (Figure S4D). The luciferase reporter gene assays, using different p62 promoter constructs with and without mutated AP-1 or κB binding sites and controls, showed that p62 expression is mainly regulated by NF-κB activity through NF-κB BS2 site and is also mediated by the three AP-1 sites in mPDAC cells, suggesting that p62 is an NF-κB and AP-1 downstream target gene (Figure 7K). Furthermore, our results showed that NF-κB was activated in the KrasG12D PDAC model in the presence of wild-type p53 function (Figures S4E and S4F), and FOXO3a was stabilized and TSC1 was activated, whereas p70/S6K was not phosphorylated in IKK2/β knockdown cells (Figures S4G and S4H), suggesting that both TSC1 and FOXO3a pathways is also involved in Kras-induced PDAC development.
To demonstrated the role of IL-1α and p62 in mutant Kras-driven cancer cells, we knocked down IL-1α and p62 expression in mPDAC cells and in LKP-13, a lung adenocarcinoma cell line derived from KrasG12D(LA1) mice (Wislez et al., 2005; Figure 8A; Figures S5A and S5B), and showed significant reduction of tumorigenic potential in the IL-1α and p62 knocked down cancer cells in orthotopic xenograft mouse and subcutaneous xenograft mouse models (Figures 8B-D). Knocking down TRAF6, which is essential for IKK2/β and JNK activation in the IL-1α and TLR pathways (Lomaga et al., 1999; Naito et al., 1999), decreased NF-κB activation in both mPDAC cells expressing KrasG12D (Figures 8E–8G). Taken together, these results suggest that IL-1α and p62 play a key role in oncogenic Kras-induced tumors. Knocking down of c-fos expression inhibited IL-1α and p62 expression and NF-κB activation (Figures 8H–8L). These results suggest that AP-1 activated by oncogenic Kras induced and sustained activation of RelA/p50 in PDAC development.
Figure 8. AP-1 Induced by Oncogenic KrasG12D Initiates Feedforward Loops of IL-1α and p62 to Induce and Sustain Constitutive NF-κB Activation and the Working Model.

(A) Knocked down expression of IL-1α and p62 in mPDAC and LKP-13 cells. The expression levels of p62 and IL-1α in mPDAC and LKP-13 cells expressing scrambled shRNA (SBshRNA), p62shRNA, and IL-1αshRNA was determined using anti-p62 or anti- IL-1α antibody in western blot with β-actin as relative protein loading controls.
(B) The resected orthotopic tumors attached to spleen in fifteen C57B6 mice injected with mPDAC-SBshRNA, mPDAC-p62shRNA, and mPDAC-IL-1αshRNA cells (n = 5 per group) were shown at week 5. S: spleen; T: tumor.
(C) Tumor weight in C57B6 mice orthotopically injected with mPDAC-SBshRNA, mPDAC-p62shRNA, and mPDAC-IL-1αshRNA cells. Columns: mean of all individual tumors in each group. Error bars: ±SD of the pancreatic tumor from five mice in each of the three groups as indicated.
(D) Percentage of C57B6 mice that developed subcutaneous tumors after injection of LKP-13-SB-shRNA, LKP-13-p62shRNA, and LKP-13-IL-1αshRNA cells. Columns: percentage of the mice that grew tumor in each group (n = 5). The statistical significance was determined by Fisher’s exact test.
(E) Expression of TRAF6 in mPDAC cells expressing scrambled Traf6 shRNA (SB-shRNA) and two Traf6-shRNA (Traf6-shRNA-1 and 2) was determined by anti-TRAF6 antibody with β-actin as loading control.
(F) NF-κB activities in the nuclear extracts of mPDAC cells expressing scrambled Traf6 shRNA (SB-shRNA) and two Traf6-shRNAs were determined by EMSA. Oct-1 DNA binding activities were determined as loading controls.
(G) Quantitation of NFκB activities in EMSA by Image Analysis Software (ImageQuant TL 7.0).
(H) c-Fos expression in mPDAC expressing a scrambled control shRNA (SB-shRNA) and three different cFos-shRNAs (cfos-shRNA-1, 2, 3) was determined with anti-cFos antibody in western blot analysis.
(I) The levels of IL-1α in mPDAC cytoplasmic extracts expressing a scrambled control shRNA (SB-shRNA) and three different cFos-shRNAs (c-fos-shRNA-1, 2, 3) were analyzed by anti-IL-1α western blot with β-actin as loading control.
(J) p62 expression in mPDAC cells expressing a scrambled control shRNA(SB-shRNA), and three different cFos-shRNAs (cfos-shRNA-1, 2, 3) were analyzed by anti-p62 western blot with β-actin as loading control.
(K) NF-κB activity from mPDAC expressing a scrambled control shRNA(SB-shRNA) and three different cFos-shRNAs (cfos-shRNA-1, 2, 3) were analyzed by EMSA. Oct-1 DNA binding activities were determined as loading controls
(L) Quantitation of NFκB activities in EMSA by Image Analysis Software (ImageQuant TL 7.0).
(M) A proposed working model illustrates the potential mechanism through which KrasG12D oncogenic signaling induces feedforward loops of IL-1α and p62 to sustain constitutive IKK2/β/NF-κB activation in PDAC development. See also Figure S5.
As illustrated in Figure 8M, our findings suggest a mechanism by which oncogenic Kras signaling pathway induced constitutive activation of NF-κB in PDAC development.
DISCUSSION
We have unequivocally demonstrated that the requirement of NF-κB pathway for PDAC development and the mechanism through which constitutive NF-κB activation and inflammatory responses were induced by oncogenic Kras during PDAC initiation. These findings suggest that the primum movens responsible for cancer-related inflammatory responses and the development of PanIN and PDAC is the mutant Kras-initiated constitutive activation of NF-κB. Importantly, our results suggest that mutant Kras may induce intrinsic inflammatory responses that promote a protumorigenic microenvironment through expressing inflammatory mediators such as chemokines and cytokines in tumor tissues, recruiting inflammatory cells, and inducing angiogenesis and tissue repair similar to those observed in hereditary pancreatitis, which has been firmly linked to the development of PDAC (Brentnall et al., 1999; Lowenfels et al., 1997). Thus, our findings further suggest that sporadic PDAC is also induced through a chronic inflammatory mechanism similar to those observed in hereditary pancreatitis related PDAC cases.
Although studies have demonstrated that mutant Ras was shown to activate NF-κB in several types of tumors in mouse models (Bassères et al., 2010; Meylan et al., 2009; Yang et al., 2010). Signaling pathways leading to the constitutive NF-κB activity in cancer cells were not clear at all. Duran et al. (2008) showed that mutant Ras induced p62 expression through AP-1 to activate IKK2/β and NF-κB. However, the requirement for p62 in NF-κB activation is not completely understood considering the reported function of p62 as an adaptor for regulating E3 ubiquitin-protein ligase TRAF6 and deubiquitination enzyme CYLD to balance the turnover of K63-polyubiquitination (Sanz et al., 2000; Wooten et al., 2005). Our results clearly demonstrated that p62 expression is not required for rapid and early NF-κB activation induced by IL-1α, but it is essential for constitutive NF-κB activation (Figure 7D; Figures S4A and S4B), which is consistent with the function of p62 as an adaptor for regulating the turnover of K63-polyubiquitinated proteins, including TRAF6, to maintain IKK2/β and NF-κB activation (Sanz et al., 2000; Wooten et al., 2005). Our results show that expression of p62 is induced by NF-κB activation during IL-1α stimulation of mouse and human PDAC cell lines (Figures 7E–7J), suggesting the existence of an autoregulatory loop whereby NF-κB regulates p62 expression, which, in turn, extends NF-κB activation.
Our results show that TSC1 and FOXO3a pathways are involved in Kras-induced PDAC (Figures S4G and S4H), consistent with the studies showing that IKK2/β phosphorylates TSC1 at Ser487 and Ser511 and FOXO3a for promoting tumorigenesis (Hu et al., 2004; Lee et al., 2007). Interestingly, expression of both IL-1α and IL-1β was inhibited in our Pdx1-Cre;KrasLSL-G12D;IKK2/βF/F mouse model (Figures 4E–4F), pointing out different regulatory mechanisms of IL-1β in pancreas as NF-κB activity inhibits the release of IL-1β from macrophage (Greten et al., 2007).
In summary, we report here constitutive NF-κB activation is required for PDAC development and proposed the mechanism for NF-κB activation by KrasG12D through AP-1-induced IL-1α overexpression. Since IL-1α overexpression correlates with poor survival in PDAC patients, and since it is found in other diseases, such as chronic inflammation autoimmune disorders (Gabay et al., 2010), pharmacologic targeting IL-1α overexpression may represent a potential therapy for PDAC.
EXPERIMENTAL PROCEDURES
Generation of Mouse Strain
The genetically engineered mouse strains used in our study were kindly and generously provided by the following laboratories: floxed IKK2/β (IKK2/βF/F) mice by Michael Karin’s laboratory (Li et al., 2003); floxed Ink4a/Arf (Ink4a/ArfF/F) mice by Ronald Depinho’s laboratory (Aguirre et al., 2003); KrasLSL-G12D mice by Tyler Jacks’ laboratory (Johnson et al., 2001); and Pdx1-Cre transgenic mice by Douglas Melton’s laboratory (Gu et al., 2002). These strains were interbred to generate the experimental cohorts, which include the following genotypes: Pdx1-Cre;KrasLSL-G12D, Pdx1-Cre;IKK2/βF/F, Pdx1-Cre;KrasLSL-G12D; IKK2/βF/F, Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F and Pdx1-Cre;KrasLSL-G12D;Ink4a/ArfF/F; IKK2/βF/F. These mutant mouse strains were genotyped by PCR as previously described by the laboratories that generated them. All animal experiments were conducted under the protocol that was approved for this study by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas MD Anderson Cancer Center.
Patient Pancreatic Ductal Adenocarcinoma and Adjacent Normal Tissues
Patient pancreatic cancer tissue microarray (TMA) was constructed and paraffin sections were obtained using the paraffin blocks from primary pancreatic ductal adenocarcinoma and paired adjacent normal tissues of 131 pancreatic cancer patients, which were collected within 1 hr after surgery under the protocol approved by the Institutional Review Board at M.D. Anderson Cancer Center, and written informed consent was obtained from patients in all cases at time of enrollment.
Cell Lines and Reagents
The human pancreatic cancer cell line AsPC-1 was purchased from the American Type Culture Collection. MDAPanc-28 was established by Marsha Frazier and Douglas B. Evans (M.D. Anderson Cancer Center). Immortalized/nontumorigenic HPDE and the hTERT-HPNE cells were described (Lee et al., 2005; Qian et al., 2005). Panc-28/ IκBαM, AsPc-1/IκBαM, HPNE/Kras, and HPDE/Kras cell lines were established in Chiao’s laboratory (Qian et al., 2005). Anti-human IL-1α and IL-1β, anti-mouse IL-1α and IL-1β neutralizing antibodies, and IL-1α were obtained from R&D Systems.
Knockdown of IKK2/β, p62, IL-1α, Traf6, and c-Fos
To silence IKK2/β, p62, and IL-1α expression, DNA sequences encoding shRNAs and a scramble sequence were chosen to clone into the FG12 lentiviral vector. The mouse primary pancreatic cancer cell lines mPDAC-1 and mPDAC-2 and human PDAC cell lines Panc28 and AsPc-1 were infected by lentivirus containing the shRNA. The infected cells were sorted by green fluorescent protein after infection 4 days. The levels of target gene knockdown by shRNA were determined using immunoblotting.
Statistical Analysis
Mean values between groups were compared by Excel. Survival curves were plotted by the Kaplan-Meier method and compared by the log-rank test using GraphPad Prism. The correlative relationships between two quantitative measurements were investigated using Spearman rank-order correlation and the chi-square statistic. Data were analyzed by the Student t test and results were considered significant at a p value < 0.05.
Supplementary Material
Significance.
Pancreatic ductal adenocarcinoma (PDAC) is the fourth most common cause of adult cancer death in the US. The 5-year survival rate has remained 1%–3% for the past 25 years. Each year approximately 42,000 cases of PDAC are diagnosed; over 80% are therapy-resistant locally advanced or metastatic disease, and median survival is less than 6 months. Thus, PDAC remains a challenge in cancer research. To understand mechanisms of pancreatic tumorigenesis, we examined IKK2/β/NF-κB activation and KrasG12D mutation, two of the signature alterations in human PDAC, using genetically engineered mouse models. Our findings establish a pathway linking dual feedforward loops of IL-1α/p62 through which IKK2/β/NF-κB is activated by KrasG12D; this suggests therapeutic targets for inhibiting KrasG12D signaling in PDAC.
Acknowledgments
We thank Dr. Michael Karin for the IKK2/βF/F mice, Dr. Douglas Melton for the Pdx1-Cre transgenic mice, Dr. Tyler Jacks for the KrasLSL-G12D mice, Dr. Ronald DePinho for the Ink4a/Arf F/F mice, and Dr. Kenneth Hess in the Department of Biostatistics, M.D. Anderson Cancer Center, for statistical analysis, Dr. Chang-gong Liu in Sequencing & Non-coding RNA Program and Dr. Wei Zhang at Genomic Core, M.D. Anderson Cancer Center, for micro-array analysis. We also thank Yu (June) Cao for technical assistance and Kathryn Hale for editorial assistance. The work was supported in part by grants from the National Cancer Institute (CA109405 and CA142674 to P.J.C.), a Cancer Center Core Supporting grant (CA16672), and Sister Institution Fund of China Medical University and Hospital and MD Anderson Cancer Center to M. C. H.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures, three tables, and Supplemental Experimental Procedures and can be found with this article online at doi:10.1016/j.ccr.2011.12.006.
ACCESSION NUMBERS
Microarray data has been deposited in the Gene Expression Omnibus (GEO) database (accession number GSE33323).
References
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basseères DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537–3546. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentnall TA, Bronner MP, Byrd DR, Haggitt RC, Kimmey MB. Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Ann Intern Med. 1999;131:247–255. doi: 10.7326/0003-4819-131-4-199908170-00003. [DOI] [PubMed] [Google Scholar]
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M, Jr, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–170. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Fujioka S, Sclabas GM, Schmidt C, Niu J, Frederick WA, Dong QG, Abbruzzese JL, Evans DB, Baker C, Chiao PJ. Inhibition of constitutive NF-kappa B activity by I kappa B alpha M suppresses tumorigenesis. Oncogene. 2003;22:1365–1370. doi: 10.1038/sj.onc.1206323. [DOI] [PubMed] [Google Scholar]
- Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232–241. doi: 10.1038/nrrheum.2010.4. [DOI] [PubMed] [Google Scholar]
- Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goöktuna SI, Neuenhahn M, Fierer J, Paxian S, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, Peck-Radosavljevic M, Leffert HL, Karin M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res. 2008;18:334–342. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc. 2009;4:1670–1680. doi: 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- Lee KM, Yasuda H, Hollingsworth MA, Ouellette MM. Notch 2-positive progenitors with the intrinsic ability to give rise to pancreatic ductal cells. Lab Invest. 2005;85:1003–1012. doi: 10.1038/labinvest.3700298. [DOI] [PubMed] [Google Scholar]
- Li ZW, Omori SA, Labuda T, Karin M, Rickert RC. IKK beta is required for peripheral B cell survival and proliferation. J Immunol. 2003;170:4630–4637. doi: 10.4049/jimmunol.170.9.4630. [DOI] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenfels AB, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK, Jr, Perrault J, Whitcomb DC International Hereditary Pancreatitis Study Group. Hereditary pancreatitis and the risk of pancreatic cancer. J Natl Cancer Inst. 1997;89:442–446. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N, Prager D. Transactivation of the interleukin-1alpha promoter by human T-cell leukemia virus type I and type II Tax proteins. Blood. 1996;87:3410–3417. [PubMed] [Google Scholar]
- Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353–362. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci USA. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng B, Ling J, Lee AJ, Wang Z, Chang Z, Jin W, Kang Y, Zhang R, Shim D, Wang H, et al. Defective feedback regulation of NF-kappaB underlies Sjogren’s syndrome in mice with mutated kappaB enhancers of the IkappaBalpha promoter. Proc Natl Acad Sci USA. 2010;107:15193–15198. doi: 10.1073/pnas.1005533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999;18:6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Niu J, Li M, Chiao PJ, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–5053. doi: 10.1158/0008-5472.CAN-04-3208. [DOI] [PubMed] [Google Scholar]
- Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–796. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- Wislez M, Spencer ML, Izzo JG, Juroske DM, Balhara K, Cody DD, Price RE, Hittelman WN, Wistuba II, Kurie JM. Inhibition of mammalian target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K-ras. Cancer Res. 2005;65:3226–3235. doi: 10.1158/0008-5472.CAN-04-4420. [DOI] [PubMed] [Google Scholar]
- Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280:35625–35629. doi: 10.1074/jbc.C500237200. [DOI] [PubMed] [Google Scholar]
- Yang J, Splittgerber R, Yull FE, Kantrow S, Ayers GD, Karin M, Richmond A. Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J Clin Invest. 2010;120:2563–2574. doi: 10.1172/JCI42358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel U, Henkel T, Silva MS, Baeuerle PA. Nuclear uptake control of NF-kappa B by MAD-3, an I kappa B protein present in the nucleus. EMBO J. 1993;12:201–211. doi: 10.1002/j.1460-2075.1993.tb05646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.