

SUMMARY
Coordinated production and remodeling of the extracellular matrix is essential during development. It is of particular importance for skeletogenesis, as the ability of cartilage and bone to provide structural support is determined by the composition and organization of the extracellular matrix. Connective tissue growth factor (CTGF, CCN2) is a secreted protein containing several domains that mediate interactions with growth factors, integrins and extracellular matrix components. A role for CTGF in extracellular matrix production is suggested by its ability to mediate collagen deposition during wound healing. CTGF also induces neovascularization in vitro, suggesting a role in angiogenesis in vivo. To test whether CTGF is required for extracellular matrix remodeling and/or angiogenesis during development, we examined the pattern of Ctgf expression and generated Ctgf-deficient mice. Ctgf is expressed in a variety of tissues in midgestation embryos, with highest levels in vascular tissues and maturing chondrocytes. We confirmed that CTGF is a crucial regulator of cartilage extracellular matrix remodeling by generating Ctgf−/−mice. Ctgf deficiency leads to skeletal dysmorphisms as a result of impaired chondrocyte proliferation and extracellular matrix composition within the hypertrophic zone. Decreased expression of specific extracellular matrix components and matrix metalloproteinases suggests that matrix remodeling within the hypertrophic zones in Ctgf mutants is defective. The mutant phenotype also revealed a role for Ctgf in growth plate angiogenesis. Hypertrophic zones of Ctgf mutant growth plates are expanded, and endochondral ossification is impaired. These defects are linked to decreased expression of vascular endothelial growth factor (VEGF) in the hypertrophic zones of Ctgf mutants. These results demonstrate that CTGF is important for cell proliferation and matrix remodeling during chondrogenesis, and is a key regulator coupling extracellular matrix remodeling to angiogenesis at the growth plate.
Keywords: CCN, CTGF, Chondrogenesis, Angiogenesis, Mutant
INTRODUCTION
Connective tissue growth factor (CTGF, CCN2) is a member of the CCN family of secreted proteins, which also includes Cyr61, NOV, WISP1, WISP2 and WISP3 (Bork, 1993; Moussad and Brigstock, 2000; Perbal, 2001). CTGF is a major inducer of extracellular matrix (ECM) production in fibrotic diseases, which are characterized by excessive collagen deposition. CTGF is overexpressed in fibrotic lesions, and the degree of overexpression correlates with severity of disease (Blom et al., 2001; Dammeier et al., 1998; Frazier et al., 1996; Grotendorst, 1997; Lasky et al., 1998; Mori et al., 1999; Stratton et al., 2001). The ability of CTGF to induce collagen deposition in pathological conditions raises the possibility that it may be a mediator of ECM production in tissues such as cartilage and bone during development. However, nothing is known about its role in normal tissues.
CTGF may act in part as a mediator of transforming growth factors β (TGFβs) and bone morphogenetic proteins (BMPs) during development. TGFβs play roles in a wide variety of developmental events, and TGFβ induces CTGF expression in many cell types because the CTGF promoter contains a TGFβ response element (Holmes et al., 2001). Moreover, CTGF contains a von Willebrand type C domain, which is thought to mediate physical interactions with growth factors such as TGFβ (Wong et al., 1997). Consistent with this, CTGF binds to BMPs and TGFβ, leading to inhibition of BMP and enhancement of TGFβ signaling (Abreu et al., 2002).
In addition to its potential role in TGFβ and BMP pathways, several lines of evidence indicate that CTGF acts independently of TGFβ superfamily members. For example, CTGF and the related protein Cyr61 have effects on gene expression that often oppose those of TGFβ (Chen et al., 2001a), and the induction of CTGF expression occurs through both TGFβ-dependent and -independent pathways (reviewed by Blom et al., 2002). In addition, a distinguishing feature of CTGF and other CCN proteins is the presence of several domains that participate in protein interactions (Bork, 1993). In addition to the von Willebrand type C domain required for TGFβ and BMP binding (Abreu et al., 2002), CCNs contain a thrombospondin (TSP) module, which enables TSP to bind to ECM proteins, matrix metalloproteinases (MMPs) and integrins (Bornstein, 2001; Lau and Lam, 1999). CTGF promotes effects on cell survival, adhesion and migration through interactions with integrins (Babic et al., 1999; Chen et al., 2001a; Jedsadayanmata et al., 1999; Leu et al., 2002). CTGF also binds to low density lipoprotein receptor-related protein (LRP), but it is as yet unclear whether this interaction facilitates CTGF signaling and/or clearance (Babic et al., 1999; Jedsadayanmata et al., 1999; Segarini et al., 2001). In addition, CTGF binds to MMPs, and inactivates VEGF through direct physical interactions (Inoki et al., 2002). Finally, the C-terminal domain of CTGF promotes cell proliferation (Brigstock, 1997). Although the constellation of proteins with which CTGF interacts in vivo is not known, the presence of multiple domains is consistent with a role for CTGF as an integrator of multiple growth factor-, integrin- and ECM-derived signals.
Because the ECM transduces signals from the microenvironment, and regulates the release of growth factors, alterations in ECM composition during development lead to dynamic changes in its signaling properties. ECM remodeling is achieved by regulating the production and degradation of specific ECM components. MMPs, which comprise a large family of enzymes with differential abilities to degrade specific ECM components, play a vital role in this process (Sternlicht and Werb, 2001). MMPs also cleave growth factors and their binding proteins, thereby activating or inhibiting specific signaling pathways. Overexpression of CTGF in fibroblasts leads to increased expression of MMP1, MMP2 and MMP3 (Chen et al., 2001b; Fan and Karnovsky, 2002), suggesting that CTGF coordinates ECM production and degradation.
The expression of CTGF in cartilage, and its ability to promote chondrogenic differentiation in vitro (Nakanishi et al., 2000), is consistent with a potential role for CTGF in ECM remodeling during skeletal development. During chondrogenesis, mesenchymal cells condense into characteristic shapes. Cells within these condensations subsequently differentiate into chondrocytes, which secrete ECM components, surrounded by a layer of perichondrial cells. As development proceeds, cells within the aggegrates exit the cell cycle and mature, leading to stratified zones of cells at progressive stages of differentiation (resting, proliferative, prehypertrophic and hypertrophic). In cartilages destined to be replaced by bone through endochondral ossification, terminally differentiated hypertrophic chondrocytes undergo apoptosis as the growth plate is invaded by blood vessels and osteoblasts. The ability of the growth plate to support angiogenesis is dependent upon the activity of MMPs, although the targets of MMP action in hypertrophic chondrocytes are not known (Vu et al., 1998; Ensig et al., 2000).
Along with a potential role in the regulation of ECM composition, a role for CTGF in angiogenesis is likely, as CTGF expression is induced by vascular endothelial growth factor (VEGF) (Suzuma et al., 2000), is expressed in endothelial and vascular smooth muscle cells, and induces neovascularization (Babic et al., 1999; Moussad and Brigstock, 2000; Shimo et al., 1999). Although these studies imply a positive role for CTGF in angiogenesis, CTGF can bind to VEGF and inhibit the ability of VEGF to induce angiogenesis (Hashimoto et al., 2002; Inoki et al., 2002). These observations suggest that CTGF may have both positive and negative effects on angiogenesis.
Despite strong evidence that CTGF promotes ECM production and angiogenesis in fibrotic disease, nothing is known about its role during development. In particular, downstream targets of CTGF action in normal tissues have not been identified. Additional questions include the extent to which CTGF collaborates with TGFβ during development, and whether CTGF and the related molecule, Cyr61, share overlapping functions, as these proteins have related activities in vitro, are co-expressed in several tissues, and serve as ligands for the same set of integrins (Perbal, 2001). To address these issues, we examined the pattern of Ctgf expression, studied its effects on ECM production, and generated Ctgf-deficient mice.
MATERIALS AND METHODS
In situ hybridization
In situ hybridization was performed as described (Hogan et al., 1994). The Ctgf probe was generated by subcloning a partial mouse cDNA IMAGE clone (ID 551901) into pBluescript (Stratagene), linearizing with EcoRI, and reverse transcribing with T7 polymerase. The Tgfb1 probe was obtained from American Type Culture Collection. The ColII and ColX probes were a generous gift from Vicki Rosen. A. McMahon kindly provided the Ihh probe.
Gene targeting
Ctgf clones were isolated from a strain 129Sv/J mouse BAC library (Incyte). The targeting construct was generated by replacing a 500 bp SmaI fragment containing exon 1, the TATA box and the transcription start site with the neomycin resistance gene under the control of a PGK promoter (PGKneopA). The targeting vector was electroporated into RW-4 ES cells (Incyte) as described (Ramírez-Solis et al., 1993). Targeted clones were injected into blastocysts by the UCLA Transgenic Mouse Facility. Chimeras were bred to Balbc/J females to test for germline transmission. The mutation has been maintained on a hybrid 129Sv/J × Balbc/J background. Genotyping was performed by Southern blot analysis of HindIII-digested genomic DNA using the external probe indicated in Fig.2A.
Fig. 2.
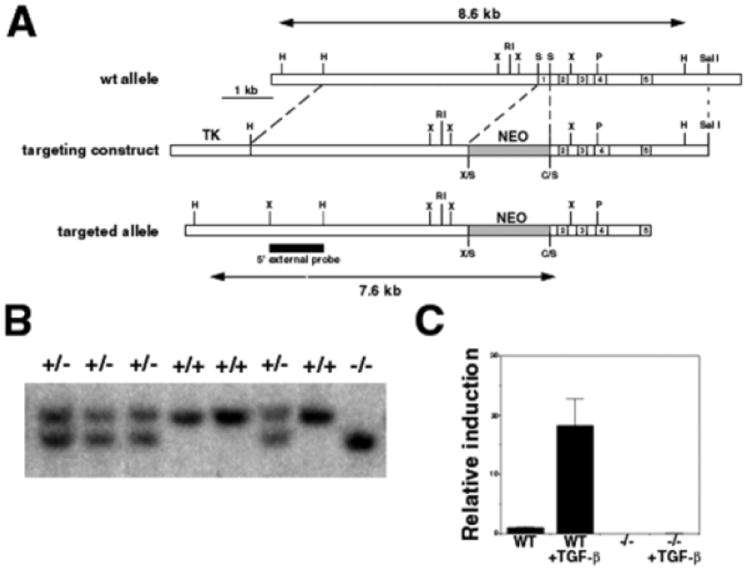
Generation of Ctgf−/− mice. (A) Map of the Ctgf locus (wild-type allele), targeting vector and mutant allele produced by homologous recombination. The HindIII fragments expected for the wild-type (8.6 kb) and mutant (7.6 kb) alleles are indicated by double-headed arrows. A, ApaI; B, BamHI; C, ClaI; H, HindIII; N, NotI; P, PstI; R, EcoRI; S, SmaI; X, XbaI. (B) Southern blot analysis of genomic DNA from a heterozygote intercross. (C) Verification that the targeted locus encodes a null allele. RT-PCR analysis of fibroblasts from wild-type and mutant embryos treated with TGFβ1 (5 ng/ml) to induce Ctgf expression. No Ctgf transcripts can be detected in mutants, even after TGFβ treatment.
Semi-quantitative RT-PCR
Embryonic fibroblasts (EFs) were prepared as described (Abbondanzo et al., 1993) and grown to confluence in DMEM containing 10% FBS. Cells were then grown in serum-free DMEM containing 50 μg/ml ascorbic acid, with or without 5 ng/ml TGFβ1 (R&D Systems) for 24 hours. RNA was harvested using the Qiashredder and Rneasy kits (Qiagen). RNA synthesis was performed using Superscript II (Gibco BRL). RT-PCR was performed using total RNA from EFs derived from mutant or wild-type neonates. The CTGF primers were 5′CTGTCAAGTTTGAGCTTTCTGG 3′ and 5′ GGACTCAAAGATGTCATTGTCC 3′ Control primers for GAPDH were 5′ ACCCAGAAGACTGTGGATGG 3′ and 5′ ATCATACTTGGCAGGTTTCTCC 3′.
Long bones at E14.5, or growth plates at later stages from individual genotyped wild-type and mutant littermates were dissected out and total RNA was prepared using TRIzol (Gibco-BRL). Levels of ColII, ColX, Cbfa1 (Runx2 – Mouse Genome Informatics), Vegf (Vegfa – Mouse Genome Informatics) and Mmp9 were examined by semi-quantitative RT-PCR, on oligo (dT)-primed cDNA (Superscript, Gibco-BRL) from growth plate total RNA using the following primers: ColII, 5′-CACACTGGTAAGTGGGGCAAGAC and 3′-GGATTGTGTTGTTTCAGGGTTCGGG; ColX, 5′-CCTGGGTTAGATGGAAA and 3′-AATCTCATAAATGGGATGGG; Vegf, 5′-GGGTGCACTGGACCCTGGGTTTAC and 3′-CCTGGCTCACCGCCTTGGCTTGTC; Cbfa1, 5′-TGACTGCCCCCACCCTCTTAG, 3′-GGCAGCACGCTATTAAATCCAAA; Mmp9 5′-AACCCTGTGTGTTCCCGTT and 3′-GGATGCCGTCTATGTCGTCT; and Gapdh 5′-CCCCTTCATTGACCTCAACT and 3′-TTGTCATGGATGACCTTGGC. Typical reactions were performed with cycles conditions of 94°C for 1 minute, 60°C for 1 minute, 72°C for 1 minute. The following numbers of cycles were used for each primer pair: Gapdh, 20, 22, 24; ColII, 22, 24, 26; ColX, 26, 30, 24; Mmp9, 26, 28, 30; Runx2, 30, 2, 34; Vegf, 28, 30, 32. RNA samples from five wild-type and five mutant littermates were examined. Each RNA sample was analyzed twice. Quantification of expression relative to Gapdh was performed using NIH image.
Skeletal analysis and histology
Cleared skeletal preparations were made as described (Yi et al., 2000). For histology, specimens were fixed with 4% paraformaldehyde or 10% neutral formalin, decalcified with Immunocal (Decal Chemical) and embedded in paraffin. Deparaffinized sections (7 μm) were stained with Toluidine Blue or safranin-o/light green. Plastic sections were fixed in neutral-buffered formalin, dehydrated and embedded undecalcified in DDK-Plast methylmethacrylate resin. Sections (4 μm) were stained with von Kossa/tetrachrome.
Immunostaining was performed on deparaffinized sections with antibodies for link protein and aggrecan (8A4, IC6, Developmental Studies Hybridoma Bank), MMP9 and MMP13 (Chemicon) or type II collagen (Research Diagnostics) at a 1:100 dilution using the Histomouse kit (Zymed). Tissue sections were pretreated with chondroitinase ABC (0.05n u/ml; Sigma) for 8A4 and IC6, or with 2.5% hyaluronidase (Calbiochem) for MMP9, MMP13, and type II collagen. Immunostaining for PECAM (Bectin Dickinson) was carried out on cryosections. Briefly, cryosections were treated with 2.5% hyaluronidase for 30 minutes at room temperature and with 3% hydrogen peroxide/PBS for 10 minutes at room temperature. After washing with PBS and blocking with 2% dry milk, 5% goat serum in PBS, incubation with anti-PECAM antibody (Zymed; 1:100) was carried out overnight at 4°C. After washing with blocking buffer, slides were incubated with rat secondary antibody for 2 hours at room temperature. Color was developed with DAB for MMP9 and PECAM, and with the Zymed kit chromogen for all other antibodies.
For analysis of osteoclasts, sections were stained for tartrate-resistant acid phosphatase (TRAP) positive cells using a TRAP staining kit (Sigma).
Cell proliferation and apoptosis
Cell proliferation was assessed by BrdU incorporation as described (Yi et al., 2000), and by PCNA immunostaining. For PCNA, 7 μm decalcified paraffin wax-embedded sections were used with a 1:100 dilution of anti-PCNA antibody as described above for PECAM staining. Cell proliferation was assessed as described (Yi et al., 2000). Cell death analysis by TUNEL analysis was performed using the Apoptosis Detection System, Fluorescein kit (Promega).
RESULTS
Ctgf expression in midgestation embryos
In situ hybridization revealed strong expression in skeletal, vascular and neural tissues. Ctgf mRNA was detected beginning at E9.5 in the nasal process, proximal regions of the second and third branchial arches, and the developing heart (Fig. 1A). Expression was also observed within the neural tube (Fig. 1B). At E10.5-E11.5, high levels of Ctgf expression persisted in the proximal branchial arches, dorsal nasal process, heart and floorplate (Figs. 1C,D). Lower levels were seen in the roofplate and dermomyotome (Fig. 1D).
Fig. 1.
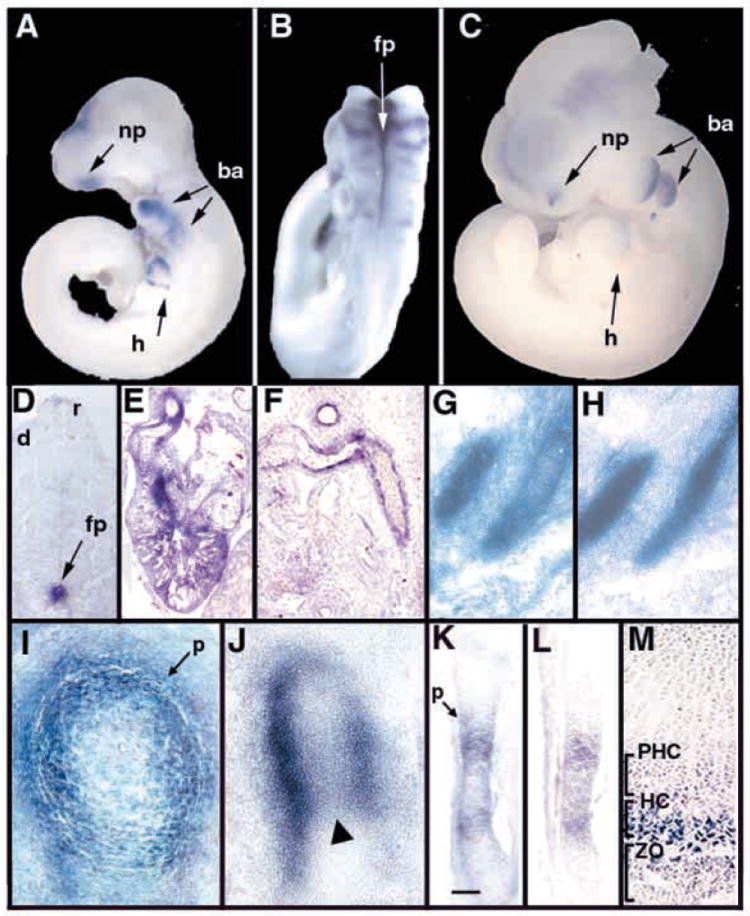
Expression of Ctgf in midgestation embryos. Whole-mount in situ hybridization of E9.5 (A,B) and an E10.5 (C) embryo treated with antisense Ctgf, demonstrating expression in heart, branchial arches, neuronal tissues and nasal process. (D) Frontal section through an E11.5 embryo showing high levels of Ctgf expression in the floorplate, and lower levels in the roofplate and dermomyotome. (E) Ctgf expression in the heart at E13.5. Transcripts are present in ventricular myofibroblasts and in fusing endocardial cushion tissue at the midline of the outflow tract. (F) Ctgf expression in the endothelium and smooth muscle layers of major blood vessels at E13.5. (G,H) Adjacent parasagittal sections through E12.5 ribs showing Ctgf expression in perichondrium (G) and collagen type II (ColII) (H) expression in proliferating chondrocytes. (I) Transverse section through an E13.5 femur showing Ctgf expression in perichondrium and in adjacent chondrocytes. (J) Sagittal section through the femur of an E13.5 embryo showing strong Ctgf expression in the perichondrium, and lower levels in chondrocytes at the center of the diaphysis (arrowhead). (K-M) Adjacent sections though an E14.5 femur showing expression of Ctgf (K), and Ihh (L). Scale bar in K: 100 μm for K,L. Ctgf is expressed in the domain of Ihh expression, as well as in prehypertrophic chondrocytes adjacent to the perichondrium. (M) Section through the proximal growth plate of a P0 femur, showing Ctgf expression in hypertrophic chondrocytes. ba, branchial arch; d, dermomyotome; fp, floorplate; h, heart; HC, hypertrophic chondrocytes; m, meninges; np, nasal process; p, perichondrium; PHC, prehypertrophic chondrocytes; r, roofplate; sc, spinal cord; ZO, zone of ossification.
Ctgf expression persists in the meninges, heart and major blood vessels from E13.5 to birth (Fig. 1E,F, and data not shown). Within the heart, Ctgf mRNA is present in ventricular myofibroblasts and in midline tissue within the fusing cushions of the outflow tract (Fig. 1E). Ctgf transcripts persist at least until birth in endothelial and smooth muscle layers of major blood vessels (Fig. 1F).
In situ hybridization experiments revealed that Ctgf is highly expressed in cartilage in midgestation embryos. During skeletogenesis, Ctgf is first expressed at E12.5 in perichondrium (Fig. 1G,H). By E13.5, Ctgf persists at high levels in perichondrium and in adjacent chondrocytes (Fig. 1I). At this stage, Ctgf can also be detected at lower levels within maturing chondrocytes at the centers of developing long bones (Fig. 1J). At E14.5, Ctgf expression persists in chondrocytes adjacent to the perichondrium, and is upregulated in maturing chondrocytes (Fig. 1K); at this stage, Ctgf expression overlaps extensively with that of Indian hedgehog (Ihh), a marker for prehypertrophic and hypertrophic chondrocytes (Bitgood and McMahon, 1995) (Fig. 1L; see also Fig. 5A,B). At this and subsequent stages, the strongest site of Ctgf expression is within the most mature population of chondrocytes. For example, by E16.5, Ctgf is highly expressed in terminally differentiated hypertrophic chondrocytes, as demonstrated by its overlapping pattern of expression with that of type X collagen (ColX), a marker for hypertrophic chondrocytes (Fig. 5F,H). Ctgf expression persists at this stage in chondrocytes adjacent to the perichondrium within the prehypertrophic zone (Fig. 5F,G). The expression of high levels of Ctgf in hypertrophic cells continues at least until birth (Fig. 1M). In summary, within developing cartilage, Ctgf is expressed initially in the perichondrium. At later stages, Ctgf is expressed within maturing chondrocytes. Transcripts persist in chondrocytes adjacent to the perichondrium at least until E16.5. However, terminally differentiating prehypertrophic and hypertrophic chondrocytes are the major sites of Ctgf expression in developing cartilage.
Fig. 5.
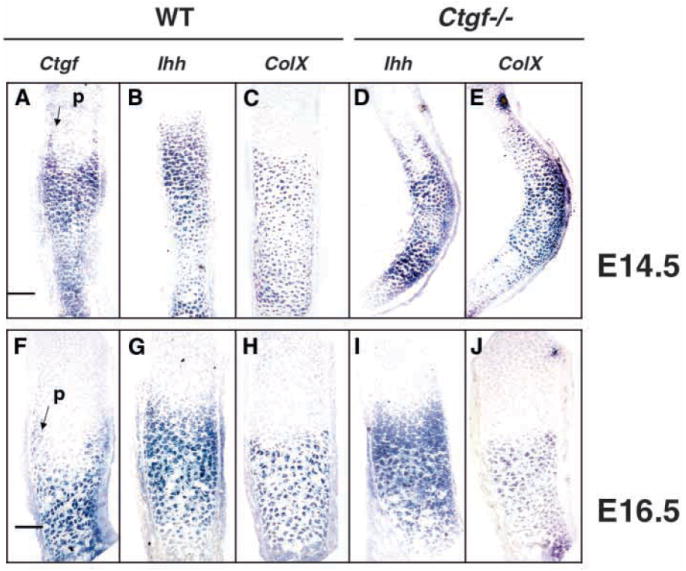
Expression of Ihh and ColX in Ctgf mutants. (A-C) Expression of Ctgf (A), Ihh (B) and ColX (C) in the radius of an E14.5 wild-type embryo. The expression patterns of Ctgf and Ihh overlap in prehypertrophic and hypertrophic chondrocytes. Ctgf is co-expressed with ColX in hypertrophic chondrocytes at this stage and in prehypertrophic chondrocytes adjacent to the perichondirum (arrow in A). (D,E) Expression of Ihh (D) and ColX (E) in the radius of an E14.5 Ctgf mutant littermate. These markers are expressed in appropriate patterns in prehypertrophic and hypertrophic chondrocytes. Scale bar: 100 μm for A-E (F-H) Expression of Ctgf (F), Ihh (G) and ColX (I) in the radius of an E16.5 wild-type embryo. Ctgf is expressed primarily in hypertrophic chondrocytes in a pattern overlapping that of ColX, but transcripts also persist in chondrocytes adjacent to the perichondrium in the prehypertrophic zone (arrow in F). (I,J) Expression of Ihh (I) and ColX (J) in the radius of an E16.5 Ctgf mutant littermate. The domains of expression of Ihh are indistinguishable in wild-type and mutant littermates, indicating that the expansion of the hypertrophic zone revealed by histological analysis is not accompanied by an expanded prehypertrophic zone. Scale bar: 50 μm for F-J.
Generation of Ctgf mutants
The expression of Ctgf in cartilage and vascular structures throughout development suggested roles in the development of these tissues. To test this hypothesis, we generated Ctgf-deficient mice (Fig. 2A,B). Ctgf heterozygotes are viable and fertile. Semi-quantitative RT-PCR analysis confirmed that the targeted allele is null. Fibroblasts from wild-type embryos exhibited a low level of Ctgf expression, which increased 20-fold upon treatment with TGFβ1 (Fig. 2C). By contrast, Ctgf transcripts were undetectable in mutant fibroblasts, even after exposure to TGFβ1. Homozygous mutants are recovered among neonates in the expected Mendelian ratio. In spite of widespread Ctgf expression in vascular tissues, histological examination and gross dissections revealed no evidence for generalized angiogenesis defects or impairment of cardiac function in Ctgf mutants (data not shown). However, Ctgf−/−mice died within minutes of birth.
Generalized chondrodysplasia in Ctgf mutants
Ctgf−/− mice die shortly after birth because of respiratory failure caused by skeletal defects. Within the axial skeleton, defects are observed along the entire vertebral column. By E14.5, vertebrae in mutants are broader than in wild-type littermates (Fig. 3A), and this phenotype persisted at birth (Fig. 3B). In newborns, the sterna of Ctgf mutants are short and bent inwards, and ossified regions of the ribs are kinked (Fig. 3C,D). The kinks in ossified regions are a consequence of prior defects in chondrogenesis, as the rib cartilage adjacent to sites of mineralization is already bent at E14.5 (Fig. 3E). The overall lengths of individual ribs are not significantly different, but the extent of ossification is reduced in mutants (Fig. 3F), and the zone of mineralizing cartilage is expanded (Fig. 3F; arrow in Fig. 3C), suggesting defective replacement of cartilage by bone during endochondral ossification. In addition, ~10% of mutants exhibit misaligned sternal fusion (Fig. 3G). Endochondral defects are also observed throughout the appendicular skeleton. By E13.5, deformation of the limb cartilage is apparent in Ctgf mutants (Fig. 3H), leading to kinks in the radius, ulna, tibia and fibula in Ctgf mutants at birth (Fig. 3I).
Fig. 3.
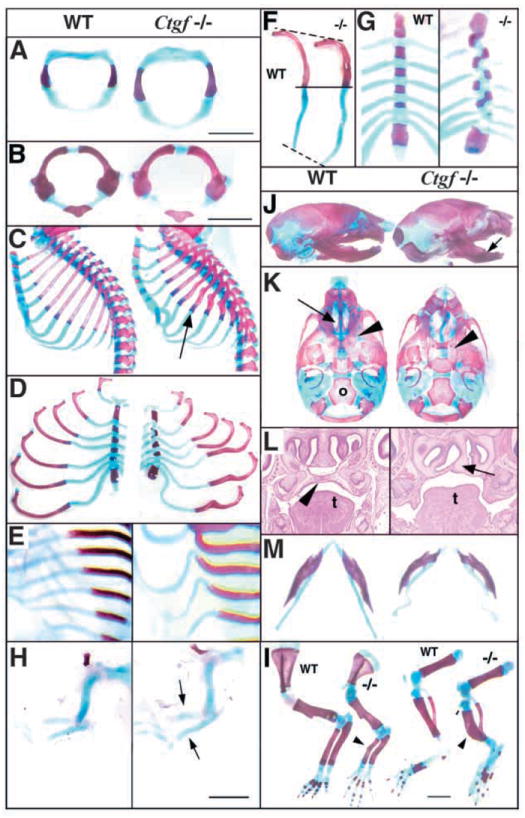
Ctgf−/− mice exhibit multiple skeletal defects. In all panels, wild-type is towards the left and Ctgf−/−is to the right. Atlases from E14.5 (A) and P0 (B) mutants are broader than those from wild-type littermates. (C) Sagittal views of neonatal rib cages showing deformation of cartilage, and kinks in bone in Ctgf−/− mice (arrow). (D) Flat mounts of rib cages show that in Ctgf mutants, ribs are kinked and the sternum is shortened. (E) E14.5 rib cages, showing that the kinks seen in neonatal mutant ribs are preceded by distorted rib cartilage. (F) Endochondral ossification is delayed in Ctgf mutants. Seventh ribs from neonatal wild-type and mutant littermates are shown. (G) Misalignment of the sternal bars is seen in ~10% of neonatal mutants. (H) Cleared skeletal preparations of E13.5 forelimbs, showing kinks in the radius and ulna (arrows) prior to ossification. (I) Cleared skeletal preparations of neonatal forelimbs and hindlimbs showing deformations (arrowheads) in the radius and ulna, and tibia and fibula. (J) Side views of neonatal skulls showing domed skull and shortened mandibles (arrow). (K) Ventral views of neonatal skulls, showing lack of elevation of the palatal shelves, leading to cleft palate (arrowhead), deformation of nasal cartilage, and consequent absence of the adjacent ethmoid bones (arrow in wild type), but no apparent defects in other membrane bones such as the occipital (o). (L) Frontal sections of E15.5 skulls, showing cleft palate (palate in wild type is indicated by arrowhead) and deformed nasal cartilage (arrow in mutant) in mutants. t, tongue. (M) Ctgf mutant neonates exhibit deformation of Meckel’s cartilage and shortened mandibles. Scale bars: 1 mm.
Within the craniofacial skeleton, the cranial vault had a domed appearance, the mandibles were shortened, and the ethmoid bones were deformed (Fig. 3J,K). All Ctgf mutants have a secondary cleft palate because of a failure in elevation of the palatal shelves (Fig. 3L), most likely as a secondary consequence of defects in the formation of endochondral elements at the base of the skull and in nasal cartilages (Fig. 3K,L). The shortened mandible is a consequence of deformations in Meckel’s cartilage (Fig. 3M). These abnormalities indicate that Ctgf-deficient cartilage has inferior mechanical properties, rendering it susceptible to deformation during development. This hypothesis provides an explanation for the enlargement of mutant vertebrae, suggesting that they become distended as a result of their inability to resist the forces of the expanding neural tube.
We performed a histological analysis to investigate these defects in more detail. At E12.5, when Ctgf mRNA is localized to the perichondrium, the sizes and morphologies of the cartilaginous condensations in Ctgf mutants and wild-type littermates are indistinguishable (Fig. 4A, and data not shown). By E14.5, chondrocytes in the midshaft regions of long bones from wild-type mice are undergoing differentiation into prehypertrophic and hypertrophic cells (Fig. 4B). No histological differences were detected in the proliferative zones of wild-type and mutant littermates at this stage. However, in Ctgf mutants, long bones are already bent near the junction between hypertrophic and nonhypertrophic cells (Fig. 4B). The hypertrophic zones did not differ in length in Ctgf mutants and wild-type littermates at this stage (Fig. 4B, and data not shown). At E16.5, when Ctgf is most highly expressed in hypertrophic chondrocytes, endochondral ossification has commenced in long bones from wild-type and mutant littermates (Fig. 4C). An enlarged and disorganized hypertrophic zone is seen in mutants, and this persists at birth (Fig. 4D).
Fig. 4.
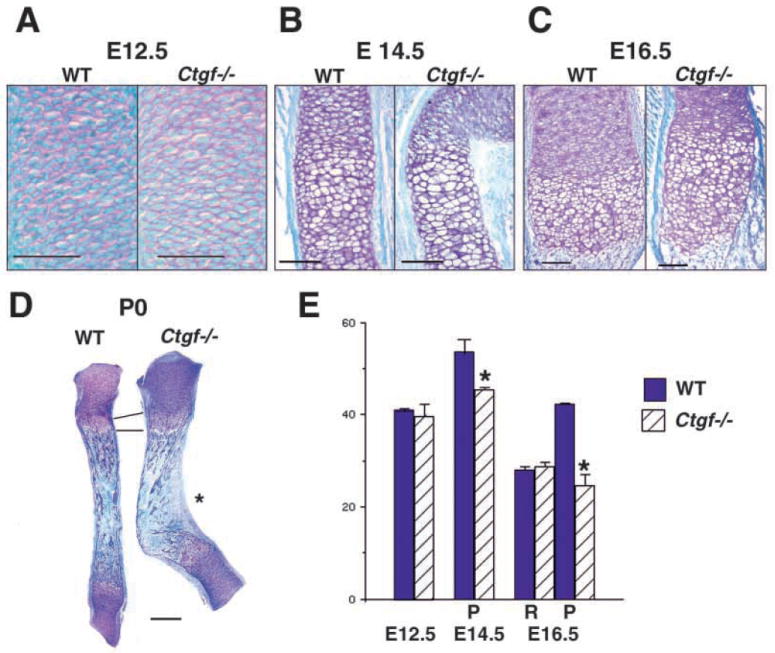
Histological and proliferative defects in Ctgf mutant cartilage. (A) Sections through wild-type and Ctgf mutant humeri at E12.5, showing no apparent differences in size or morphology. Scale bar: 50 μm. (B) Sections through wild-type and Ctgf mutant E14.5 radii at the metaphysis. Hypertrophic cells are present in wild-type and mutant littermates. Scale bar: 100 μm. (C) Sections through growth plates of E16.5 wild-type and Ctgf mutant radii demonstrate that the growth plate is expanded in mutants. The junction between the zones occupied by prehypertrophic and hypertrophic chondrocytes is disorganized in mutants. Scale bar: 100 μm. (D) Radii from newborn wild-type and Ctgf mutant littermates, demonstrating persistence of the enlarged hypertrophic zone. The epiphyses appear normal in mutants. The concave surface of the kink in mutants is a site of membrane bone formation (asterisk). Scale bar: 300 μm. (E) Reduced rates of chondrocyte cell proliferation in Ctgf mutants. Quantification of PCNA labeling is shown in the graph, with values expressed as % labeled nuclei. Cells in five adjacent sections, each spanning 40 μm, were scored by an observer blinded to the genotype. Statistical significance was assessed by Student’s t-test. *P<0.01; P, proliferative zone; R, resting zone.
Thus, loss of Ctgf leads to distorted cartilages. The presence of these defects prior to ossification, along with high levels of Ctgf expression seen in differentiating chondrocytes, indicate a primary role for Ctgf in cartilage. Moreover, although Ctgf is expressed in perichondrium beginning at E12.5, histological differences are not apparent until E14.5, coincident with the expression of Ctgf in maturing chondrocytes. These results suggest that Ctgf is involved in late stages of chondrocyte proliferation and/or differentiation.
Deficient cell proliferation in Ctgf−/− chondrocytes
CTGF promotes chondrocyte proliferation in vitro (Nakanishi et al., 2000), and Ctgf−/− mice exhibit morphological and histological features consistent with proliferative defects. Therefore, proliferation was examined by staining for proliferative cell nuclear antigen (PCNA). These analyses revealed a proliferative defect in neonatal Ctgf−/− growth plates (Fig. 4E). No differences were noted at E12.5. However, by E14.5, when Ctgf is highly expressed in prehypertrophic chondrocytes (Fig. 1K,L), the percentage of proliferating chondrocytes was decreased in mutants. This proliferative defect was more pronounced at E16.5 (Fig. 4E). Therefore, Ctgf appears does not regulate cell proliferation at early stages of chondrogenesis, but appears to be required at later stages.
TUNEL staining was performed to determine whether altered rates of apoptosis might contribute to the cartilage deformations and/or expansion of the hypertrophic zone in mutants. In both wild-type and mutant growth plates, apoptosis is confined to terminal chondrocytes (data not shown). Therefore, apoptosis does not appear to contribute to the defective mechanical properties of Ctgf mutant cartilage, and the expansion of the hypertrophic zone in mutants cannot be attributed to an inability of mutant chondrocytes to undergo apoptosis.
Ctgf is highly expressed in prehypertrophic chondrocytes at E14.5 and promotes chondrocyte proliferation at this stage (Fig. 1K,L, Fig. 4E). Ihh, which coordinates the progression of chondrocytes to hypertrophy and promotes cell proliferation (Long et al., 2001; St-Jacques et al., 1999), is expressed in a pattern overlapping that of Ctgf. Therefore, to determine whether Ctgf might affect chondrocyte proliferation by regulating Ihh levels, we examined Ihh expression by in situ hybridization and semi-quantitative RT-PCR. We also examined the expression of ColX because CTGF promotes ColX expression in chondrocytes in vitro (Nakanishi et al., 2000). Semi-quantitative RT-PCR analysis of growth plates at E14.5 revealed no apparent differences in levels of Ihh or ColX expression between Ctgf mutants and wild-type littermates (data not shown). In situ hybridization studies also indicated that the expression of these markers is not altered in Ctgf mutants. In wild-type mice at E14.5, Ihh is expressed in prehypertrophic and hypertrophic chondrocytes, while ColX expression is restricted to hypertrophic chondrocytes (Fig. 5A-C). At this stage, Ctgf expression overlaps with that of Ihh and ColX, indicating that Ctgf is expressed primarily in hypertrophic chondrocytes (Fig. 5A-C). In E14.5 Ctgf mutant littermates, Ihh and ColX are similarly expressed in prehypertrophic and hypertrophic chondrocytes (Fig. 5D,E). At E16.5 in wild-type mice, the pattern of Ctgf expression overlaps extensively with that of ColX in hypertrophic chondrocytes; Ctgf transcripts persist in chondrocytes adjacent to the perichondrium in the prehypertrophic zone (Fig. 5F-H). By E16.5, the hypertrophic zone is expanded in Ctgf mutants. At this stage, Ihh and ColX were appropriately expressed in the prehypertophic and hypertrophic regions, respectively (Fig. 5I,J). Owing to the distorted shapes of the Ctgf mutant skeletal elements, we were unable to determine unequivocally whether the zones of expression of these markers were expanded in mutants. However, RT-PCR analysis revealed no differences in levels of ColX expression (data not shown). Therefore, although the hypertrophic zone in Ctgf mutants is enlarged by E16.5, this is not accompanied by obvious expansions of the domains of Ihh or ColX expression (Fig. 5G,I). The basis for these observations is unknown at present. It is possible that the subtle expansions in the domains of Ihh and/or ColX expression collectively lead to the expanded growth plates seen in Ctgf mutants.
In summary, Ctgf is required for normal rates of chondrocyte proliferation in vivo. Proliferative defects were detected beginning at E14.5, coincident with the upregulation of Ctgf in prehypertrophic and hypertrophic chondrocytes. However, the decreased rate of cell proliferation in Ctgf mutants does not appear to be due to a decrease in Ihh mRNA levels, indicating that Ctgf acts downstream of Ihh, and/or by an independent mechanism. Finally, although CTGF promotes chondrocyte differentiation in vitro (Nakanishi et al., 2000), cleared skeletal preparations, histological examination, and analysis of Ihh and ColX expression revealed no apparent alterations in chondrocyte progression to hypertrophy in Ctgf mutants.
Defective extracellular matrix production in Ctgf mutants
Cartilage ECM components are the primary determinants of its elastic and tensile properties. The deformed cartilages seen in Ctgf mutants suggested that CTGF is required for synthesis of normal levels of cartilage ECM components. Therefore we examined whether abnormalities in ECM content might contribute to the defective properties of Ctgf mutant cartilage. As previously discussed, no clear differences were seen in ColX mRNA levels in midgestation Ctgf mutants and wild-type littermates (Fig. 5, and data not shown). Similar results were obtained when the expression of type II collagen (ColII), the most abundant collagen present in cartilage, was examined by semi-quantitative RT-PCR and in situ hybridization from E12.5-17.5 (data not shown). Examination of neonates also revealed no obvious differences in the amount or distribution of collagen types II and X in cartilage matrix in Ctgf mutants (Fig. 6A,B). Therefore, although CTGF is a major regulator of type I collagen production during fibrotic responses, and induces the transcription of types II and X collagens in vitro (Nakanishi et al., 2000), CTGF does not appear to be a major regulator of their expression in vivo.
Fig. 6.
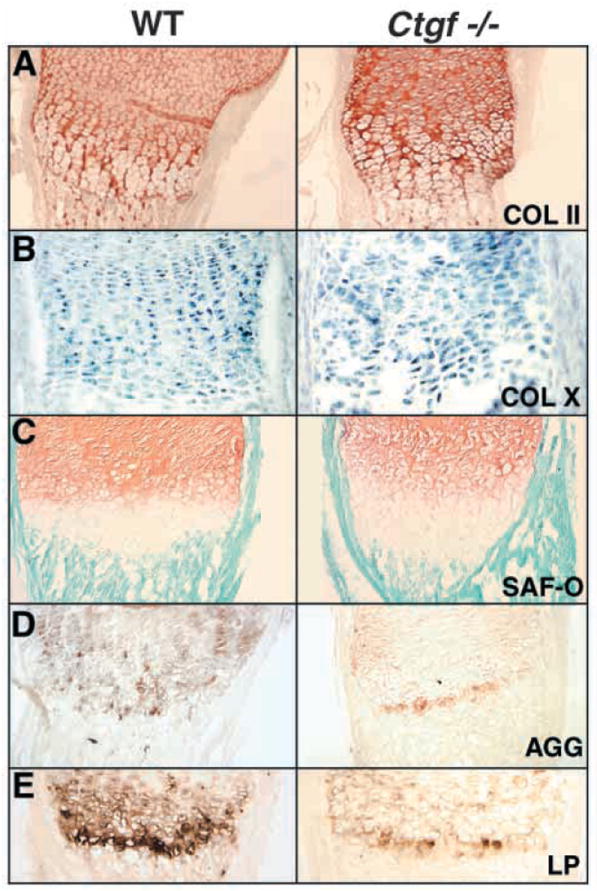
Expression of ECM components is altered in Ctgf mutants. (A) Immunostaining for type II collagen protein is at comparable intensities in P0 growth plates of Ctgf mutants and wild-type littermates. (B) Levels of type X collagen mRNA are indistinguishable in wild-type and Ctgf mutant growth plates. (C) Safranin-o staining demonstrates that proteoglycan levels are normal in the resting and proliferative zones, and that the hypertrophic zone, which is not stained intensely by safranin-O, is expanded in mutants. (D,E) Expression of aggrecan (D) and link protein (E) is reduced in P0 growth plates of Ctgf mutants.
We also examined proteoglycan levels, as CTGF induces proteoglycan synthesis in vitro (Nakanishi et al., 2000). Safranin-o staining, a measure of overall proteoglycan content, revealed no apparent differences between wild-type and mutant littermates in the reserve and proliferative zones, but confirmed that the hypertrophic zone, which does not stain intensely with safranin-o, is expanded in mutants (Fig. 6C). However, levels of aggrecan, the main cartilage proteoglycan (Fig. 6D), and link protein, which stabilizes aggregates of aggrecan and hyaluronin (Fig. 6E), are reduced in Ctgf−/− growth plates, confirming that Ctgf-deficient growth plate cartilage exhibits defects in ECM content. Therefore, Ctgf is required for the expression of wild-type levels of specific cartilage ECM components in vivo, and the inferior mechanical properties of Ctgf mutant cartilage can be attributed to the reduced expression of these components.
Defective growth plate angiogenesis and osteopenia in Ctgf mutants
Histological examination revealed a number of defects in neonatal growth plates of Ctgf mutants. Consistent with the proliferative defects detected by PCNA staining, longitudinal columns are disorganized within the hypertrophic zones in mutants (Fig. 7A). Staining by the von Kossa method revealed apparently normal mineralization of the hypertrophic cartilage matrix (Fig. 7A). Mineralized bone collars, which normally form in the perichondrium adjacent to prehypertrophic and hypertrophic chondrocytes, are lengthened in Ctgf mutants, consistent with the expansion of the hypertrophic zone (Fig. 7B).
Fig. 7.
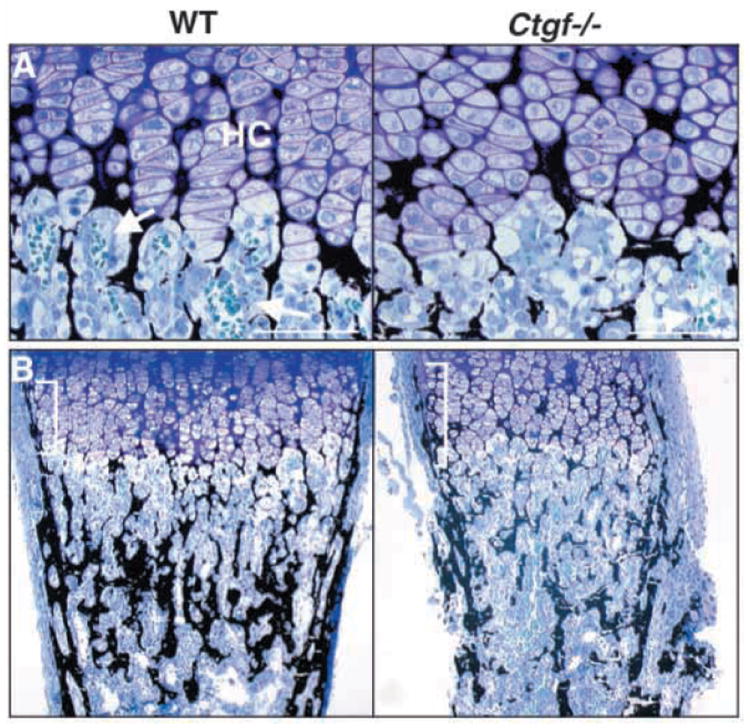
Impaired angiogenesis and osteopenia in growth plates of Ctgf mutants. (A) Plastic sections through the growth plates of P0 femora stained by the method of von Kossa. The ECM of Ctgf mutants is mineralized (black stain), but hypertrophic chondrocyte columns (HC) are disorganized, and there are few capillaries (arrows) invading the cartilage matrix. A single capillary can be seen in the vicinity of the mutant growth plate. (B) von Kossa-stained plastic sections through neonatal femora from wild-type and Ctgf−/−mice demonstrate that mutants are osteopenic; the amount of mineralization (black stain) is greatly reduced in mutants. The bone collar (brackets) adjacent to the expanded hypertrophic zone is lengthened in mutants, but is thinner than in wild-type littermates. Scale bars: 40 μm.
As impairment of angiogenesis leads to an enlarged zone of hypertrophy (Gerber et al., 1999; Haigh et al., 2000; Vu et al., 1998), and Ctgf mutants exhibit expanded hypertrophic zones, we examined whether growth plate angiogenesis is defective. In wild-type neonates, abundant capillaries are seen invading the mineralized hypertrophic cartilage. However, few intact capillaries are visible in growth plates of mutants (Fig. 7A). Immunostaining for PECAM confirmed that growth plate angiogenesis is defective in Ctgf mutants; the fine network of capillaries, well developed in the ossification zones of wild-type neonates, is less extensive in Ctgf mutants, although blood vessels are present within intertrabecular spaces (Fig. 8A).
Fig. 8.
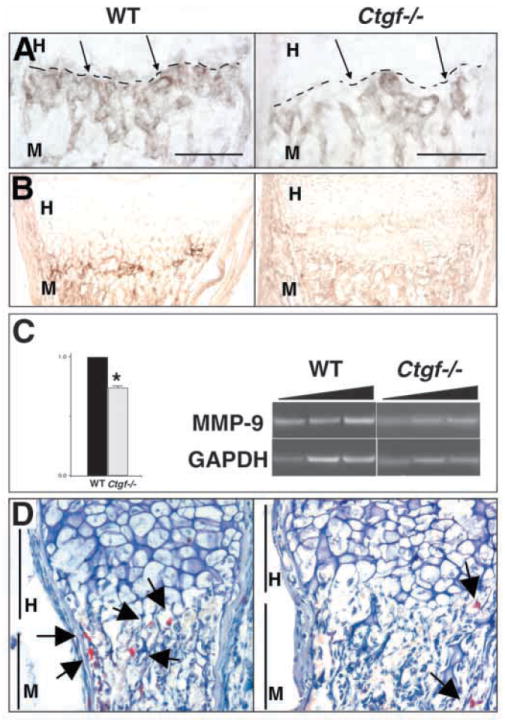
Defective expression of angiogenic factors in Ctgf mutant growth plates. Growth plates of neonatal radii are shown in all panels. (A) PECAM immunostaining demonstrates the existence of blood vessels in the intertrabecular spaces of the metaphysis, but the fine network of capillaries seen at the ossification zone (broken line) in wild-type animals is not as extensive in Ctgf mutants. Scale bar: 100 μm. (B) MMP9 immunostaining is reduced in neonatal mutants. (C) Mmp9 RNA levels are reduced in neonatal mutants. The triangular bars above the photograph of the gel represent increasing numbers of amplification cycles (see Materials and Methods). Data are from three pairs on Ctgf mutants and wild-type littermates. Expression of Mmp9 was normalized to Gapdh in each reaction. *P<0.05. (D) TRAP-positive cells (arrows) are present in normal levels in the bone marrow of Ctgf mutants, but in reduced levels at the cartilage-bone junction, indicating a defect in recruitment of osteoclasts/chondroclasts to the hypertrophic region. MMP13 protein is present at diminished levels in hypertrophic chondrocytes in Ctgf mutants. H, hypertrophic zone; M, metaphysis.
Defective growth plate angiogenesis is associated with decreased trabecular bone density (Gerber et al., 1999). Consistent with a defect in growth plate angiogenesis, the bone collar appears thinner, and less trabecular bone is present in Ctgf mutants (Fig. 7B). A primary role for CTGF in osteoblast function is also possible as CTGF is expressed in osteoblasts and promotes their proliferation and differentiation in vitro (Nishida et al., 2000; Xu et al., 2000a). Additional studies will be required to discriminate between direct and indirect roles for CTGF in osteoblasts.
CTGF regulates the availability of local factors required for growth plate angiogenesis
Angiogenesis at the growth plate requires localized proteolytic modification of the ECM to permit invasion by endothelial cells, and MMPs play essential roles in this process (Vu et al., 1998; Zhou et al., 2000). MMP9 is required for growth plate angiogenesis, and is expressed in osteoclasts/chondroclasts (Reponen et al., 1994). MMP9 immunostaining in wild-type neonates is most intense at the hypertrophic cartilage-bone junction. By contrast, MMP9 immunostaining at this junction is diminished in growth plates of Ctgf mutants (Fig. 8B). Semi-quantitative RT-PCR analysis confirmed that Mmp9 mRNA levels are reduced in the growth plates of mutants (Fig. 8C). To determine whether the decrease in MMP9 levels in mutant growth plates reflects a generalized loss of osteoclasts, staining for tartrate-resistant acid phosphatase (TRAP) activity, a marker for cells of the osteoclast lineage, was performed. In wild-type neonates, TRAP-positive cells are distributed throughout the bone marrow, and co-localize with MMP9-expressing cells at the cartilage-bone junction (Fig. 8D). By contrast, in Ctgf mutants, although TRAP-positive cells are found at normal levels in bone marrow (data not shown), few are seen at the cartilage-bone junction (Fig. 8D). Therefore, CTGF is important for efficient infiltration of the calcified cartilage matrix by MMP9-expressing osteoclasts/chondroclasts.
VEGF produced by hypertrophic cartilage promotes angiogenesis, is activated by MMP-mediated degradation of the cartilage matrix and is chemotactic for osteoclasts (Gerber et al., 1999; Haigh et al., 2000). VEGF protein is expressed at low levels in maturing chondrocytes at E14.5, and at high levels in hypertrophic chondrocytes of the neonatal wild-type growth plate (Carlevaro et al., 2000; Engsig et al., 2000). By contrast, VEGF immunostaining per cell is diminished in the expanded hypertrophic zone in newborn Ctgf mutants (Fig. 9A). This decrease in VEGF expression is seen only in the hypertrophic cartilage; expression in osteoblasts (Horner et al., 2001) is at normal levels (data not shown). We used semi-quantitative RT-PCR to examine whether the reduced VEGF immunostaining in Ctgf mutants is due to decreased VEGF mRNA levels (Fig. 9B,C). At E14.5, when Vegf is expressed at low levels in perichondrium and maturing chondrocytes (Zelzer et al., 2002), no differences in levels of Vegf expression can be detected in long bones of Ctgf mutants and wild-type littermates. However, by birth, when Vegf mRNA is highly expressed in hypertrophic chondrocytes, levels of Vegf mRNA are reduced in growth plates of Ctgf mutants, despite the enlargement of the hypertrophic zone (Fig. 9B,C).
Fig. 9.
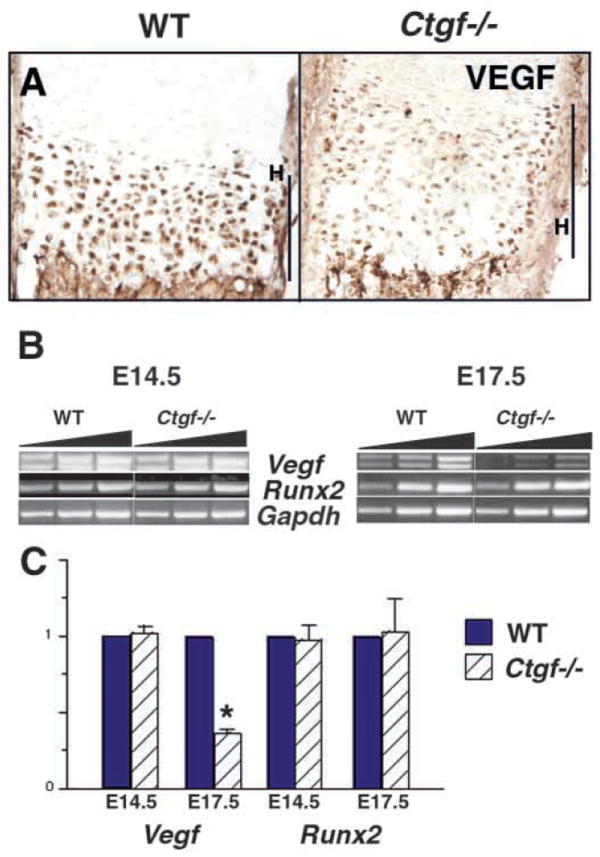
VEGF expression is reduced in the hypertrophic zones of Ctgf mutant growth plates. (A) VEGF immunostaining is reduced in the hypertrophic zones of newborn Ctgf mutants. (B) Semi-quantitative RT-PCR analysis of Vegf and Runx2 expression. Representative data from a single Ctgf mutant and wild-type littermate at E14.5 and at E17.5 is shown. The triangular bars above the photograph of the gel represent increasing numbers of amplification cycles (see Materials and Methods). Expression of Runx2 and Vegf was normalized to Gapdh in each reaction. (C) Expression of Runx2 and Vegf in Ctgf mutants relative to wild-type littermates. Data are from five pairs of Ctgf mutants and wild-type littermates. Expression of Runx2 is not altered in mutants at E14.5 or E17.5. Vegf transcripts are present at indistinguishable levels in Ctgf mutants and wild-type littermates at E14.5, but at reduced levels in mutants at E17.5; the upper and lower bands correspond to the 165 and 120 Vegf isoforms, respectively. * P<0.05. H, hypertrophic zone.
Expression of VEGF in hypertrophic chondrocytes is dependent on Cbfa1/Runx2 (Zelzer et al., 2001). However, no differences in levels of Cbfa1/Runx2 expression were observed in mutant growth plates, suggesting that CTGF acts downstream of Cbfa1/Runx2, or in an independent pathway to induce and/or maintain Vegf mRNA expression in hypertrophic chondrocytes (Fig. 9B,C). In summary, VEGF is a target of CTGF action in hypertrophic cartilage. Reduced expression of this factor can account for the expanded zone of hypertrophy, reduced numbers of MMP9-expressing cells at the growth plate and diminished angiogenesis observed in Ctgf mutants.
DISCUSSION
A large body of evidence links CCN proteins to many diseases, including fibrosis and tumorigenesis (Ayer-Lelievre et al., 2001; Denton and Abraham, 2001; Xu et al., 2000b). However, the roles of CCN proteins in normal developmental processes have received little attention. This study demonstrates that CTGF is important for multiple aspects of chondrogenesis. CTGF regulates chondrocyte proliferation, ECM synthesis and angiogenesis.
CTGF stimulates DNA synthesis in chondrocytes in vitro (Nakanishi et al., 2000), and chondrocyte proliferation is impaired in Ctgf−/− mice (Fig. 4E, Fig. 7A). Interestingly, in spite of high levels of expression in perichondrium at E12.5, no differences in rates of proliferation can be detected at this stage. Differences are first detected at E14.5, when Ctgf expression occurs at the highest levels in prehypertrophic and hypertrophic chondrocytes. This suggests that Ctgf acts in a paracrine manner to control chondrocyte proliferation.
Defects in ECM content in Ctgf mutants confer defective mechanical properties on mutant cartilage. CTGF induces collagen and proteoglycan synthesis in chondrocytes in vitro (Nakanishi et al., 2000). Interestingly, no differences in types II and X collagen expression were seen in mutants. Therefore, although CTGF is a potent inducer of collagen synthesis in chondrocytes in vitro (Nakanishi et al., 2000), it is not required for collagen synthesis in vivo. Compensatory pathways may restore types II and X collagen levels in Ctgf mutants. The related CCN family member Cyr61 (Ccn1) is of particular interest in this regard. Cyr61 is expressed in chondrocytes and induces the synthesis of collagen and other ECM components in vitro (Wong et al., 1997). Therefore, Ctgf and Cyr61 may have overlapping roles in cartilage.
That CTGF is required in vivo for ECM production is demonstrated by the severely reduced levels of aggrecan and link protein in the growth plates of Ctgf mutants (Fig. 6D,E). Parallels between the phenotypes of Ctgf mutants and mice deficient in link protein (Crtl1) highlight the essential role played by CTGF as a regulator of ECM content in cartilage. Link protein is an ECM component, and is essential for the acquisition of tensile strength in cartilage (Morgelin et al., 1994). Crtl1 and Ctgf mutants have similar constellations of defects: shortened mandibles, enlarged vertebrae, and bends and kinks in the same subset of long bones. Finally, chondrocyte columns are disorganized and endochondral ossification is delayed in both strains (Watanabe and Yamada, 1999). However, there are important differences between Ctgf and Crtl1 mutants. Crtl1−/− mice exhibit more severe reductions in proteoglycan levels, and a greater disorganization of the growth plate. Moreover, Ctgf mutants exhibit defects in cell proliferation and enlarged hypertrophic zones not seen in Crtl1 mutant mice. Therefore, some, but not all, of the defects in Ctgf mutant cartilage are caused by decreased proteoglycan content.
Our results show that CTGF is important for efficient recruitment of MMP9-expressing cells to the growth plate (Fig. 8B-D). The crucial role that MMPs play in ECM remodeling in skeletal tissues is illustrated by the skeletal phenotypes of MMP-deficient mice (Vu et al., 1998; Zhou et al., 2000). Recruitment of MMP9-expressing chondroclasts/osteoclasts to the growth plate is dependent on VEGF (Engsig et al., 2000). The paucity of these cells at the growth plates of Ctgf mutants is probably due, at least in part, to the decreased expression of VEGF in Ctgf−/− hypertrophic cartilage.
There are several mechanisms through which the reduced levels of MMP9 seen in Ctgf mutants can lead to growth plate defects. MMP9 degrades collagens and proteoglycans expressed in cartilage and is thus required for ECM remodeling (D’Angelo et al., 2001; Sternlicht and Werb, 2001). Therefore, loss of MMP expression would impair cartilage ECM turnover. This is consistent with the suspected role of CTGF as a key mediator of fibrotic responses, where matrix degradation and synthesis must occur simultaneously (Martin, 1997). In addition, MMPs control angiogenesis, cell migration and differentiation by cleaving cell surface molecules, growth factors and their binding proteins (Sternlicht and Werb, 2001). For example, MMP9 can activate latent TGFβ (D’Angelo et al., 2001; Yu and Stamenkovic, 2000). The reduced levels of MMP9 in growth plates of Ctgf mutants is thus expected to lead to changes in the distribution and activities of growth factors such as TGFβ.
CTGF may control MMP9 expression in several ways. MMP expression can be induced by integrin-mediated interactions. CTGF, by altering ECM composition, may alter integrin-induced MMP9 expression. CTGF can bind directly to integrins to induce MMP transcription (Chen et al., 2001a). CTGF could also affect levels of MMP9 post-translationally by altering its retention and/or degradation. This is especially interesting given that direct associations occur between CTGF and LRP (low density lipoprotein receptor-related protein), and between MMP9 and LRP (Hahn-Dantona et al., 2001; Segarini et al., 2001), raising the possibility that CTGF controls clearance of MMPs by altering their degradation via LRP-mediated endocytosis.
We show that CTGF acts as a cartilage matrix-associated molecule that couples hypertrophy to growth plate angiogenesis and trabecular bone formation (Figs 7-9). CTGF promotes neovascularization through integrin-mediated signaling (Babic et al., 1999), and direct engagement of integrins is therefore one mechanism through which CTGF may act in the growth plate. CTGF can also regulate angiogenesis by modulating MMP expression, as MMPs directly activate integrins on endothelial cells to induce angiogenic responses (Sternlicht and Werb, 2001).
Our results also show that CTGF plays an important role in VEGF localization in hypertrophic chondrocytes (Fig. 9). VEGF is required for growth plate angiogenesis (Gerber et al., 1999; Haigh et al., 2000). The mechanism by which VEGF expression in the growth plate is controlled is not well understood. The transcription factor CBFA1/RUNX2 is required for VEGF expression in hypertrophic cartilage (Zelzer et al., 2001). The observation that CBFA1/RUNX2 levels are not altered in Ctgf mutants suggests that CTGF acts downstream of CBFA1/RUNX2, or in an independent pathway. The transcription factor hypoxia inducible factor 1α(HIF1α) is expressed by hypertrophic chondrocytes and is required, but not sufficient, for VEGF expression (Schipani et al., 2001). HIF1α-independent pathways are also essential, and one of these may involve TGFβ, as HIF1α and SMAD3 form a complex that synergistically induces VEGF expression (Sanchez-Elsner et al., 2001). CTGF may interact with TGFβ to induce VEGF expression, since CTGF binds directly to TGFβ, and enhances the ability of TGFβ to interact with its receptors (Abreu et al., 2002). CTGF may also act independently of TGFβ to induce VEGF expression. For example, CTGF induces adhesive signaling in fibroblasts through integrins, leading to activation of p42/44 MAPKs (Chen et al., 2001a), and the p42/44 MAPK pathway has been shown to be required for VEGF expression in fibroblasts (Milanini et al., 1998). These results raise the possibility that CTGF induces VEGF expression via activation of p42/44. Whether a similar pathway controls VEGF expression in hypertrophic chondrocytes is not yet known.
Secreted proteins controlling VEGF expression in the growth plate have not been previously identified. In endothelial cells, VEGF induces CTGF expression (Suzuma et al., 2000). Taken together with our results, CTGF and VEGF may therefore participate in a positive-feedback loop in hypertrophic chondrocytes. In addition to this transcriptional control, CTGF appears to act post-translationally by binding to VEGF, and impairing VEGF-induced angiogenesis (Inoki et al., 2002). These findings suggest that, in addition to its role as an inducer of VEGF transcription, CTGF plays a role in regulating VEGF activity. CTGF may sequester VEGF in an inactive form in the hypertrophic ECM through direct physical association, and may regulate the release of active VEGF to induce maximal angiogenic activity.
In summary, CTGF is important for chondrocyte proliferation, acquisition of tensile strength by cartilage, ECM remodeling and growth plate angiogenesis. A role for multiple members of the CCN family in angiogenesis and chondrogenesis is likely. For example, both CTGF and Cyr61 promote neovascularization and chondrogenesis in vitro (e.g., Chen et al., 2001a; Kireeva et al., 1997). Mice that lack Cyr61 die in midgestation because of defects in nonsprouting angiogenesis within the placenta (Mo et al., 2002). Thus, Cyr61 and CTGF have similar activities in vitro and are co-expressed, but regulate distinct aspects of angiogenesis in vivo.
The related family member WISP3/CCN6 may also share overlapping functions with CTGF. Although the sites of WISP3 expression and its in vitro activities are unknown, loss of WISP3 in humans causes progressive pseudorheumatoid dysplasia, a disease characterized by degeneration of articular cartilage (Hurvitz et al., 1999). Therefore, multiple members of the CCN family may be required for angiogenesis and the formation and maintenance of cartilage. Analysis of double mutants will provide insights into the roles of these genes in other developmental processes.
Acknowledgments
We thank Lester Lau for sharing unpublished data; and Drs Judith Lengyel, Dan Cohn, Gary Grotendorst, Eddy DeRobertis and Patricia Segarini, and members of the laboratory for discussions and comments on the manuscript. This work was supported by NIH AR44528 (K.M.L.), a seed grant from the UCLA Johnsson Cancer Center (K.M.L.), and the BioSTAR project (K.M.L.), and NIH AR45879 (Fibrogen). B.S.Y. was supported by a USPHS National Research Service Award (GM07185).
References
- Abbondanzo S, Gadi I, Stewart C. Derivation of embryonic stem cell lines. Methods Enzymol. 1993;225:803–823. doi: 10.1016/0076-6879(93)25052-4. [DOI] [PubMed] [Google Scholar]
- Abreu G, Ketpura N, Reversade B, De Robertis E. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β. Nat Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayer-Lelievre C, Brigstock D, Lau L, Pennica D, Perbal B, Yeger H. Report and abstracts on the first international workshop on the CCN family of genes. Mol Pathol. 2001;54:105–120. doi: 10.1136/mp.54.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic AM, Chen C-C, Lau L. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin αvβ3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol. 1999;19:2958–2966. doi: 10.1128/mcb.19.4.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitgood MJ, McMahon AP. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev Biol. 1995;172:126–138. doi: 10.1006/dbio.1995.0010. [DOI] [PubMed] [Google Scholar]
- Blom I, van Dijk A, Wieten L, Duran K, Ito Y, Kleij L, deNichilo M, Rabelink T, Weening J, Aten J, et al. In vitro evidence for differential involvement of CTGF, TGFbeta, and PDGF-BB in mesangial response to injury. Nephrol Dial Transplant. 2001;16:1139–1148. doi: 10.1093/ndt/16.6.1139. [DOI] [PubMed] [Google Scholar]
- Blom I, Goldschmeding R, Leask A. Gene regulation of connective tissue growth factor: new targets for antifibrotic therapy. Matrix Biol. 2002;21:473–482. doi: 10.1016/s0945-053x(02)00055-0. [DOI] [PubMed] [Google Scholar]
- Bork P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993;327:125–130. doi: 10.1016/0014-5793(93)80155-n. [DOI] [PubMed] [Google Scholar]
- Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigstock DR, Steffen CL, Kim GY, Vegunta RK, Diehl JR, Harding PA. Purification and characterization of novel heparin-binding growth factors in uterine secretory fluids. J Biol Chem. 1997;272:20275–20282. doi: 10.1074/jbc.272.32.20275. [DOI] [PubMed] [Google Scholar]
- Carlevaro M, Cermelli S, Cancedda R, Descalzi Cancedda F. Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: auto-paracrine role during endochondral bone formation. J Cell Sci. 2000;113:59–69. doi: 10.1242/jcs.113.1.59. [DOI] [PubMed] [Google Scholar]
- Chen C-C, Chen N, Lau L. The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J Biol Chem. 2001a;276:10443–10452. doi: 10.1074/jbc.M008087200. [DOI] [PubMed] [Google Scholar]
- Chen C-C, Mo F-E, Lau L. The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J Biol Chem. 2001b;276:47329–47337. doi: 10.1074/jbc.M107666200. [DOI] [PubMed] [Google Scholar]
- D’Angelo M, Billings P, Pacifici M, Leboy P, Kirsch T. Authentic matrix vesicles contain active metalloproteinases (MMP) J Biol Chem. 2001;276:11347–11353. doi: 10.1074/jbc.M009725200. [DOI] [PubMed] [Google Scholar]
- Dammeier J, Brauchle M, Falk W, Grotendorst G, Werner S. Connective tissue growth factor: a novel regulator of mucosal repair and fibrosis in inflammatory bowel disease? Int J Biochem Cell Biol. 1998;30:909–922. doi: 10.1016/s1357-2725(98)00046-6. [DOI] [PubMed] [Google Scholar]
- Denton C, Abraham D. Transforming growth factor-beta and connective tissue growth factor: key cytokines in scleroderma pathogenesis. Curr Opin Rheumatol. 2001;13:505–511. doi: 10.1097/00002281-200111000-00010. [DOI] [PubMed] [Google Scholar]
- Engsig M, Chen Q-J, Vu T, Pedersen A-C, Therkidsen B, Lund L, Henriksen K, Lenhard T, Foged N, Werb Z, et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. 2000;151:879–889. doi: 10.1083/jcb.151.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W-H, Karnovsky M. Increased MMP-2 expression in connective tissue growth factor over-expression vascular smooth muscle cells. J Biol Chem. 2002;277:9800–9805. doi: 10.1074/jbc.M111213200. [DOI] [PubMed] [Google Scholar]
- Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol. 1996;107:404–411. doi: 10.1111/1523-1747.ep12363389. [DOI] [PubMed] [Google Scholar]
- Gerber H-P, Vu T, Ryan A, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;55:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- Grotendorst G. Connective tissue growth factor: a mediator of TGF-β action on fibroblasts. Cytokine Growth Factor Rev. 1997;8:171–179. doi: 10.1016/s1359-6101(97)00010-5. [DOI] [PubMed] [Google Scholar]
- Hahn-Dantona E, Ruiz J, Bornstein P, Strickland D. The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9_ by mediating its cellular catabolism. J Biol Chem. 2001;276:15498–15503. doi: 10.1074/jbc.M100121200. [DOI] [PubMed] [Google Scholar]
- Haigh J, Gerber H-P, Ferrara N, Wagner E. Conditional inactivation of VEGF-A in areas of collagen2a1 expression results in embryonic lethality in the heterozygous state. Development. 2000;127:1445–1453. doi: 10.1242/dev.127.7.1445. [DOI] [PubMed] [Google Scholar]
- Hashimoto G, Inoki I, Fujii Y, Aoki T, Ikeda E, Okada Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J Biol Chem. 2002;277:36288–66295. doi: 10.1074/jbc.M201674200. [DOI] [PubMed] [Google Scholar]
- Hogan BLM, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Holmes A, Abraham D, Sa S, Shiwen X, Black C, Leask A. CTGF and SMADs, Maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem. 2001;276:10594–10601. doi: 10.1074/jbc.M010149200. [DOI] [PubMed] [Google Scholar]
- Horner A, Bord S, Kelsall A, Coleman N, Compston J. Tie2 ligands angiopoietin-1 and angiopoietin-2 are coexpressed with vascular endothelial cell growth factor in growing human bone. Bone. 2001;28:65–71. doi: 10.1016/s8756-3282(00)00422-1. [DOI] [PubMed] [Google Scholar]
- Hurvitz J, Suwairi W, Van Hul W, El-Shanti H, Superti-Furga A, Roudier J, Holdermaum D, Pauli R, Herd J, Van Hul E, et al. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nat Genet. 1999;23:94–98. doi: 10.1038/12699. [DOI] [PubMed] [Google Scholar]
- Inoki I, Shiomi T, Hashimoto G, Enomoto H, Nakamura H, Makino K-i, Ikeda E, Takata S, Kobayashi K, Okada Y. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002;16:219–221. doi: 10.1096/fj.01-0332fje. [DOI] [PubMed] [Google Scholar]
- Jedsadayanmata A, Chen C-C, Kireeva ML, Lau LF, Lam S. Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/mouse connective tissue growth factor is mediated through integrin αIIbβ3. J Biol Chem. 1999;274:24321–24327. doi: 10.1074/jbc.274.34.24321. [DOI] [PubMed] [Google Scholar]
- Kireeva ML, Latinkic B, Kolesnikova TV, Chen C-C, Yang G, Abler A, Lau L. Cyr61 and Fisp12 are both ECM-associated signaling molecules: activities, metabolism, and localization during development. Exp Cell Res. 1997;233:63–77. doi: 10.1006/excr.1997.3548. [DOI] [PubMed] [Google Scholar]
- Lasky J, Ortiz L, Tonthat B, Hoyle G, Corti M, Athas G, Lungarella G, Brody A, F M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am J Physiol. 1998;275:L365–L371. doi: 10.1152/ajplung.1998.275.2.L365. [DOI] [PubMed] [Google Scholar]
- Lau L, Lam S. The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res. 1999;248:44–57. doi: 10.1006/excr.1999.4456. [DOI] [PubMed] [Google Scholar]
- Leu S-J, Lam S-T, Lau L. Proangiogenic activities of CYR61 CCN1 mediated through integrins αvβ3 and α6β1 in human umbilical vein endothelial cells. J Biol Chem. 2002;277:46248–46250. doi: 10.1074/jbc.M209288200. [DOI] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon A. Genetic manipulation of hedgehog signaling in the endorchondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Martin P. Wound healing-aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- Milanini J, Viñals F, Pouysségur J, Pagès G. p42/44 MAP kinase module plays a key role in the transcriptional regulation of the vascular endothelial growth factor gene in fibroblasts. J Biol Chem. 1998;273:18165–18172. doi: 10.1074/jbc.273.29.18165. [DOI] [PubMed] [Google Scholar]
- Mo F-E, Muntean A, Chen C-C, Stolz D, Watkins S, Lau L. CYR61 (CCN1) is essential from placental development and vascular integrity. Mol Cell Biol. 2002;22:8709–8720. doi: 10.1128/MCB.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgelin M, Heinegard D, Engel J, Paulsson M. The cartilage proteoglycan aggregate: assembly through combined protein-carbohydrate and protein-protein interactions. Biophys Chem. 1994;50:113–128. doi: 10.1016/0301-4622(94)85024-0. [DOI] [PubMed] [Google Scholar]
- Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T, Takehara K. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. J Cell Physiol. 1999;181:153–159. doi: 10.1002/(SICI)1097-4652(199910)181:1<153::AID-JCP16>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Moussad EE, Brigstock D. Connective Tissue Growth Factor: What’s in a Name? Mol Genet Metab. 2000;71:276–292. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Nishida T, Shimo T, Kobayashi K, Kubo T, Tamatani T, Tezuka K, Takigawa M. Effects of CTGF/Hcs24, a product of a hypertrophic chondrocyte-specific gene, on the proliferation and differentiation of chondrocytes in culture. Endocrinology. 2000;141:264–273. doi: 10.1210/endo.141.1.7267. [DOI] [PubMed] [Google Scholar]
- Nishida T, Nakanishi T, Asano M, Shimo T, Takigawa M. Effects of CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, on the proliferation and differentiation of osteoblastic cells in vitro. J Cell Physiol. 2000;184:197–206. doi: 10.1002/1097-4652(200008)184:2<197::AID-JCP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Perbal B. NOV (nephroblastoma overexpressed) and the CCN family of genes: structural and functional issues. Mol Pathol. 2001;54:57–79. doi: 10.1136/mp.54.2.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Solis R, Davis AC, Bradley A. Gene targeting in embryonic stem cells. In: Wassarman PM, DePamphilis ML, editors. Guide to Techniques in Mouse Development. Vol. 225. San Diego: Academic Press; 1993. pp. 855–878. [DOI] [PubMed] [Google Scholar]
- Reponen P, Sahlberg C, Munaut C, Thesleff I, Tryggvason K. High expression of 92-kD ype IV collagenase (gelatinase B) in teh osteoclast lineage during mouse development. J Cell Biol. 1994;124:1091–1102. doi: 10.1083/jcb.124.6.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Elsner T, Botella L, Velasco B, Corbi A, Attisano L, Bernabéu C. Synergistic cooperation between hypoxia and transforming growth factor-β pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- Schipani E, Ryan E, Didrickson S, Kobayashi K, Knight M, Johnson R. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segarini P, Nesbitt J, Li D, Hayes L, Yates JI, Carmichael D. The low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor is a receptor for connective tissue growth factor (CTGF) J Biol Chem. 2001;276:40659–40667. doi: 10.1074/jbc.M105180200. [DOI] [PubMed] [Google Scholar]
- Shimo T, Nakanishi T, Nishida T, Asano M, Kanyama M, Kuboki T, Tamatani T, Tezuka K, Takemura M, Matsumma T, et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem. 1999;126:137–145. doi: 10.1093/oxfordjournals.jbchem.a022414. [DOI] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon A. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht M, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton R, Shiwen X, Martini G, Holmes A, Leask A, Haberberger T, Martin G, Black C, Abraham D. Iloprost suppresses connective tissue growth factor production in fibroblasts and in the skin of scleroderma patients. J Clin Invest. 2001;108:241–250. doi: 10.1172/JCI12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuma K, Naruse K, Suzuma I, Takahara N, Ueki K, Aiello L, King G. Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, Flt1, and phosphatidylinositol 3-kinase-Akt-dependent pathways in retinal vascular cells. J Biol Chem. 2000;275:40725–40731. doi: 10.1074/jbc.M006509200. [DOI] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/Gelatinase B is a key Regulator of Growth Plate Angiogenesis and Apoptosis of Hypertrophic Chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Yamada Y. Mice lacking link protein develop dwarfism and craniofacial abnormalities. Nat Genet. 1999;21:225–229. doi: 10.1038/6016. [DOI] [PubMed] [Google Scholar]
- Wong M, Kireeva ML, Kolesnikova TV, Lau LF. Cyr61, product of a growth factor-inducible imediate-early gene regulates chondrogenesis in mouse limb bud mesenchymal cells. Dev Biol. 1997;192:492–508. doi: 10.1006/dbio.1997.8766. [DOI] [PubMed] [Google Scholar]
- Xu J, Smock S, Safadi FF, Rosenzweig AB, Odgen PR, Marks SC, Jr, Owen TA, Popoff SN. Cloning the full length cDNA for rat connective tissue growth factor: implications for skeletal development. J Cell Biochem. 2000a;77:103–115. doi: 10.1002/(sici)1097-4644(20000401)77:1<103::aid-jcb11>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Xu L, Corcoran R, Welsh J, Pennica D, Levine A. WISP-1 is a Wnt-1- and β-catenin-responsive oncogene. Genes Dev. 2000b;14:585–595. [PMC free article] [PubMed] [Google Scholar]
- Yi SE, Daluiski A, Pederson R, Rosen V, Lyons KM. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development. 2000;127:621–630. doi: 10.1242/dev.127.3.621. [DOI] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Zelzer E, Glotzer D, Hartmann C, Thomas D, Fukai N, Soker S, Olsen B. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev. 2001;106:97–106. doi: 10.1016/s0925-4773(01)00428-2. [DOI] [PubMed] [Google Scholar]
- Zelzer E, McLean W, Ng Y-S, Fukai N, Reginato A, Lovejoy S, D’Amore P, Olsen B. Skeletal defects in VEGF120/120 mice reveal multiple roles for VEGF in skeletogenesis. Development. 2002;129:1893–1904. doi: 10.1242/dev.129.8.1893. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Apte S, Soininen R, Cao R, Baaklini G, Rauser R, Wang J, Cao Y, Tryggvason K. Impaired endochondral ossificiation and angiogenesis in mice deficient in membrane-type matrix metalloporteinase I. Proc Natl Acad Sci USA. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]