

Abstract
With recent approval of the first dendritic cell (DC) vaccine for patient use, many other DC vaccine approaches are now being tested in clinical trials. Many of these DC vaccines employ tumor cell lysates (TLs) generated from cells cultured in atmospheric oxygen (~20% O2) that greatly exceeds levels found in tumors in situ. In this study, we tested the hypothesis that TLs generated from tumor cells cultured under physiological oxygen (~5% O2) would be more effective as a source for DC antigens. Gene expression patterns in primary glioma cultures established at 5% O2 more closely paralleled patient tumors in situ and known immunogenic antigens were more highly expressed. DCs treated with TLs generated from primary tumor cells maintained in 5% O2 took up and presented antigens to CD8 T cells more efficiently. Moreover, CD8 T cells primed in this manner exhibited superior tumoricidal activity against target cells cultured in either atmospheric 20% O2 or physiologic 5% O2. Together, these results establish a simple method to greatly improve the effectiveness of DC vaccines in stimulating the production of tumoricidal T cells, with broad implications for many of the DC-based cancer vaccines being developed for clinical application.
Keywords: Hypoxia, Dendritic Cells, Vaccine, Glioma, Immunotherapy
Introduction
Therapeutic vaccination utilizing dendritic cells (DCs) pulsed with tumor-associated antigens is a promising approach for cancer immunotherapy (1). The United States Food and Drug Administration recently approved a peptide-pulsed DC vaccine for the treatment of prostate cancers resistant to hormone ablation (2, 3). Tumor cells are frequently used as a source of antigen for DC-based vaccines. There are 385 clinical trials currently registered that utilize tumor cells as the source of vaccine antigen (4). Vaccines using tumor cells and tumor cell lysates (TLs) have several advantages including targeting multiple, patient-specific tumor antigens. Vaccination with TL-pulsed DCs has demonstrated encouraging results in early stage clinical trials in numerous malignancies, but there is clearly a need for additional improvement (5).
It remains unclear how tissue culture might affect anti-tumor immune responses evoked by tumor cell vaccines. Primary human glioma cells cultured in serum-containing media were genetically and phenotypically different from the primary tumor; culture of the same cells in serum-free conditions more closely reflected the primary tumor and enriched for a tumor stem cell phenotype (6). Moreover, in a murine model, glioma cell lysates generated in serum-free conditions were more effective than those derived from serum-containing media when employed for TL-pulsed DC vaccines (7). Traditionally, tumor cell vaccines are derived from cultures maintained at atmospheric oxygen (~ 20.95%; hereafter 20% O2), far from the average oxygen tension of less than 6% O2 (range 0.1–10%) measured in glioblastoma in situ (8). Lowering oxygen causes alterations gene expression, cell metabolism, proliferation, survival (9–12), and increases the expression of tumor stem cell markers CD133, Nestin, and SOX2 (13–15). We previously demonstrated that glioma and breast carcinoma TL vaccines derived at 5% O2 had superior therapeutic efficacy relative to 20% O2 in murine models. However, this previous study did not address DC vaccines and it remained uncertain if the results would translate to stimulating human leukocytes using primary human tumors, which are often heterogeneous and unpredictable. We therefore investigated the mechanisms by which oxygen may change the immunogenicity of serum-free glioma cultures derived from glioblastoma patient surgical resections.
Material and Methods
Glioma cell culture and lysates
Surgically resected glioblastomas (supplementary table S1) were enzymatically dissociated and cultured in neural stem cell media for all experiments (16); tumors were not cultured in serum. Unless noted, all tumors were initially grown in 20% O2 then subsequently moved to 5% O2 for a minimum of 30 days prior to experiments as described previously (17). 5% O2 was maintained by regulated nitrogen injection into a Thermo Scientific Forma Series II incubator. Cultures were maintained in the indicated oxygen tension uninterrupted for at least 48 h prior to use. To generate TLs, cells were pelleted, washed with PBS, then killed by five freeze thaw cycles and complete lysis was confirmed by trypan blue exclusion.
Microarray
Total RNA was isolated from snap frozen tissue at resection, or cultured cells from the primary tumor at grown at 5% or 20% O2. Expression levels of 18,401 genes were analyzed using a Whole-Genome Gene Expression DASL Assay (Illumina, San Diego, CA). For detailed methods, see supplemental material.
Quantitative RT-PCR
Extracted RNA was analyzed by real time PCR using SYBR Green one step PCR master mix (Qiagen). The following conditions were used for PCR in an ABI PRISM 7500 thermocycler: 95°C for 15 min; 94°C for 30s, 55°C for 30s, 68°C for 30s in a total of 40 cycles; 72°C for 10 min; and 10°C until collected. Relative quantification of gene expression was calculated and normalized to GAPDH expression levels using the 2−ΔΔCt method (18). Primers are listed in supplementary table S2.
Flow cytometry
5 × 105 cells were stained with: anti-CD133/2 (clone 293C3) (Miltenyi), EphA2 (clone 371805), SOX2 (clone 245610), Nestin (clone 196909), HER-2/neu (clone 191924) (R&D Systems), and IL13Rα2 (clone B-D13) (SantaCruz Biotechnology). Following three washes, CD133/2 was stained with a secondary anti-mouse-647; IL13Rα2 and EphA2 were stained with secondary anti-mouse-PE along with their respective isotype control and analyzed using a FACS Canto II. For DC phenotyping, 5 × 105 DCs were stained with CD80-FITC (clone L307.4), CD86-PE (clone IT2.2), CD83-PE (clone HB15E), HLA-DR-APC (clone G46-6), and HLA-ABC-FITC (clone W6132) (BD Biosciences), incubated at 4°C for 30 min, washed, fixed, then analyzed on a FACS Caliber. Intracellular staining was performed according to the manufacturer’s protocol (BD Biosciences) using 2 mM GolgiStop for 4 h prior to staining.
TL-pulsed DCs and DC uptake assay
Monocytes were purified using CD14 magnetic beads (Miltenyi Biotech) from the peripheral blood of healthy HLA-A2+ donors (supplementary table S3), plated at a concentration of 5 × 105 in 24-well plates and matured as previously described (19). On day 6, DCs were pulsed with whole TLs derived in either 5% or 20% O2 containing 100 µg of protein, and maturated prior to experiments. To assess TL uptake, tumor cells were labeled with 10 mM Carboxyfluorescein succinimidyl ester (CFSE), lysed, and pulsed onto iDC harvested at day 6 of culture. 24 h later, DCs were analyzed by flow cytometry.
Cytotoxic T lymphocyte (CTL) assays
1 × 106 HLA-A2+ PBMCs from normal donors were added to lysate-pulsed DCs with 50 U IL-2 and incubated seven days. For re-stimulation, a second set of pulsed DCs was added to the co-culture for four days. On day 11, peripheral blood mononuclear cells (PBMCs) were co-cultured with 2 × 104 CFSE-labeled HLA-A2+ glioma cells cultured in 20% O2 for 6 h, then analyzed by flow cytometry to determine cytotoxicity as described (20). For the blocking assay, 1.25 µg anti-HLA-ABC (clone G46-2.6) was added to target cells for 25 min before washing and adding PBMCs. A competition CTL assay was also conducted whereby target cells were pre-cultured in two oxygen levels and pooled as follows: 1.5 × 104 glioma cells cultured in 5% O2 were stained with 10 mM/mL of CellTrace™ Violet (Invitrogen) and added to 1.5 × 104 glioma cells cultured in 20% O2 stained with 10 mM/mL CFSE. Pooled target cells were co-cultured with PBMCs in 20% O2 for 6 h, then analyzed by flow cytometry whereby the targets were distinguished via Violate or CFSE.
CMV assay
5 × 105 HLA-A2+ iDCs were pulsed with whole TLs derived in 5% or 20% O2 TLs containing 100 µg of protein with or without addition of 10 µg of pp65495–503 (NLVPMVATV) and matured as described above. Following maturation, DCs were washed 3 times, and 5 × 105 PBMCs from CMV sera-positive donors were added to DCs. CMV sera-negative PBMCs were used as a control. Cells were incubated for 48 h, then supernatant was analyzed by cytometric bead array (BD Biosciences) for IFNγ. Cells were cultured for an additional 24 h, stained with anti-CD8, pp65 pentamer (ProImmune), and then stained intracellularly for IFNγ.
Statistical analysis
Statistical comparisons were made by ANOVA, followed by post hoc comparisons using a 2-tailed t-test. All tests were performed with Prism 4 software (Graph Pad Software, Inc). P values <0.05 were considered significant.
See supplemental material for additional materials and methods.
Results and Discussion
To compare tissue cultures to the primary tumor in situ, mRNA expression levels were profiled in replicate from two glioblastomas and cultured cells derived from the same tumor grown in 5% and 20% O2. There was a significant difference in the expression of 3,333 genes between the 20% O2 culture and the in situ tumor in both patients (Supplementary Fig. S1). Of these, 77 genes were differentially expressed between the 5% and 20% O2 cultures (Fig. 1), trending towards expression levels observed in situ in 5% O2 cultures. Differential expression of CD133 (PROMININ-1) that was detected by microarray was further validated by PCR, flow cytometry and western blot (Fig 2A–C). It is uncertain that enrichment in CD133 expression reflected selective growth of CD133+ cells because studies have shown that reducing oxygen rapidly induces CD133 expression on CD133− cells (21). We also investigated the expression of genes of interest that were not statistically different on the microarray. A trend towards increased expression of stem cell markers Sox2 and nestin was apparent in 5% O2 but this failed to reach statistical significance (Fig 2B; p>0.05). Importantly for immunotherapy, 5% O2 increased expression of EphA2 and IL13Rα2, both of which are overexpressed in a high percentage of glioblastomas and appear viable as immunotherapeutic targets (Fig 2A–C). Malignant glioma patients vaccinated with peptides derived from EphA2 and IL13Rα2 exhibited clinical responses (22) and spontaneously occurring EphA2-reactive CD8 T cells have been documented in long-term survivors (23). Collectively, these experiments demonstrated that cells cultured in 5% O2 better reflect gene expression on the tumor in situ, are enriched for markers of “stemness” and higher expression of glioma-associated antigens.
Figure 1. Tissue culture gene expression compared with tumor in situ.

Heat maps showing log base 2-transformed data for each experiment with a p-value < 0.001 and a fold change >3 for the comparison of 5% versus 20% O2.
Figure 2. Primary glioma cell lines cultured in 5% oxygen express higher levels of glioma-associated antigens and makers of stemness.
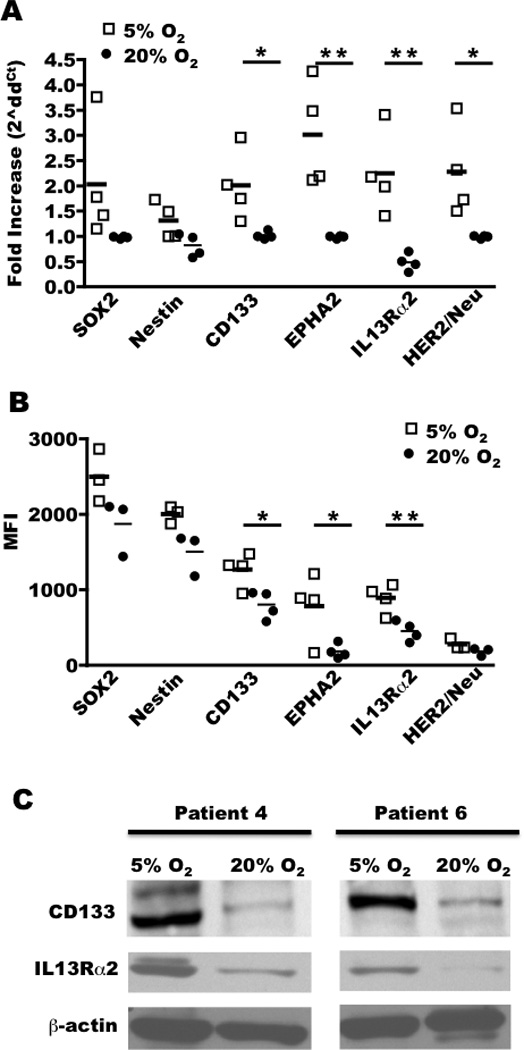
A, cells from four gliomas (patients 3, 4, 6, and 7) were cultured in 5% or 20% O2 and analyzed for mRNA expression by real time PCR (n=4/group) and B, protein by flow cytometry where mean fluorescent intensity (MFI) is shown (n=3–4/group) or C, by western blot. * P < 0.05; ** P < 0.01
We next asked if oxygen would alter the ability of TLs to prime tumoricidal T cells. HLA-A2+ PBMCs from normal donors were primed by autologous DCs pulsed with TLs derived from three different glioma patients. Primed PBMCs were assessed for their ability to lyse the same HLA-A2+ glioma cells that were used for priming. PBMCs primed by 5% O2 TLs demonstrated superior tumoricidal activity against glioma target cells cultured in standard 20% O2 in 3/3 patients (Fig. 3A). Blockade of MHC I-TCR interactions by pre-treatment of target cells with an anti-MHC I antibody prevented specific lysis, implicating CTLs as the main effectors in this assay (Fig. 3B).
Figure 3. Tumor Lysates from 5% oxygen prime CTLs with superior tumoricidal function.
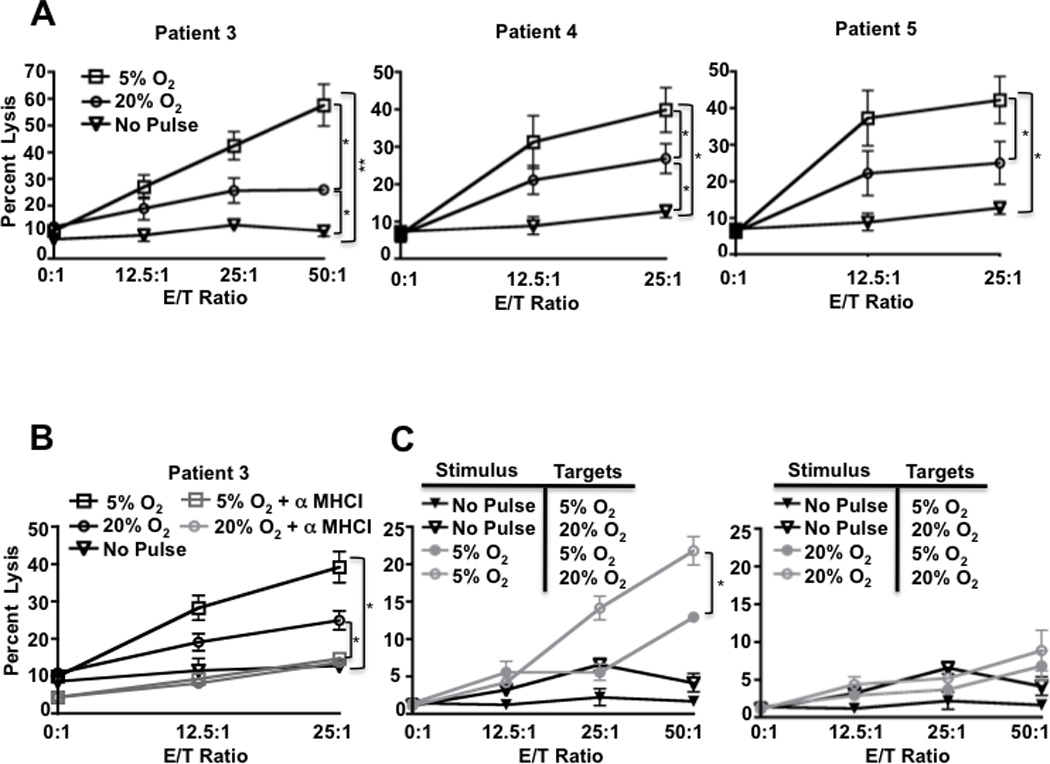
A, PBMCs were stimulated with DCs pulsed with TL from 5% or 20% O2 and incubated with HLA-A2 matched target glioblastoma cells (Patients 3 and 4) or an ependymoma (Patient 5). B, to determine if the increased tumoricidal response was MHC-I-dependent, pan-MHC I blocking antibody was added to target cells prior combining with PBMC. C, DCs pulsed with TL from 5% or 20% O2 from patient 3 and incubated with patient 3 target cells cultured in 20% or 5% O2. All experiments were repeated at least twice with similar results. Error bars, ± SEM. * P < 0.05; ** P < 0.01.
A plausible explanation for the enhancement in CTL priming would be improvement of DC maturation by 5% O2 TLs. The expression of co-stimulatory and MHC molecules on DCs was measured after pulsing with TLs from 20% or 5% O2. There were no appreciable differences in expression of CD83, CD80, CD86, HLA-ABC, or HLA-DR (Supplementary Fig. S2). The levels of DC-elaborated IL-6, IL-8, IL-10, and IL-12p70 were measured 48 hours after lysate pulsing, revealing no significant differences between 5% and 20% O2 lysates (Supplementary Fig. S3). Therefore, the adjuvant activity of 5% O2 TLs was not likely due to enhancing DC maturation or cytokine expression, suggesting other mechanisms.
We also interrogated the effect of oxygen on the target cells by establishing a competitive cytotoxicity assay whereby CTLs were co-cultured with targets pre-cultured in 5% and 20% O2. Target cells cultured in 5% O2 were significantly more resistant to CTL killing than targets grown in 20% O2, only showing susceptibility to CTLs primed with TLs from 5% O2 at high effector:target ratios (Fig 3C). We reproducibly observed suppression of the ability of CTLs primed by 20% O2 TLs to kill any target in the presence of targets pre-cultured in 5% O2 (compare Fig 3A to 3C). We speculate that the target cells pre-cultured in 5% O2 may have expressed factors that inhibited CTL killing, which is supported by increased TGFβ1/latentancy-associated peptide (LAP) complex cell surface expression on a subset of target cells cultured in 5% O2 (Supplementary Fig S4). The differences measured in target cell susceptibility to CTL killing were not due to differential labeling efficiency (supplementary Fig S5). Despite the ability of target cells grown in 5% O2 to directly suppress CTL killing, when these cells were lysed and processed in DCs (wherein suppressive factors could be degraded or overridden), they reproducibly primed CTLs with enhanced ability to kill target cells at conventional and physiologic oxygen. Accordingly, CTLs primed by TLs from 5% O2 would be expected to have more tumoricidal function in vivo where oxygen is limited.
Experiments were conducted to determine whether the superior CTL priming achieved with 5% O2 TLs could be due to changes in lysate uptake. Immature DCs were pulsed with CFSE-labeled glioma TLs from cells grown in 5% or 20% O2. Flow cytometry-based detection of CFSE+HLA-DR+ cells (marking DCs loaded with lysate) revealed that 5% O2 TLs were uptaken by a significantly greater percentage of DCs compared to TLs derived in 20% O2 (Fig. 4A). Thus, as of yet unidentified factor(s) in the 5% O2 TLs increased the fraction of DCs that engulfed TLs in tissue culture.
Figure 4. Tumor lysates from 5% oxygen cultures increase CD8 T cell priming.
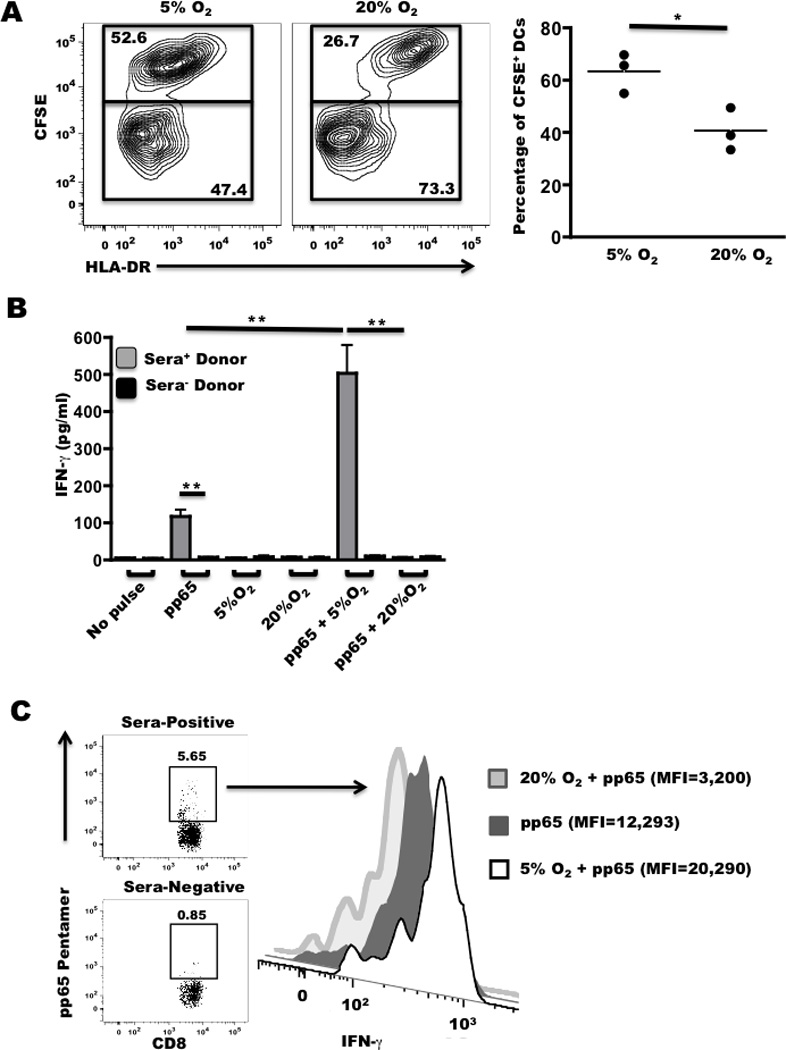
A, Dendritic cells were pulsed with CFSE-labeled TLs isolated from three patients derived in 5% or 20% O2, stained with a-HLA-DR and analyzed by flow cytometry. PBMCs were co-cultured with DCs pulsed with 5% or 20% O2 TLs +/− pp65 peptide for, B, 48 h and analyzed for secreted IFNγ or C, 72 h to determine IFNγ production on a per-cell basis in CD8+pp65 pentamer+ cells as indicated by gating strategy shown in sera-positive donor. All experiments were repeated at least twice with similar results. Error bars, ± SEM. * P < 0.05; ** P < 0.01.
We then tested the hypothesis that 5% O2 TLs should exhibit intrinsic adjuvant properties independent of glioma antigen expression by mixing the lysates with pp65 derived from Cytomegalovirus (CMV). CMV-derived pp65495–503 is an HLA-A2-restricted immunodominant epitope to which CMV sera-positive patients typically have CD8 T cell memory responses (reviewed in (24)). PBMCs from sera-positive donors were co-cultured with TL or TL/ pp65495–503 loaded DCs. PBMCs from sera-negative donors were used to control for CD8 T cell activation independent of pp65495–503. Soluble IFNγ in the tissue culture supernatant was quantified as a measure of CD8 T cell activation (Fig. 4B). There was no significant difference in IFNγ elaborated between the non-pulsed and TL-pulsed PBMCs from sera-positive donors, demonstrating negligible reactivity to possible CMV antigens in the TLs themselves (25). As expected, PBMCs from sera-positive donors primed with pp65495–503 alone (no TLs) elaborated IFNγ at levels ten fold above background, whereas sera-negative donor PBMCs did not respond to pp65495–503. PBMCs primed with pp65495–503 mixed with 5% O2 TLs elaborated five-fold the amount of IFNγ compared to pp65495–503 alone. In marked contrast, 20% O2 TLs significantly suppressed pp65-dependent IFNγ secretion (Fig. 4B). To confirm that pp65-reactive CD8 T cells were the main source of IFNγ, flow cytometry was conducted to assess IFNγ expression specifically in CD8+pp65-pentamer+ cells (Fig. 4C). Consistent with measured soluble IFNγ, CD8+pp65-pentamer+ cells primed by 5% O2 TLs plus pp65495–503 produced more IFNγ on a per-cell basis relative to all other groups. Taken together, these data demonstrate that 5% O2 TLs have intrinsic adjuvant activity that is independent of the amount of glioma antigen expressed. It is noteworthy that these findings paralleled what we recently reported using established murine glioma cell lines; specifically that 5% O2 TLs increased the presentation of exogenous ovalbumin on MHC I and enhanced CD8 T cell activation, whereas 20% O2 TLs were suppressive to CD8 T cell priming and alternatively promoted antibody responses (17).
In summary, our data demonstrate that tissue culture oxygen functions as a master “immunologic switch” by simultaneously regulating the expression of glioma antigens and factors that modulate DC-mediated priming of CD8 T cells. Growing primary glioma cells in 1–7% O2 increased cancer stem cell markers in several previous studies (13–15) and this study, suggesting that the T cells primed might be better suited to eradicate cancer stem cells in situ. By selecting for expression of glioma antigens that are more abundant on the tumor in situ, we propose that oxygen can be exploited to prime CD8 T cells with greater specificity to antigens that are actually presented MHC molecules in situ.
Most of the experiments conducted were done by reverting cultures initially grown in 20% O2 to 5% O2, suggesting that the immunogenicity of standard tumor cell vaccines could be “rescued” by conversion to 5% O2. However, the enhancement in immunogenicity afforded by low oxygen was not an artifact of conversion from 20% O2 to 5% O2 because cells cultured directly from surgical resections in 5% O2 exhibited: i) increased expression of CD133, IL-13Rα2, and Epha2 (supplementary Fig. S6), ii) enhanced uptake by DCs (Fig. 4A), and intrinsic adjuvant activity as shown by increasing pp65495–503 cross priming (Fig. 4B–C). The same trends in Fig 4 were also observed by reverting cultures initially grown in 20% O2 to 5% O2 (data not shown). Thus, the changes in immunogenicity are reversible and are present regardless of the initial oxygen tension used to establish the cell culture. Tumor cells grown in physiologic oxygen are inherently more capable of priming CD8 T cells by a mechanism that appears independent of increasing MHC I/II, CD80/83/86, IL-6, IL-8, IL-10, or IL12p70 expression in DCs. A deeper investigation into the mechanisms by which oxygen regulates tumor cell immunogenicity in the context of T cell priming needs to be undertaken to establish a molecular basis for our results. We propose that vaccines made by expansion of primary tumor cells in physiologic oxygen will serve as a powerful system to induce clinically useful anti-tumor immune responses.
Supplementary Material
Acknowledgments
This work was supported by grants from the American Cancer Society RSG-09-189-01-LIB (JRO), State of Minnesota, Minnesota Partnership for Biotechnology and Medical Genomics (JRO), Randy Shaver Cancer Research and Community Fund (JRO), Children’s Cancer Research fund (JRO), NIH MSTP grant T32 GM008244 (BMA), and NIH R01 CA72669 (BRB).
Footnotes
Author Contributions: M.R.O., B.M.A, A.J.L., A.S., B.R.B., and J.R.O designed research; M.R.O., B.M.A, A.J.L., P.T.G, A.S., P.T.R., X.L., I.F.P., W.C., and M.A.H. performed research and analyzed data; M.R.O, B.M.A, A.S., B.R.B., and J.R.O edited the manuscript.
References
- 1.Ridgway D. The first 1000 dendritic cell vaccinees. Cancer Invest. 2003;21:873–886. doi: 10.1081/cnv-120025091. [DOI] [PubMed] [Google Scholar]
- 2.DeFrancesco L. Landmark approval for Dendreon's cancer vaccine. Nat Biotechnol. 28:531–532. doi: 10.1038/nbt0610-531. [DOI] [PubMed] [Google Scholar]
- 3.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 4.Apetoh L, Ghiringhelli F, Tesniere A, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 5.Le DT, Pardoll DM, Jaffee EM. Cellular vaccine approaches. Cancer J. 16:304–310. doi: 10.1097/PPO.0b013e3181eb33d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 7.Pellegatta S, Poliani PL, Corno D, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer research. 2006;66:10247–10252. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]
- 8.Evans SM, Judy KD, Dunphy I, et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res. 2004;10:8177–8184. doi: 10.1158/1078-0432.CCR-04-1081. [DOI] [PubMed] [Google Scholar]
- 9.van den Brenk HA, Moore V, Sharpington C, Orton C. Production of metastases by a primary tumour irradiated under aerobic and anaerobic conditions in vivo. Br J Cancer. 1972;26:402–412. doi: 10.1038/bjc.1972.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sciandra JJ, Subjeck JR, Hughes CS. Induction of glucose-regulated proteins during anaerobic exposure and of heat-shock proteins after reoxygenation. Proc Natl Acad Sci U S A. 1984;81:4843–4847. doi: 10.1073/pnas.81.15.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 12.Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71–77. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platet N, Liu SY, Atifi ME, et al. Influence of oxygen tension on CD133 phenotype in human glioma cell cultures. Cancer Lett. 2007;258:286–290. doi: 10.1016/j.canlet.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 14.McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ. Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Mol Cancer Res. 2009;7:489–497. doi: 10.1158/1541-7786.MCR-08-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu A, Oh S, Wiesner SM, et al. Persistence of CD133+ cells in human and mouse glioma cell lines: detailed characterization of GL261 glioma cells with cancer stem cell-like properties. Stem Cells Dev. 2008;17:173–184. doi: 10.1089/scd.2007.0133. [DOI] [PubMed] [Google Scholar]
- 17.Olin MR, Andersen BM, Zellmer DM, et al. Superior efficacy of tumor cell vaccines grown in physiologic oxygen. Clin Cancer Res. 2010;16:4800–4808. doi: 10.1158/1078-0432.CCR-10-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Mailliard RB, Wankowicz-Kalinska A, Cai Q, et al. alpha-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004;64:5934–5937. doi: 10.1158/0008-5472.CAN-04-1261. [DOI] [PubMed] [Google Scholar]
- 20.Olin MR, Hwa Choi K, Lee J, Molitor TW. Gammadelta T-lymphocyte cytotoxic activity against Mycobacterium bovis analyzed by flow cytometry. J Immunol Methods. 2005;297:1–11. doi: 10.1016/j.jim.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Griguer CE, Oliva CR, Gobin E, et al. CD133 is a marker of bioenergetic stress in human glioma. PLoS One. 2008;3:e3655. doi: 10.1371/journal.pone.0003655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 29:330–336. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueda R, Low KL, Zhu X, et al. Spontaneous immune responses against glioma-associated antigens in a long term survivor with malignant glioma. J Transl Med. 2007;5:68. doi: 10.1186/1479-5876-5-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moss P, Khan N. CD8(+) T-cell immunity to cytomegalovirus. Hum Immunol. 2004;65:456–464. doi: 10.1016/j.humimm.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 25.Cobbs CS, Harkins L, Samanta M, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002;62:3347–3350. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.