

Abstract
The single-subunit RNA polymerases are a widespread family of proteins found in phage, mitochondria, and chloroplasts. Unlike the phage RNAPs, the eukaryotic RNAPs require accessory factors to melt their promoters and diverge from the phage RNAPs in the regions where functions associated with promoter melting in the latter have been mapped, suggesting that promoter melting mechanisms in the eukaryotic RNAPs diverge from those in the phage enzymes. However, here we show that an element in the yeast mitochondrial RNAP, identified by sequence alignment with the T7 phage RNAP, fulfills a similar role in promoter melting as does the T7 RNAP ‘intercalating hairpin’. The yeast mitochondrial RNAP intercalating hairpin appears to be as important in promoter melting as is the mitochondrial transcription factor, MTF1, and both a structurally integral hairpin and MTF1 are required to achieve high levels of transcription on a duplex promoter. Deletions in the hairpin also relieve MTF1 inhibition of promoter escape on pre-melted promoters, likely because such deletions disrupt interactions with the upstream edge of the transcription bubble. These results are consistent with recent structural and functional studies of human mitochondrial RNAP and further reveal the surprising extent of mechanistic conservation between the eukaryotic and phage-encoded members of the single-subunit RNAP family.
Two large families of RNAPs carry out mRNA, tRNA, and ribosomal RNA synthesis in all cells. One is the family of multi-subunit RNAPs that includes the eukaryotic pol I, II, and III enzymes, as well as the multi-subunit RNAPs of the eubacteria and archaea(1). The other is the family of RNAPs encoded by many bacteriophage, and which also have homologs in mitochondria and chloroplasts and are both nuclear and plastid encoded(2). The latter are usually designated as the single-subunit RNAPs, but while it is correct that these RNAPs almost invariably function as a single subunit during RNA chain elongation, the mitochondrial and chloroplast RNAPs require an additional factor(s) during transcription initiation and these factors have been shown to be required for promoter melting(3, 4). The well-studied phage T7 RNAP, and its characterized phage RNAP homologs, melt their promoters without accessory factors by using at least two mechanisms: (1) the introduction of a sharp bend in the promoter, centered on the region that is opened(5–7), and (2) the intercalation of a β-hairpin between the template (T) and non-template (NT) strands at the upstream edge of the melted region in the initiation complex(8–10).
This intercalating hairpin occurs in a region which, at the sequence level, is very poorly conserved in the single subunit RNAP family(2) and, combined with the differences in factor requirements for promoter melting by the phage vs. eukaryotic RNAPs, this would suggest that the phage and eukaryotic enzymes use distinct mechanisms for promoter melting. However, to test this we created deletions in two regions of the yeast mitochondrial (Mt) RNAP suggested by ambiguous sequence alignments as potential locations of an element that could be functionally and structurally homologous to the phage RNAP intercalating hairpin. We find that deletions in one of these regions generates RNAPs that are transcriptionally active and capable of promoter specific binding, but cannot melt a duplex promoter. A region from human MtRNAP which aligns with this putative intercalating hairpin in yeast MtRNAP was recently shown to also be important in allowing initiation from duplex, but not pre-melted templates(11), suggesting that this promoter melting mechanism is utilized by most, if not all, members of the single-subunit RNAP family.
Experimental Procedures
Mutant RNAP genes were prepared with the Stratagene Quick-Change directed mutagenesis kit using vectors described previously(12). Yeast MtRNAP and MTF1 were expressed in E. coli BL21(DE3) and purified as described previously(12), except that bacterial cultures, following IPTG induction, were transferred to 16 °C for overnight protein expression because we found that low-temperature expression increased the yield of soluble MtRNAP and MTF1.
-
1
Transcription reactions (25 μl volume) were carried out at room temperature for 15 minutes in 4 mM MgCl2, 20 mM Tris-Cl pH 8.0, 5 mM DTT, and 50 mM NaCl with synthetic promoter templates at 2 μM, RNAPs and MTF1 (when present) at 1 μM, NTPs at 0.5 mM, and with 0.1 μCi/μl of 3000 Ci/mM α-32P ATP to label the transcripts. Transcription reactions were resolved by denaturing page and imaged/quantified on a Molecular Dynamics Storm Phosphorimager as described(12). The following synthetic promoter templates were used in these reactions:
-
Duplex promoter non-template (NT) strand (underlined base corresponds to +1; annealed to its complement):
AATTCATTTATTTATTATTATATAAGTAATAAAGAATAGTTTTATATACTAATAATAATATAG
-
Bubble promoter NT strand (annealed to same T strand as the duplex promoter to create a −4 to +2 mismatch in the bolded region):
AATTCATTTATTTATTATTATATGCAGCTTAAAGAATAGTTTTATATACTAATAATAATATAG
-
Het1 to Het5 promoters were created by annealing a common 33 base T strand to the following 33 base NT strands to generated promoters with mismatches in the −4 to +1 region ranging from 1 to 5 base-pairs in size (underlined base=+1, bolded bases=mismatches):
T-strand: CGCGTAAAACTATTCTTTATTACTTATATCGCG
Het1 NT strand: CGCGATATGAGTAATAAAGAATAGTTTTACGCG
Het2 NT strand: CGCGATATGCTAATAAAGAATAGTTTTACGCG
Het3 NT strand: CGCGATATGCATAATAAAGAATAGTTTTACGCG
Het4 NT strand: CGCGATATGCAGAATAAAGAATAGTTTTACGCG
Het5 NT strand: CGCGATATGCAGCATAAAGAATAGTTTTACGCG
Het1D NT strand: CGCGATATAAGTACTAAAGAATAGTTTTACGCG
Het2D NT strand: CGCGATATAAGTGCTAAAGAATAGTTTTACGCG
-
Other promoters used in this study were generated by annealing different NT strands, as described in the text, to the T strand shown above.
MTF1 binding experiments
1 μM MtRNAPs were mixed with equimolar MTF1 in 50 mM Tris-Cl pH 8.0, 1 mM EDTA, 1mM DTT and 500 mM NaCl in 50 μl volumes, and then spun through an Amicon minicon ultrafiltration device with a 100 kD MW cutoff filter in a minifuge at 8000 rpm for 10 minutes. Following this the retenates and filtrates were recovered and enough buffer was added to the retenate to bring its volume to 50 μl and both retenates and filtrates were analyzed by denaturing PAGE.
Permanganate reactivity experiments were carried out as described(9) with the 33 bp duplex promoter template labeled with γ32P-ATP at either the 5′-end of the T or NT strand, and at the same reagent concentrations and buffer conditions used for the transcription reactions but with limiting NTP mixes as specified in individual figure legends.
Electrophoretic mobility shift analysis (EMSA) experiments were carried out with promoters prepared with a 33 base T-strand identical to that used to prepare the Het1-Het5 promoters, but labeled with fluoresceine at its 5′-end and with either a fully complementary 33 base NT strand (‘Duplex’ promoter) or an NT strand in which the −4 to +2 region was changed to GCAGCT to create a 6 base-pair mismatch (‘Bubble’ or ‘Pre-Melted’ promoter). Reactions contained constant amounts of MtRNAP with or without MTF1 and were incubated with varying concentrations of promoter DNA, with promoter always in excess of MtRNAP (specific concentrations are indicated in individual figure legends), in 20 mM Tris-Cl pH 8.0, 75 mM NaCl, 5 mM DTT, 1 mM EDTA, 10% glycerol, and .001% w/v bromophenol blue. After a 30 min. incubation at room temperature, 10 μl reaction aliquots were resolved on 4–15% Native PAGE gels run in 1X TAE buffer and visualized on a Molecular Dynamic Strom imager with excitation at 450 nM and emission detection at 520 nM. Kd values were determined by fitting the fraction of MtRNAP bound to the concentration of free promoter DNA using the program Origin70 and a Hill equation with the Hill coefficient set to 1.0.
Results
Deletions centered on scMtRNAP residues 641/642 disrupt transcription of duplex, but not pre-melted, templates
The N-terminal regions of distantly related members of the single-subunit RNAP family averages exhibit only ~20% average sequence identity(2) making the identification of corresponding functional elements—if present—uncertain, and when we aligned the T7RNAP sequence with those of yeast and human mitochondrial RNAPs, the T7RNAP intercalating hairpin (aa 228-246) was aligned with either residues 616-634 or 633-651 of the yeast enzyme and residues 590-608 or 604-622 of the human enzyme (fig. 1). To determine if either of these alignments identify an element in the yeast mitochondrial polymerase that corresponds functionally to the T7RNAP intercalating hairpin, we generated deletions in the yeast enzyme and characterized the ability of these deletion mutants to transcribe either fully duplex templates or pre-melted (“bubble”) promoter templates in which base-pairs from −4 to +2 were mismatched due to changes in the NT strand from consensus. The results were unambiguous: the mutant with residues 621-624 deleted (ΔPVTK) could transcribe both the duplex and bubble templates (fig. 2a, lanes 1, 5, 13), but the mutants with deletions of residues 641-643 (ΔHNG) or 642-644 (ΔNGS) could transcribe the bubble template (lanes 2, 3, 6. 7) but not the duplex template (lanes 13, 14). This suggested that residues around position 642 in the yeast RNAP form a structure with a function analogous to the T7RNAP intercalating hairpin, which is required for melting the promoter during initiation.
Figure 1. Poor sequence conservation in the N-terminal regions of the single-subunit RNAPs results in ambiguous alignments in these regions.

A. Graphic alignment of the yeast mitochondrial and T7 phage RNAP sequences. Sequence identity in C-terminal regions of the RNAPs ranges from 20% to 50% and averages 30%, but in the N-terminal regions, which include the T7RNAP intercalating hairpin, sequence identity is less than 20%. B. Alignment of the T7RNAP intercalating hairpin with scMtRNAP or human MtRNAP varies depending on the program used. T-coffee and MT4 align the intercalating hairpin (in purple lettering) with scMtRNAP residues 641- 644 (red), whereas structural models generated with MOE and GENO3D align the hairpin with residues 621-624.
Figure 2. Deletion of scMtRNAP residues 631-633 or 632-634 does not abrogate promoter specific binding, but eliminates the ability of the RNAP to transcribe duplex, but not-premelted, promoters.

A. Transcription of either a fully duplex (lanes 9–16) or ‘bubble’ mitochondrial promoter template (containing a heteroduplex in the −4 to +2 segment; lanes 1–8), either without (lanes 1–4; 9–12) or with added MTF1 (lanes 5–8; 13–16) shows that all of the RNAPs can transcribe the bubble template, either with or without MTF1, and that none of RNAPs can transcribe the duplex template in the absence of MTF. However, both WT and ΔPVTK RNAPs can transcribe the duplex template with MTF1, while both the ΔHNG and ΔNGS RNAPs cannot. B: EMSA with fluorescently labeled promoters and WT, ΔHNG ΔNGS RNAPs reveals that all 3 can bind promoter templates and form a ternary complex with promoter and MTF1. Lane 1: WTRNAP with bubble promoter; 2: as in lane 1 but with MTF1added (note supershift of mtRNAP:promoter complex due to MTF1binding); 3: as in 2, but with a duplex promoter; 4: As in 1 but with ΔHNG RNAP; 5: As in 4, but with MTF1added; 6: As in 5, but with a duplex promoter; 7: As in 1 but with ΔNGS RNAP; 8: As in 7 but with MTF1added; 9: As in H, but with duplex promoter. C: Competition experiment shows promoter binding by ΔHNG and ΔNGS is specific. EMSA experiments were run with bubble promoter and MTF1and the indicated RNAPs in either the absence (lanes 1–3), or presence of 5x excess specific unlabeled promoter DNA (lanes 4–6) or non-promoter DNA (lanes 7–9).
However, it is also possible that these mutations perturbed the structure of the RNAP and disrupt interactions with the promoter or MTF1, which could also specifically disrupt initiation from duplex promoters. To test this, we carried out EMSA experiments with fluorescently labeled duplex and bubble promoters with the deletion mutants, WT RNAP, and with or without MTF1. Addition of yeast mtRNAP to a reaction with a 33 bp fluorescent bubble promoter results in formation of a slowly migrating band on a native 8–25% polyacrylamide gel (fig. 2B, lane 1). When MTF1 is added to these reactions this band is observed to super-shift (lane 2), indicating formation of a promoter:RNAP:MTF1 complex (addition of MTF1 alone at the concentration used in fig, 2 to labeled bubble or duplex promoter does not result in a shifted band, though at when MTF1 is added at >5 uM, a slowly migrating but diffuse band is observed, suggesting that MTF1 alone binds DNA weakly; fig. 1, supplemental data). A band migrating at the same position is observed with a duplex promoter and with mtRNAP and MTF1 present (lane 3). The ΔHNG and ΔNGS RNAPs also form complexes migrating at the same positions with MTF1 and either bubble or duplex promoters (lanes 5, 6, 8, 9) but, in the absence of MTF1, multiple weak bands are seen in the reactions with the deletion mutants and the bubble promoter (lanes 3, 7). We have also observed such multiple bands with WT RNAP and bubble promoters when the RNAP is in excess of the promoter and MTF1 is not present (fig. 2 supplemental data; the experiments in fig 2B were done with promoter in excess of RNAP), suggesting that excess RNAP can form non-specific complexes with the bubble promoter in the absence of MTFI and that this tendency is exacerbated with the deletion mutants.
We therefore determined whether promoter binding by these mutants is indeed sequence specific in competition experiments. Limiting amounts of WT, ΔHNG, or ΔNGS RNAPs were mixed with labeled bubble promoter and with either no competitor (fig. 3B, lanes 1–3) or five-fold excess of unlabeled bubble promoter (lanes 4–6) or an unlabeled bubble DNA with a non-promoter sequence (lanes 7–9). For all 3 RNAPs the promoter, but not the non-promoter, DNA was observed to compete for formation of the labeled promoter:RNAP:MTF1 complex. These results indicate that the failure of the ΔHNG or ΔNGS RNAPs to transcribe duplex templates is not due to general loss of transcriptional activity, nor to loss of promoter specific or MTF1 interaction ability, but is instead due specifically to loss of promoter melting activity.
Figure 3. Quantitative EMSA reveals that ΔHNG and ΔNGS RNAPs bind bubble promoters with comparable affinity to WT RNAP, but bind duplex promoters more weakly.

A. Indicated RNAPs at 10−7 M were mixed with 10−7 M MTF1 and fluorescently labeled bubble promoter at concentrations that varied from 10−6 M (lanes 1, 6, 11) to 0.63x10−7 M (lanes 5, 10, 15) in serial 2-fold dilutions and resolved on native 6% PAGE. B. As in A, but in the absence of MTF1. B. As in A, but without MTF1 present and with bubble promoter varied from 10−5 M (lanes 1, 6, 11) to 0.63x10−6 M (lanes 5, 10, 15). C. As in A, but with duplex promoter varying from 4x10−6 M (lanes 1, 6, 11) to 0.25x10−6 M (lanes 5, 10, 15). D. As in C, but with RNAPs and MTF1 at 10−5 M and promoter varying from 10−4 M (lanes 1, 6, 11) to 0.63x10−5 M (lanes 5, 10, 11). Note: In panels B and C, gels are shown over-exposed to allow visualization of the weak and/or multiple complex bands in the ΔHNG/ΔNGS reactions.
Deletions centered on scMtRNAP residues 641/642 weaken binding to duplex, but not pre-melted, promoters
Though experiments at high RNAP and DNA concentrations indicated that the deletion mutants could form specific complexes with either bubble or duplex promoters and MTF1, we undertook quantitative EMSA experiments to determine if the deletions had any effects on promoter affinity. With a bubble promoter and MTF1, the deletion mutants formed promoter complexes with apparent affinities only 2–3 fold less than the WT enzyme (fig. 3a; Table 1). In the absence of MTF1, the apparent affinity of the WT RNAP for a bubble promoter is decreased by ~30-fold while the deletion mutants exhibit multiple shifted bands, indicating formation of heterogeneous complexes on this promoter in the absence of the transcription factor (fig. 3b). To estimate promoter affinity for the deletion mutants under these conditions, the amount of promoter present in all the shifted bands was summed and set as the concentration of bound complex. The apparent affinity calculated in this way was similar for the mutant and WT RNAPs (table 1). With a duplex promoter and MTF1, the apparent affinity of the WT RNAP was ~3-fold less than with the bubble promoter, however the apparent affinities of the ΔHNG and ΔNGS mutants were, respectively, 14- and 28-fold less than for the bubble promoter (fig. 3c, d; table 1). This indicates that the deletions do not markedly affect affinity for a pre-melted promoter (though they do form more heterogeneous complexes on such a promoter in the absence of MTF1), but do weaken binding to fully duplex promoters. The EMSA measured Kd values we obtain for yeast MtRNAP binding to duplex promoters are higher than those measured recently using fluorescence anisotropy(13). This may reflect the different methods used to measure these values, but may also be due to differences in binding buffer conditions, as we used chloride as the counter anion in our binding buffer and did not have Mg++ present, while the previous study used actetate and glutamate as the counter anions and included Mg++.
Table 1.
Apparent Kd values for binding of the indicated RNAPs to the indicated promoter DNAs in either the presence or absence of MTF1, as indicated. Error range are ± s.e. for n=3.
| RNAP | Bubble Promoter | Bubble Promoter+MTF | Duplex Promoter+MTF |
|---|---|---|---|
| WT | 4.3±0.8×10−6 | 1.6±0.1×10−7 | 4.8±0.5×10−7 |
| ΔHNG | 5.3±1.0×10−6 | 3.4±0.4×10−7 | 4.9±0.9×10−6 |
| ΔNGS | 8.3±1.1×10−6 | 4.3±0.6×10−7 | 1.2±0.4×10−5 |
Deletions centered on scMtRNAP residues 641/642 abrogate promoter melting ability
To determine directly if the deletions disrupt promoter melting, we used permanganate footprinting at promoter and RNAP concentrations where, as judged from the EMSA experiments, both the WT and deletion mutants should bind the promoter. In complexes formed with the WT enzyme and a duplex promoter there is minimal permanganate reactivity in the absence of MTF1 (fig. 4, lane 1) but, upon addition of MTF1, reactivity is seen at T-strand bases −2/−3 and weaker reactivity is seen at +1/+2 (lane 3), and on the NT strand around −1 (lane 9). Addition of NTPs allowing transcript extension to +3 results in strengthening of cleavage on the T strand (lane 4) and the appearance of reactivity at +3 on the NT strand (lane 10). Addition of NTPs allowing extension to +6 enhances T-strand reactivity at +1/+2 but does not visibly alter reactivity on the NT strand (lane 12). In contrast to this, we could observe no permanganate reactivity in complexes formed with the either of the deletion mutants (lanes 13–26) under any of these conditions, indicating that the deletions disrupt the ability of the RNAP to melt the promoter.
Figure 4. Deletion of scMtRNAP residues 631-633 or 632-634 abrogates the ability of these RNAPs to melt a duplex promoter.

Duplex promoter 33P-labeled at the 5′-end of the template (T; lanes 1–6, 13–19) or non-template (NT; lanes 7–12, 20–26) was mixed with MTF1 and either WT (lanes 3–5, 9–11), ΔHNG (lanes 13–15, 20–22) or ΔNGS (lanes 16–18, 23–25) RNAPs and then treated with KMnO4 in either the absence (lanes 3, 9, 13, 16, 20, 23) or presence of NTPs allowing RNA extension to 3 (lanes 4, 10, 14, 17, 21, 24) or 6 (lanes 5, 11, 15, 18, 22, 25) bases in length. Numbering of bands is relative to +1 transcription start site and G+A ladders are run in lanes 6, 12, 19, and 26.
MTF1 and the intercalating hairpin make approximately similar and synergistic contributions to promoter melting
The results of fig. 2 show that yeast MtRNAP requires both an intact intercalating hairpin and MTF1 to initiate transcription on a fully duplex template, but provide no information on the relative contribution of these two elements to promoter melting. To evaluate this, we tested transcription by the WT and deletion mutant RNAPs, in either the presence or absence of MTF1, on a series of promoters in which melting is increasingly facilitated by progressive introduction of 1 to 5 mismatched base-pairs starting promoter position −4 and extending downstream to +1. On a promoter with a single mismatch (“Het1”; fig. 5A, lane 1) and in the absence of MTF1, runoff transcription by WT RNAP is low but detectable (~1 runoff transcript synthesized every 200 minutes) but runoff transcription by the ΔHNG or ΔNGS RNAPs is undetectable (lanes 6, 11). A promoter with two mismatches enhances transcription by the WT enzyme by ~5 fold (lane 2), but transcription by the deletion mutants is ~3-fold lower (lanes 7, 12), corresponding to approximately one transcript every 100 minutes. Upon introduction of 3, 4, or 5 mismatches (“Het 3, 4, 5”) we observe similarly high levels of transcription with all 3 RNAPs (lanes 3–5, 8–10, 13–15). Addition of MTF1 to the reactions with the ΔHNG or ΔNGS RNAPs increases transcription of the Het2 promoter by ~8-fold (fig. 5b; lanes 7, 12) and results in detectable transcription of the Het1 promoter (lanes 6, 11). However, high levels of runoff transcription on the Het1 and Het2 promoters are seen only when both MTF1 and an integral hairpin are present (lanes 1, 2).
Figure 5. MTF1 and 631-634 region make approximately equal and synergistic contributions to promoter melting.

A: Promoters containing either 1 (“Het1”; lanes 1, 6, 11), 2 (“Het2”; lanes 2, 7, 12), 3 (“Het3”; lanes 3, 8, 13), 4 (“Het4”; lanes 4, 9, 14) or 5 (“Het5”, lanes 5, 10, 15) mispaired bases extending downstream from −4 were transcribed with either WT (lanes 1–5), ΔHNG (lanes 6–10), or ΔNGS (lanes 11–15) RNAPs in the absence of MTF1. B: As in panel A but with MTF1 added. C: Quantification of the results in panels A and B expressed as number of runoff transcripts per minute per RNAP. Quantification was done by multiplying the fraction of total radioactivity in a given gel lane (including unincorporated radioactivity) incorporated into runoff (19–21 nt) transcripts by the ATP concentration (500 μM) and dividing by the reaction time, MtRNAP concentration (1 μM), and number of A bases (9) in the runoff transcript.
The amount of runoff transcription observed in these assays is a complex function that reflects the efficiency of promoter melting and initiation of transcription, of progression through initial (abortive) transcription, promoter escape, elongation and RNAP cycling following runoff. We therefore also carried out assays in which only ATP was present, so as to limit progression of transcription beyond the +2 template position (fig. 6; the promoter used in these experiments initiates ‘AAU’). Under such conditions, in addition to synthesizing dimers, the RNAP will make poly-A ladders of indefinite length. In the absence of MTF1, the WT RNAP exhibits low levels of transcription on the Het1 template, which increases as the heteroduplex region is increased from 1 to 5 base-pairs (fig. 6A, lanes 1–5). In comparison to WT RNAP, and in the absence of MTF1, the deletion mutants exhibit lower levels of transcription on the Het1/Het2 promoters and, to a lesser extent on the Het3/4 promoters, and levels of transcription similar to WT are observed only on the Het5 promoter (lanes 5–15). Addition of MTF1 increases transcription 10–20 fold on the Het1/Het2 promoters for the WT enzyme (fig. 6B, lanes 1, 2), by 3-fold on the Het3 promoter (lane 3), and has little effect with the Het4/5 promoters (lanes 4, 5). With the deletion mutants, MTF1 causes a 2–3 fold increase in transcription on the Het1/2 promoters (lanes 6, 7, 11, 12). But has little effect on transcription from the Het3-5 promoters (lanes 8–10, 13–15).
Figure 6. Deletion of scMtRNAP residues 631-633 or 632-634 affects dinucleotide synthesis as well as runoff transcription.

A: Promoters containing either 1 (“Het1”; lanes 1, 6, 11), 2 (“Het2”; lanes 2, 7, 12), 3 (“Het3”; lanes 3, 8, 13), 4 (“Het4”; lanes 4, 9, 14) or 5 (“Het5”, lanes 5, 10, 15) mispaired bases extending downstream from −4 were transcribed with either WT (lanes 1–5), ΔHNG (lanes 6–10), or ΔNGS (lanes 11–15) RNAPs in the absence of MTF1 were transcribed in the presence ATP only. B: As in panel A but with MTF1 added. C: Quantification of the results in panels A and B expressed as number of runoff transcripts per minute per RNAP Quantification done as described in fig. 5C after correcting for the number of A bases in a dinucleotide vs. runoff transcript.
Finally, we determined whether the placement of the mismatched base pairs has an effect on transcription. Since promoters with 1 or 2 (Het1 or Het2) mismatches at −4 and −3 were most sensitive to the effects of a hairpin deletion or MTF1 (promoters with more mismatches could be transcribed by both deletion mutants and in either the presence or absence of MTF1), we tested promoters in which either the +2 or both the +1 and +2 positions were mismatched (designated Het1D and Het2D and corresponding to promoters with 1 or 2 mismatches at the downstream, rather than upstream, end of the transcription bubble). In the absence of MTF1 neither the WT MtRNAP nor the deletion mutants showed significant transcription of the Het1D promoter (fig. 7a, lanes 1, 3, 5). WT MtRNAP transcribed the Het2D promoter at 10-fold higher levels than Het1D (fig. 7a, lane 2 and fig. 7b), but transcription of Het2D by the deletion mutants remained low (fig. 7a, lanes 4, 6). Addition of MTF1 increased transcription by WT MtRNAP of the Het1D promoter to a level similar to that seen for Het2D (lanes 7, 8), and similarly increased transcription of Het2D by the deletion mutants (lanes 10, 12). Transcription of the Het1D promoter by the deletion mutants remained low even with MTF1 (lanes 9, 11).
Figure 7.
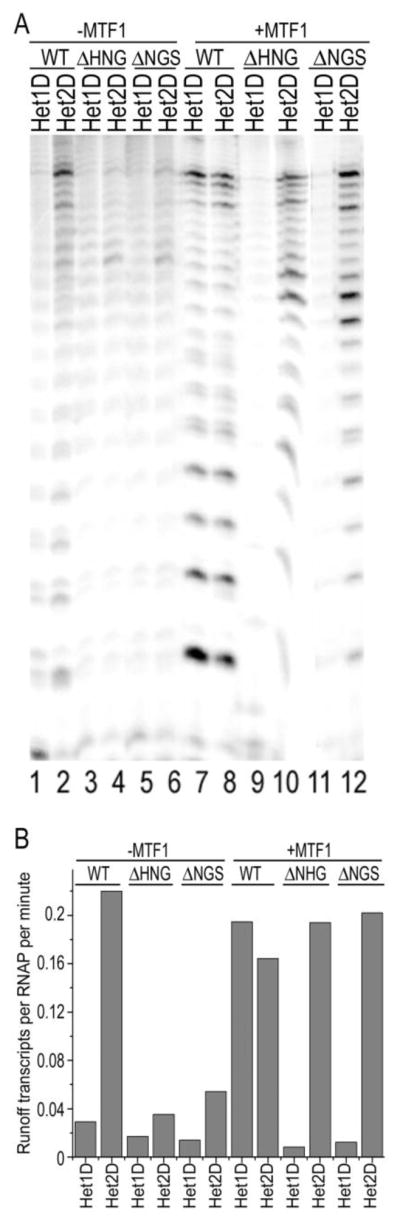
MTF1 and the intercalating hairpin make equal and synergistic contributions to transcription from promoters with mismatches at +1 and +2. A. Promoters containing mismatches at +2 (Het1D; odd numbered lanes) or +1 and +2 (Het 2D; even numbered lanes) transcribed with WT (lanes 1, 2, 7, 8), ΔHNG (lanes 3, 4, 9, 10) or ΔNGS (lanes 5, 6, 11, 12) MtRNAPs in either the absence (lanes 1–6) or presence (lanes 7–8) of MTF1. B: Rates of runoff transcription on the indicated promoters and by the indicated MtRNAPs with or without MTF1.
Overall, these results indicate that: (1) Both MTF1 and the intercalating hairpin individually contribute to promoter melting. In the absence of MTF1, the WT RNAP transcribes the Het1 or Het2 promoters at ~3-fold higher levels than the deletion mutants, and transcribes the Het2D promoter at ~6-fold greater levels than the deletion mutants. Addition of MTF1 enhances deletion mutant transcription of the Het1/2 or Het2D promoters by 2–8 fold. (2) The contributions of the hairpin and MTF1 to melting are similar. On the Het1D/2D promoters addition of MTF1 to the WT+Het1D reaction increases transcription by ~6-fold, and in the absence of MTF1 transcription of Het2D by the WT MtRNAP is also ~6-fold greater than by the deletion mutants. With the Het1/2 promoters the contribution of MTF1 to melting is modesty greater than the contribution of the hairpin since addition of MTF1 to the deletion mutant reactions increases transcription by ~2-fold more than does the use of WT MtRNAP vs. ΔHNG or ΔNGS MtRNAPs. (3) The effects of the hairpin and MTF1 are synergistic: high levels of transcription on the Het1/2 or Het1D promoters are seen only when both an integral hairpin and MTF1 are present.
ΔHNG and ΔNGS relief of MTF1 inhibition of transcription on pre-melted promoters likely reflects disruption of interactions with NT nucleotides −2/−3
Inspection of figure 5 reveals that the deletions have an effect on transcription patterns beyond the defect in transcription of duplex promoters. With the WT RNAP it is seen that, in the presence of MTF1, the levels of runoff transcript decrease as the heteroduplex region in the promoter increases (fig. 5B, lanes 1–5). This is not seen with the deletion mutants (fig. 5B, lanes 6–10). The MTF1 dependent decrease in runoff transcription on pre-melted promoters has been observed previously and attributed to inhibition of promoter escape due to a too strong interaction with the melted promoter and MTF1(3, 14). It is relieved by deletions in the MtRNAP N-terminal domain that weaken the interaction with MTF1(3, 14). To determine if ΔHNG and ΔNGS also relieved MTF1 inhibition of transcription of escape on bubble templates to as great an extent as the much large N-terminal deletions we compared transcription on duplex and bubble templates with these mutants to those by RNAPs with residues 1-358 (ΔN1) or 1-264 (ΔN2) deleted. A homology model based on the human MtRNAP structure(11) (supplement fig. S3) reveals that the larger deletion removes the entire N-terminal extension that is absent from T7RNAP, while the smaller deletion removes part of this domain, but that neither removes elements that are structurally similar to T7RNAP. A more extensive deletion of yeast MtRNAP residues 1-380 has been reported to reduce promoter binding and disrupt transcription(14), and the homology model suggests that this deletion may remove part a region of the MtRNAP that is analogous, and possibly homologous, to the T7RNAP N-terminal domain.
Though yeast MtRNAP N-terminal deletions are reported to weaken MTF1 binding, under the conditions of these assays, MTF1 binds the ΔN1 and ΔN2 initiation complexes as shown by the fact it allows both RNAPs to initiate transcription from duplex templates (compare fig. 8A lanes 7, 9 to 17, 19), and by the fact that MTF1 binds to ΔN1 and ΔN2 initiation complexes by EMSA (supplemental fig. S4). On the bubble template, the presence of MTF1 reduces transcription by the WT enzyme (compare lanes 2 and 12) and scans of lanes 2 and 12 reveal the large proportionate increase in abortive transcription in the presence of MTF1, indicative of an inhibition of promoter escape. Neither the hairpin nor the N-terminal deletion mutations show such inhibition and inspection of the transcript patterns (fig. 8A) and scans of the relevant gel lanes (fig. 8B) indicate that, qualitatively, the 3 residue hairpin deletions relieve this inhibition as effectively as the large N-terminal deletions. Quantitatively, the proportions of abortive transcripts support this conclusion as MTF1 increased the percent incorporation into 3–8mers on the bubble promoters by 2–3 fold for WT MtRNAP, but had no effect on the fractional incorporation into 3–8mers for the 4 mutant MtRNAPs (Table II)
Figure 8. Deletion of scMtRNAP residues 631-633 or 632-634 relieves MTF1 inhibition of transcription on pre-melted promoters but does not cause defects in MTF1 binding.

A. Duplex (‘Dup’; odd numbered lanes) or Bubble (heteroduplex from −4 to −2; even numbered lanes) were transcribed in either the absence (lanes 1–10) or presence (lanes 11–20) of MTF1 with either WT (lanes 1, 2, 11, 12), ΔHNG (lanes 3, 4, 13, 14), ΔNGS (lanes 5, 6, 15, 16), ΔN1 (lanes 7, 8, 17, 18) or ΔN2 RNAPs (lanes 9, 10, 19, 20). B. Scans of the indicated gel lanes from panel A (bubble promoter reactions only). C. Equimolar mixtures of MTF1 and either WT (lanes 1, 2), ΔN1 (lanes 3, 4), ΔN2 (lanes 5, 6), ΔHNG (lanes 7, 8), ΔNGS (lanes 9, 10), or No RNAP (lanes 11, 12) were filtered through membranes with 100 kD MW cutoffs. Retenates (“R”) are shown in odd numbered lanes and filtrates (“F”) in even numbered lanes.
Table 2.
Percent incorporation of α32P-AMP into abortive transcripts on the indicated promoters and by the indicated RNAPs. Values are ± s.d. for n=3. Partially single-stranded promoters with −4 and −2 NT strands coorespond, respectively, to promoters with NT strands that extend to −4 (promoter 1 in fig. 9C) or −2 (promoter 3 in fig. 9C).
| Promoter | ||||||
|---|---|---|---|---|---|---|
| Bubble (−4 to −2 heteroduplex) | Partially single-stranded (−4 NT strand) | Partially single-stranded (−2 NT strand) | ||||
| MtRNAP | −MTF1 | +MTF1 | −MTF1 | +MTF1 | −MTF1 | +MTF1 |
| WT | 18±1.5 | 43±3.2 | 14±1.5 | 24±1.9 | 19±4 | 39±1.5 |
| ΔHNG | 8.2±0.9 | 8.4±1.0 | 18±1.9 | 13±0.5 | 20±0.6 | 14±2.5 |
| ΔNGS | 7.8±0.7 | 8.1±1.3 | 15±1.7 | 10±1.0 | 18±2.0 | 14±1.3 |
We next asked whether relief of MTF1 inhibition by the hairpin deletion enzymes worked through a mechanism similar to that of the N-terminal deletions, by a decrease in affinity for MTF1. Equimolar amounts of MTF1 and MtRNAP were mixed to final concentrations of 1 μM and subjected to filtration through membranes with 100 kD cutoffs that allowed free MTF1 to flow through, but not MtRNAP or MtRNAP:MTF1 complexes. With WT RNAP essentially all of the MTF1 was retained with the RNAP (fig. 7C, lanes l, 2) while in the absence of any RNAP all of the MTF1 was found in the filtrate (lanes 11, 12). The ΔN1 and ΔN2 RNAPs both show reduced retention of MTF1 (lanes 3–6) with the larger ΔN1 deletion having a more severe effect. The ΔHNG and ΔNGS RNAPs however, show no defect in MTF1 binding and exhibit retention like the WT enzyme (lanes 7–10)
Since the hairpin deletions did not affect the apparent affinity of the MtRNAP for MTF1, we asked whether this relief might be due to loss of interactions with the upstream fork of the transcription bubble. We reasoned that, if this were the case, then deletion of the relevant DNA elements from a pre-melted promoter might similarly relax MTF1 inhibition of the WT MtRNAP. We therefore tested transcription of templates in which the complementary NT strand extended from −12 to −4, −3, or −2 (fig. 9). On templates where the NT stranded extended to −2 or −3, the presence of MTF1 markedly increased the proportion of abortive transcripts in WT reactions, with the increase in abortives being greater when the NT strand was extended to −2 than −3 (compare lanes 2, 3 to 6, 7 in fig 9A). However, when the NT strand extended only to −4, the presence of MTF1 caused almost no increase in abortives (compare lanes 1 and 5). Nor was the difference in the effect of MTF1 due to differences in how many base pairs have to be melted with the −4 vs. −2 NT strand. When we used an NT strand that was complementary to the T strand only from −12 to −5, but that also had a 6 nt non-complementary tail, we observed MTF1 dependent increases in abortives similar to those seen with the −2 NT strand (compare lanes 8 and 4 in fig. 9A). In contrast, with ΔHNG or ΔNGS the proportions of abortives on all promoters were similarly low in either the absence or presence of MTF1 (fig. S5). Quantification of transcription on partially single-stranded promoters in which the NT strand extends to either −4 (PNT-4) or −2 (PNT-2) confirms that MTF1 increases abortive transcription by the WT RNAP on PNT-2 but not PNT-4, and that abortive transcription by the hairpin deletion mutants is unaffected by MTF1 on either promoter (Table II).
Figure 9. Removal of NT strand nucleotides −2/−3 relieves MTF1 inhibition of runoff transcription on pre-melted promoters.

A: Transcription of partially single-stranded promoters in which the NT strand extends from −4 to −2 (promoter structures as shown in panel C) by WT MtRNAP in either the absence (lanes 1–4) or presence (lanes 5–8) of MTF1. B. Scan of lanes 1–8 from panel A. C: Structure of promoters used in reactions in panels A and B.
We conclude that removal of NT nucleotides −2/−3 disrupts interactions responsible for the effect of MTF1 in inhibiting promoter escape and increasing abortive transcription. Since modeling based on the T7RNAP IC structure indicates that the MtRNAP intercalating hairpin would interact with these same nucleotides (fig. S3), this suggests that the relief of MTF1 inhibition by the hairpin deletions is due to a similar mechanism.
Discussion
It is important to define the degree of conservation of structure and mechanism in the single-subunit RNAP family. Initial characterization can suggest a greater degree of mechanistic divergence than is borne out by deeper analysis. For example, the very different sequences and sizes of mitochondrial and phage promoters(12), the requirement of yeast MtRNAP for an auxillary factor (MTF1) for transcription initiation, the observation that this factor is released from the RNAP upon transition to elongation(15), and the detection of weak sequence similarity between MTF1 and E. coli σ70 suggested not only that mitochondrial and phage RNAPs used very different promoter recognition mechanisms, but also that MTF1 functioned like a sigma factor to endow a core MtRNAP with promoter specificity(16). In fact, more recent studies have shown that MtRNAPs are capable of promoter specific transcription in the absence of these auxillary factors(3, 17), and that yeast MtRNAP contains a ‘promoter recognition loop’ similar in size, secondary structure, position, and function to a homologous element in the phage RNAPs(12). The recently described structure of a human MtRNAP also reveals a similar promoter recognition loop(11), indicating that such an element may be common to most or all single-subunit RNAPs.
Similarly, poor sequence conservation in the single-subunit RNAPs in the region encompassing the intercalating hairpin, and the fact that mitochondrial, but not phage, RNAPs require additional factors for promoter melting would suggest that melting mechanisms are very different in different members of this family. However, our results reveal that the yeast MtRNAP contains an element that appears functionally analogous to that hairpin. Homology models of the yeast MtRNAP based on the human MtRNAP structure (fig. S3) show this element superimposes on a disordered loop in the N-terminal region of the human enzyme that is proposed to be similar in structure to the phage intercalating hairpin, and is also indicated to play a role in promoter melting(11), again indicating that this element may be structurally and functionally conserved in all or most single-subunit RNAPs. Evaluation of transcriptional activity on promoters that are pre-melted to varying extents by introduction of different numbers of mismatched base-pairs into the −4 to +2 region indicates that MTF1 and the intercalating hairpin individually make similar contributions to promoter melting, and that both are required for high levels of transcriptional activity on duplex promoters or promoters with only one or two base-pair mismatches.
Deletions in the yeast MtRNAP hairpin had another, unexpected, effect on transcription: they eliminated the MTF1 induced inhibition of transcript release observed with full-length MtRNAP on pre-melted promoters. This inhibition can also be relieved by deletions in the MtRNAP N-terminal domain that weaken MTF1 binding, and this has been interpreted to mean that the combination of a pre-melted promoter and MTF1 interaction results in a too stable set of interactions that inhibit escape of the MtRNAP from the promoter (interestingly, human MtRNAP does not exhibit escape inhibition on pre-melted promoters in the presence of its transcription factor(18, 19) However, we found that the deletions in the intercalating hairpin did not weaken MtRNAP:MTF1 interaction, or at least not the same extent as the N-terminal deletions, though their effects on relief of MTF1 inhibition were comparable. Instead we found that MTF1 inhibition could also be relieved by deletion of the −2/−3 nucleotides of the NT strand, indicating that MtRNAP interactions with the upstream region of the transcription bubble could also inhibit promoter escape, and that the hairpin deletions may relieve this inhibition by disrupting these interactions.
Intriguingly, not only is the mitochondrial RNAP homologous to phage RNAP, but the catalytic subunit of the mitochondrial DNAP has also been shown to be most closely related to the T7 phage DNAP(20), and the human ‘twinkle’ gene has been shown to encode a mitochondrial primase/helicase which is most closely related to the T7 gene 4 primase/helicase(21) (a further functional similarity is that the T7 and mitochondrial RNAPs both prime leading strand DNA replication as well as synthesizing mRNAs(22, 23)). To a first approximation, mitochondria may be described as using a DNA transcription and replication apparatus derived from a phage. This may have happened because the structurally simpler phage enzymes are easier to import and assemble into functional complexes in the mitochondrion than would be, for example, a multi-subunit bacterial or nuclear RNAP. However, the reason for the conservation of such relatively fine scale structure-function features such as the promoter recognition loop or intercalating hairpin across the evolutionary distances that separate yeast and human mitochondria is surprising and, as yet, without a fully satisfactory adaptive explanation.
Supplementary Material
Acknowledgments
Funding Source: This work was supported by NIH GMS52522 (to RS) and CONACYT J48770 (to LGB).
Abbreviations used
- RNAP
RNA Polymerase
- Mt
mitochondrial
- Mtf1
Yeast mitochondrial transcription factor
- IC
initiation complex
- WT
wild type
- T-strand
Template strand
- NT-strand
non-template strand
- EMSA
electrophoretic mobility shift analysis
Footnotes
Supporting information available. Figures showing: (1) Low-affinity non-specific DNA binding by MTF1 alone, (2) Multimer formation on bubble promoters with excess WT MtRNAP in the absence of MTF1, (3) Molecular models of yeast MtRNAP showing putative locations of residues deleted in these studies. (4) EMSA showing MTF1 association with N-terminal deletion mutant MtRNAP IC. (5) Effect of MTF on ΔHNG and ΔNGS transcription from partially single-stranded promoters with varying NT strand lengths. This material is available free of charge via the internet at http://pubs.acs.org.
Contributor Information
Gilberto Velazquez, Email: gilvelazquez83@gmail.com.
Qing Guo, Email: bcqguo@hotmail.com.
Liping Wang, Email: wangl1@uthscsa.edu.
Luis G. Brieba, Email: lgbrieba@ira.cinvestav.mx.
Rui Sousa, Email: sousa@uthscsa.edu.
References
- 1.Jun SH, Reichlen MJ, Tajiri M, Murakami KS. Archaeal RNA polymerase and transcription regulation. Crit Rev Biochem Mol Biol. 46:27–40. doi: 10.3109/10409238.2010.538662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cermakian N, Ikeda TM, Miramontes P, Lang BF, Gray MW, Cedergren R. On the evolution of the single-subunit RNA polymerases. J Mol Evol. 1997;45:671–681. doi: 10.1007/pl00006271. [DOI] [PubMed] [Google Scholar]
- 3.Matsunaga M, Jaehning JA. Intrinsic promoter recognition by a “core” RNA polymerase. J Biol Chem. 2004;279:44239–44242. doi: 10.1074/jbc.C400384200. [DOI] [PubMed] [Google Scholar]
- 4.Sologub M, Litonin D, Anikin M, Mustaev A, Temiakov D. TFB2 is a transient component of the catalytic site of the human mitochondrial RNA polymerase. Cell. 2009;139:934–944. doi: 10.1016/j.cell.2009.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ujvari A, Martin CT. Evidence for DNA bending at the T7 RNA polymerase promoter. J Mol Biol. 2000;295:1173–1184. doi: 10.1006/jmbi.1999.3418. [DOI] [PubMed] [Google Scholar]
- 6.Tang GQ, Patel SS. T7 RNA polymerase-induced bending of promoter DNA is coupled to DNA opening. Biochemistry. 2006;45:4936–4946. doi: 10.1021/bi0522910. [DOI] [PubMed] [Google Scholar]
- 7.Tang GQ, Patel SS. Rapid binding of T7 RNA polymerase is followed by simultaneous bending and opening of the promoter DNA. Biochemistry. 2006;45:4947–4956. doi: 10.1021/bi052292s. [DOI] [PubMed] [Google Scholar]
- 8.Stano NM, Patel SS. The intercalating beta-hairpin of T7 RNA polymerase plays a role in promoter DNA melting and in stabilizing the melted DNA for efficient RNA synthesis. J Mol Biol. 2002;315:1009–1025. doi: 10.1006/jmbi.2001.5313. [DOI] [PubMed] [Google Scholar]
- 9.Brieba LG, Sousa R. The T7 RNA polymerase intercalating hairpin is important for promoter opening during initiation but not for RNA displacement or transcription bubble stability during elongation. Biochemistry. 2001;40:3882–38890. doi: 10.1021/bi002716c. [DOI] [PubMed] [Google Scholar]
- 10.Cheetham GM, Jeruzalmi D, Steitz TA. Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature. 1999;399:80–83. doi: 10.1038/19999. [DOI] [PubMed] [Google Scholar]
- 11.Ringel R, Sologub M, Morozov YI, Litonin D, Cramer P, Temiakov D. Structure of human mitochondrial RNA polymerase. Nature. 478:269–273. doi: 10.1038/nature10435. [DOI] [PubMed] [Google Scholar]
- 12.Nayak D, Guo Q, Sousa R. A promoter recognition mechanism common to yeast mitochondrial and phage t7 RNA polymerases. J Biol Chem. 2009;284:13641–13647. doi: 10.1074/jbc.M900718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang GQ, Deshpande AP, Patel SS. Transcription factor-dependent DNA bending governs promoter recognition by the mitochondrial RNA polymerase. J Biol Chem. 286:38805–38813. doi: 10.1074/jbc.M111.261966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paratkar S, Deshpande AP, Tang GQ, Patel SS. The N-terminal domain of the yeast mitochondrial RNA polymerase regulates multiple steps of transcription. J Biol Chem. 286:16109–16120. doi: 10.1074/jbc.M111.228023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mangus DA, Jang SH, Jaehning JA. Release of the yeast mitochondrial RNA polymerase specificity factor from transcription complexes. J Biol Chem. 1994;269:26568–26574. [PubMed] [Google Scholar]
- 16.Jang SH, Jaehning JA. The yeast mitochondrial RNA polymerase specificity factor, MTF1, is similar to bacterial sigma factors. J Biol Chem. 1991;266:22671–22677. [PubMed] [Google Scholar]
- 17.Gaspari M, Falkenberg M, Larsson NG, Gustafsson CM. The mitochondrial RNA polymerase contributes critically to promoter specificity in mammalian cells. Embo J. 2004;23:4606–4614. doi: 10.1038/sj.emboj.7600465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lodeiro MF, Uchida AU, Arnold JJ, Reynolds SL, Moustafa IM, Cameron CE. Identification of multiple rate-limiting steps during the human mitochondrial transcription cycle in vitro. J Biol Chem. 285:16387–16402. doi: 10.1074/jbc.M109.092676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shutt TE, Lodeiro MF, Cotney J, Cameron CE, Shadel GS. Core human mitochondrial transcription apparatus is a regulated two-component system in vitro. Proc Natl Acad Sci U S A. 107:12133–12138. doi: 10.1073/pnas.0910581107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filee J, Forterre P, Sen-Lin T, Laurent J. Evolution of DNA polymerase families: evidences for multiple gene exchange between cellular and viral proteins. J Mol Evol. 2002;54:763–773. doi: 10.1007/s00239-001-0078-x. [DOI] [PubMed] [Google Scholar]
- 21.Korhonen JA, Gaspari M, Falkenberg M. TWINKLE Has 5′ -> 3′ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J Biol Chem. 2003;278:48627–48632. doi: 10.1074/jbc.M306981200. [DOI] [PubMed] [Google Scholar]
- 22.Fuller CW, Richardson CC. Initiation of DNA replication at the primary origin of bacteriophage T7 by purified proteins. Site and direction of initial DNA synthesis. J Biol Chem. 1985;260:3185–3196. [PubMed] [Google Scholar]
- 23.Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006;24:813–825. doi: 10.1016/j.molcel.2006.11.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.