

Abstract
Hexokinase (HK), the rate-limiting enzyme in glycolysis, regulates cell survival either by promoting metabolism and/or inhibiting apoptosis. HKI and HKII, isoforms with mitochondrial targeting sequences, were studied in renal epithelial cells after ATP depletion, an insult that activates GSK3β and Bax, induces mitochondrial membrane injury and causes apoptosis. Stress decreased total ATP content, caused HKII to dissociate from mitochondria, released mitochondrial apoptosis inducing factor (AIF) and cytochrome c, activated caspase-3 and reduced cell survival. Compared to control, HKI or II over-expression improved survival after stress without preventing GSK3β or Bax activation, increasing ATP content, or reducing mitochondrial fragmentation. HKI or HKII over-expression increased mitochondria-associated, isoform-specific HK content and decreased mitochondrial membrane injury and apoptosis. In vivo, HKII localized to the murine proximal tubule. Ischemia reduced total HKII content and caused mitochondrial HKII dissociation in proximal but not distal tubules. In HK over-expressing cells, HKII and Bax did not interact before or after stress. However, HK over-expression increased organelle-associated HK during stress and decreased mitochondrial Bax accumulation, outer membrane injury and apoptosis in a manner that appears to be independent of its effect on renal epithelial cell ATP content, suggesting that HK and Bax compete for similar mitochondrial membrane binding sites.
Keywords: acute kidney injury, apoptosis, proximal tubule, renal ischemia-reperfuison
INTRODUCTION
Hexokinase (HK) converts glucose to glucose-6-phosphate, the rate-limiting step in glycolysis. Since the kidney contains abundant amounts of HK, it has been assumed that its primary role is to promote cell metabolism (1). However, two HK isoforms, HK I and II have mitochondrial amino-terminal targeting sequences that control “on” and “off” HK shuttling at the outer mitochondrial membrane (2, 3), a key intracellular site that regulates aerobic metabolism and apoptosis. Although the isoform-specific renal distribution of HK I and II is unknown, total HK content is greater in the medulla than in the cortex (4, 5). Interestingly, in cancer cell lines, including renal cell carcinoma, abundant levels of HK I or II (6, 7) confer marked resistance to chemotherapy or other pro-apoptotic stimuli including UV irradiation and oxidative stress (8). In these cancer cells, protection by HK has been primarily attributed to its benefits on cell metabolism (6, 8).
Recent evidence suggests that HK directly regulates apoptosis. Specifically, HK appears to interfere with members of the BCL2 protein family that mediate mitochondrial membrane injury, a hallmark of apoptotic cell death (9, 10). Although the binding site at the outer mitochondrial membrane is presently debated (see Discussion; (11, 12)), it has been suggested that mitochondrial-associated HK affords protection against apoptosis caused by members of the BCL2 protein family, whereas dissociated HK might not (9, 13, 14). Of the BCL2 proteins, pro-apoptotic Bax is an attractive target for HK in renal epithelial cells, since metabolic stress activates Bax, resulting in the exposure of the Bax carboxy-terminal 6A7 epitope (15), oligomerization (16, 17) and translocation to mitochondria (17). Furthermore, Bax is rapidly activated by renal ischemia in vivo (18). Bax activation and translocation are partly mediated by Akt (protein kinase B) and glycogen synthase kinase 3-beta (GSK3β), potent kinases that serine phosphorylate and either activate or inactivate Bax (19). Since GSK3β is activated in renal epithelial cells subjected to metabolic stress (20) or renal ischemia in vivo (18), we hypothesized that mitochondrial-bound HK ameliorates “Bax attack” by either inhibiting GSK3β activation, limiting Bax activation, interacting with Bax or competing for Bax binding at the outer mitochondrial membrane. Also, whether HK isoforms regulate renal epithelial cell survival by promoting metabolism or inhibiting apoptosis is not known.
In the present study, renal epithelial cells were exposed to metabolic stress in the absence of glucose, an ideal model for separating the effects of HK on cell metabolism from its anti-apoptotic effects at the level of the outer mitochondrial membrane. We report that metabolic stress causes progressive mitochondrial HK II displacement and a fall in total cellular HK II content concomitant with GSK3β and Bax activation, mitochondrial membrane permeabilization and cell death. Renal ischemia in vivo also results in mitochondrial HK II dissociation and a decrement in total HK content. In cultured cells, enhancing HK I or II expression, the major renal HK isoforms, decreases mitochondrial Bax accumulation and improves cell survival without inhibiting GSK3β or Bax activation or altering total cell ATP content after stress. HK II is a relevant isoform, since it localizes to the proximal tubule, a major site for ischemic injury. This study supports the hypothesis that HK decreases apoptosis after metabolic stress by antagonizing “Bax attack” of the outer mitochondrial membrane.
RESULTS
Effect of metabolic stress on HK II distribution in vitro and in vivo
In intact control cells examined by immunofluorescence microscopy at baseline, HK II co-localizes with Cox IV, a mitochondrial marker (Fig 1, left panel). After 60 minutes of ATP depletion followed by 15 minutes recovery, dramatic HK II re-distribution away from mitochondria is observed (right panel). Given the relatively large volume of cytosol, this approach could not accurately assess HK II content in this compartment. Therefore, to determine whether the loss of mitochondrial-associated HK II is due to HK II degradation and/or re-distribution, both total and cytosolic levels of HK II after metabolic stress were assessed by immunoblot analysis in cell lysates and digitonin-permeabilized cells, respectively (Fig 2). Compared to baseline, total HK II content progressively decreased after ATP depletion and 15 min recovery (top panel), suggesting that metabolic stress promotes HK II degradation. Concomitant with the decrease in total HK II, a transient increase in “cytosolic” HK II was detected after ATP depletion (middle panel).
Fig 1. Metabolic stress causes HK II to dissociate from mitochondria.
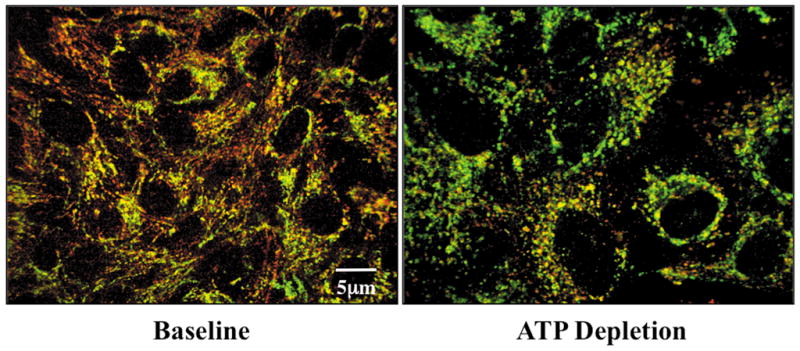
HK II (red) and Cox IV (green), an outer mitochondrial membrane protein, by immunofluorescence at baseline and following 15 min recovery from 60 min ATP depletion.
Fig 2. Effect of metabolic stress on cell HK II distribution.
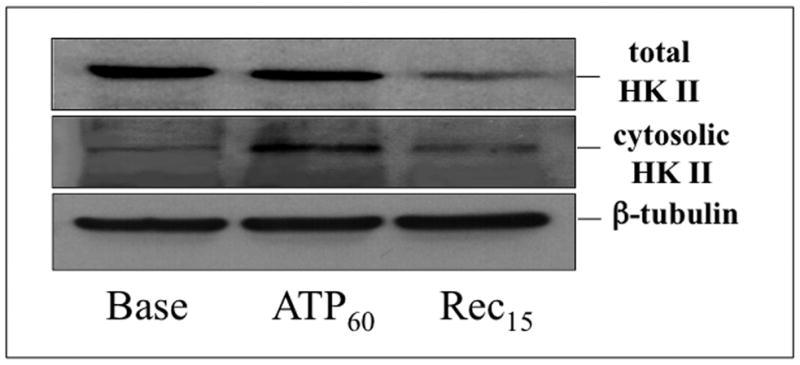
Total (upper panel) and cytosolic (middle panel) HK II content assessed by immunoblot analysis in cell lysates and in soluble extracts of digitonin-treated cells respectively, at baseline (Base), after 60 min ATP depletion (ATP60), and 15 min recovery (Rec15); β-tubulin serves as a loading control (lower panel).
In perfusion fixed renal tissue, HK II localizes almost exclusively to the proximal tubule (Fig 3A). A sharp demarcation in HK staining is evident at the corticomedullary junction (upper panels). HK II is abundant in the cortex (middle panels) but is virtually absent in distal tubules (lower panels). As observed in renal cells in culture at baseline, HK II extensively co-localized with mitochondrial specific F1F0-ATPase in proximal tubular cells in vivo (Fig 3B, upper right inset). HK II staining was also prominent in the proximal tubule brush border (middle panel, upper row). After ischemia however, overall intracellular HK II staining intensity was markedly reduced as compared to baseline (upper vs. lower middle panels) and minimal mitochondrial HK II was detected (lower right inset). Interestingly, brush border HK II staining was no longer visible although abundant HK II was detected in the lumens of post-ischemic proximal tubules. Compared to baseline, ischemia caused marked loss of immunoreactive renal HK II content (Fig 4). In fact, densitometric analysis revealed a 40% decrement in renal HK II content after ischemia (P<0.05; n=4; Fig 4B). In contrast, sham ischemia did not significantly alter HK II content (data not shown; P >0.05). These results show that renal ischemia in vivo causes marked HK-II re-distribution and intracellular loss in a manner that parallels the effects of ATP depletion in vitro.
Fig 3. Effect of renal ischemia in vivo on mitochondrial-associated HK II.


(A) Phase contrast, HK II immunofluorescence (red), and merged images of the corticomedullary region (“CM junction”; upper panels); renal cortex (middle panels) and medulla (lower panels) of normal murine renal tissue. HK II localizes to the proximal tubule; (B) HK II (red) partially co-localizes with mitochondrial F1F0 ATPase (green) at baseline as indicated by orange-yellow staining (right upper panel); HK II dissociates from mitochondrial after 40 min renal artery occlusion as shown by separation of the red and green signals (right lower panels); original magnification 630x. Arrow indicates brush border with prominent HK II staining. Solid arrow shows HK II in the lumen of a proximal tubule. Inset shows mitochondrial staining at higher magnification to show differences in mitochondrial HK II before vs. after renal ischemia. These images are representative of at least three separate studies.
Fig 4. Renal ischemia in vivo alters HK II.

(A) Effect of 40 min unilateral renal artery occlusion on HK II content in renal homogenates (‘R’ or right kidney) compared to baseline (‘L’ or left kidney) in four representative animals; mitochondrial F1F0 ATPase, serves as loading control; (B) densitometric analysis of HK II in unilateral sham ischemia (n=6) vs. unilateral renal ischemia; n=6; P < 0.05 vs. baseline or sham.
Effect of HK I or II over-expression on mitochondrial HK content, mitochondrial membrane injury, caspase 3 activation, and cell survival after metabolic stress
Compared to empty vector, proximal tubule cells exposed to adenovirus expressing full-length human HK I exhibited increased mitochondrial-associated HK I expression without altering that of HK II (Fig 5A, upper panel). Similarly, over-expression of HK II did not alter organelle-associated HK I content (middle panel). However, increased HK I or HK II expression reduced mitochondrial membrane injury after metabolic stress. Compared to control (empty vector), cells over-expressing either HK I (Fig 5B, upper panels) or HK II (lower panels) released less apoptosis inducing factor (AIF) from mitochondria after stress (Fig 5B; lanes 5 & 6 vs. 2 & 3 of top panel in each pair). In control, pro-caspase 3 the inactive precursor of caspase 3, progressively decreased (Fig 5C, top panel, lanes 1–3), confirming that metabolic stress induces enzyme activation in renal epithelial cells (1). Unlike control, HK II over-expression maintains caspase 3 predominantly in its inactive form despite ATP depletion (top panel – lanes 4–6), indicating that HK limits outer membrane injury. While only 64.5 ± 1.4% of control cells survived after stress, survival reached 84.8 ± 8.1 % of HK I and 92.8 ± 2.4% of HK II over-expressing cells (Fig 5D; P < 0.05 vs. empty vector; n=4). Exposure of cells to adenovirus containing either the HK I or II isoform increased total cell HK I or HK II content, respectively (Fig 5D, inset) without altering the expression level of the other isoform (not shown).
Fig 5. Effect of HK I or II over-expression on mitochondrial associated HK, organelle injury, caspase 3 activation and cell survival after stress.
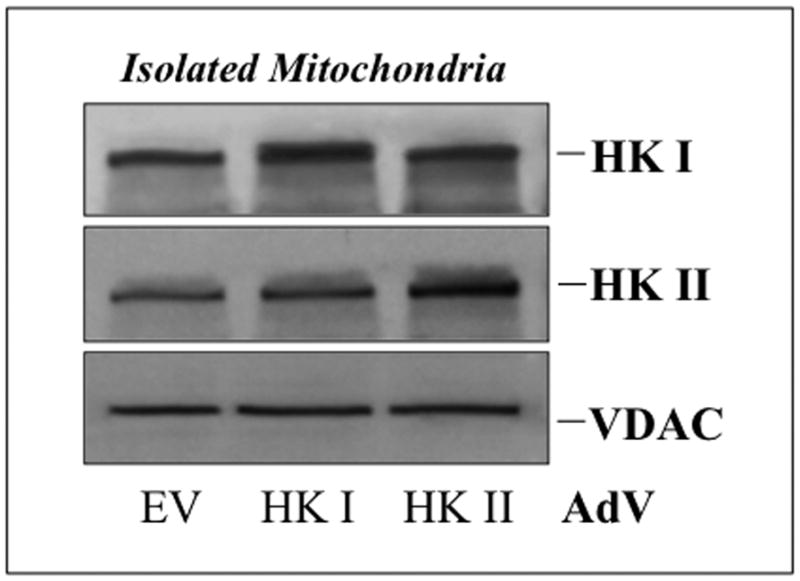
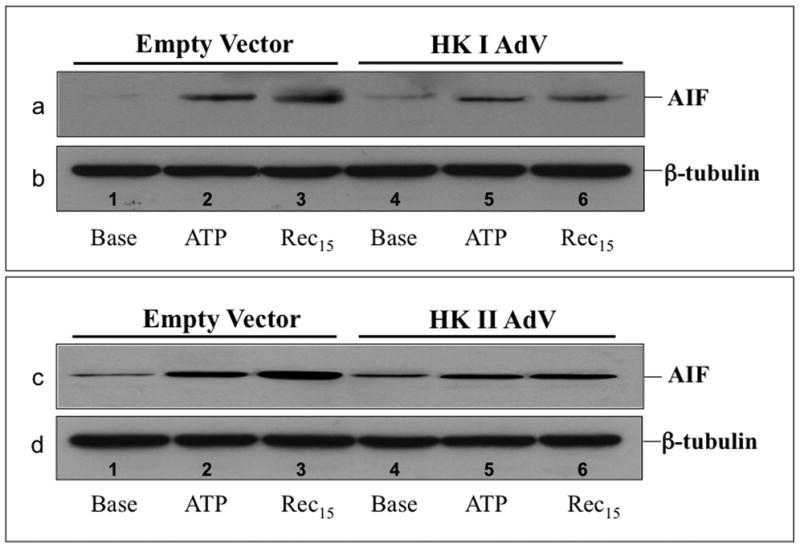
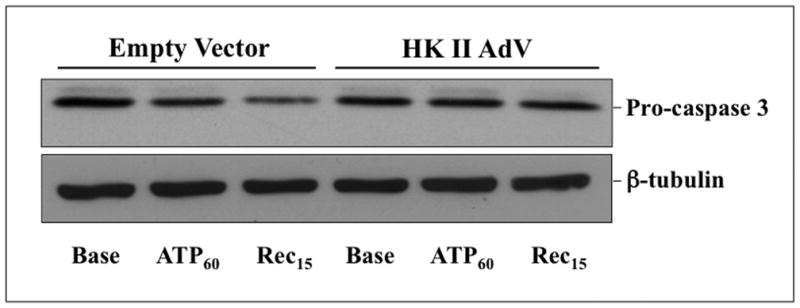
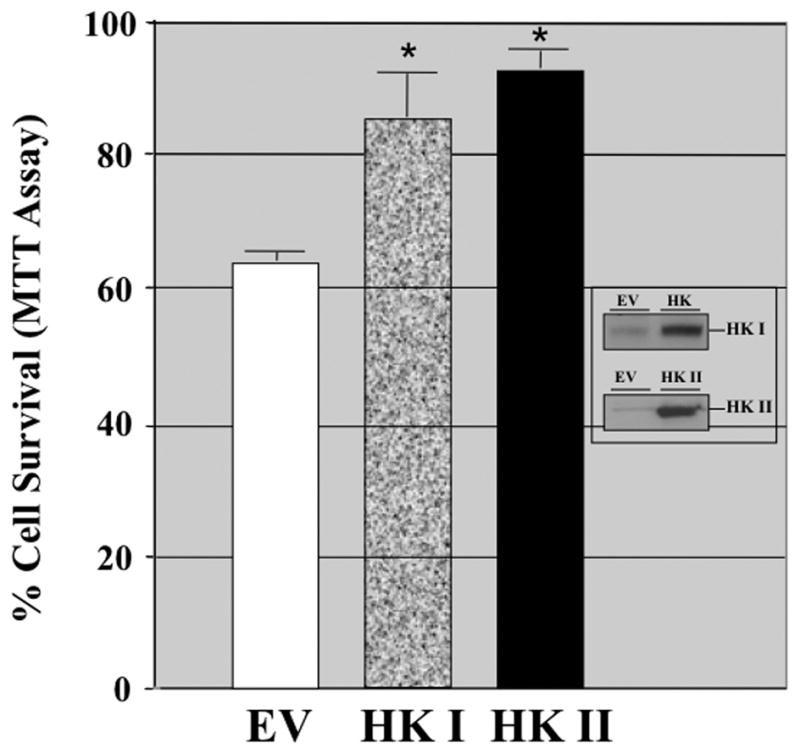
(A) HK I and HK II content in isolated mitochondria harvested from cells that express either HK I HK II or empty vector (EV); (B) mitochondrial membrane injury assessed by leakage of apoptosis inducing factor (AIF) into the cytosol of digitonin-permeabilized cells (upper panel) at baseline (Base), after 60 min ATP depletion (ATP60), and following 15 min recovery (Rec15) in empty vector vs. HK I (panel a) or II (panel c) over-expressing cells (HK I or II AdV); β-tubulin loading control (lower panel); (C) content of pro-caspase 3, the inactive form of the apoptotic enzyme at baseline (Base), after 60 min ATP depletion (ATP60), and following 15 min recovery (Rec15) in HK II over-expressing vs. empty vector cells; (D) Survival assessed by the MTT assay in empty vector (EV) vs. HK II over-expressing cells (HK II AdV) after 2 hr ATP depletion followed by 6 hr recovery (Rec6hr); immunoblot analysis confirming HK II over-expression without altering HK I content (inset);
Effect of HK II expression on ATP content before and after metabolic stress
Exposure to two metabolic inhibitors in the absence of medium glucose resulted in a rapid fall in total ATP content to approximately 10% of the baseline value (Fig 6) as previously reported by our laboratory (21). During recovery, total ATP content progressively increased after removing both inhibitors and adding glucose to the medium. No significant differences in total ATP content between control and HK II over-expressing cells were evident before, during or after metabolic stress, suggesting that increasing HK II content in the presence of metabolic inhibitors does not alter total adenine nucleotide content.
Fig 6. Effect of HK II over-expression on cell ATP content after metabolic stress.
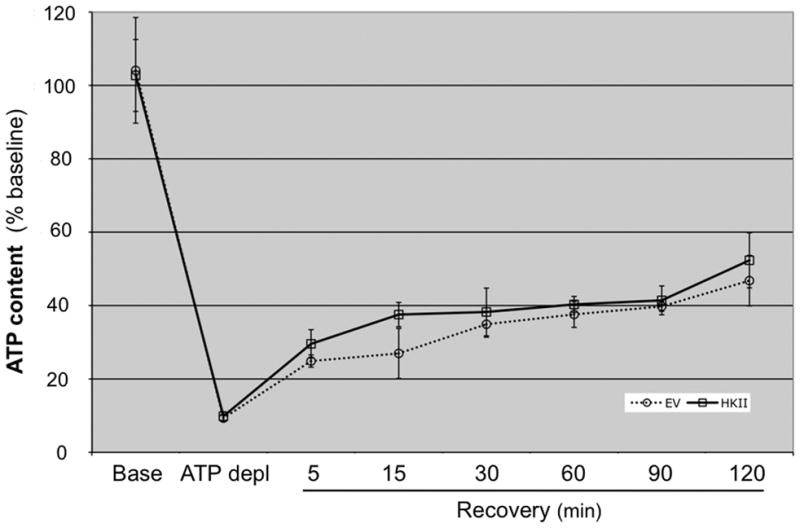
Luciferase assay measurements of total ATP content at baseline (Base), after 60 min ATP depletion (ATP Depl), and after 5–120 min recovery (Rec5 - Rec120) in cells treated with either empty vector (EV) or HK II adenovirus (HK II). Data are expressed as mean ± SE; n=6.
Effect of HK II over-expression on mitochondrial morphology
At baseline, mitochondria appear elongated or “filamentous” both in cells incubated with adenovirus containing either empty vector or HK II (Fig 7, panels A,B). ATP depletion caused mitochondria to become punctate (panels C,D). HK II expression failed to prevent this change, suggesting that cytoprotection does not operate at the level of organelle dynamics.
Fig 7. Effect of HK II over-expression on mitochondrial morphology.

Morphology of mitochondria stained with Mitotracker Green FM at baseline (panels A,B) or after 30 min ATP depletion (panels C,D) in renal cells infected with either control AdV (‘empty vector’) or HK II (‘HK II AdV’).
Effect of HK II on GSK3β and Bax activation
To measure the potential effect of HK II expression on GSK3β activation during metabolic stress, p-ser9 GSK3β, the inactive form of this kinase, was serially measured in lysates from control and HK II over-expressing cells before and after metabolic stress (Fig 8). In control cells, GSK3β is activated (i.e., p-ser9 GSK3β, content markedly decreases) during ATP depletion and is then rapidly inactivated (i.e., p-ser9 GSK3β, content increases) during recovery (top panel; HK II minus). The pattern of GSK3β activation and inactivation is virtually identical in HK II over-expressing cells (top panel; HK II plus), showing that HK II acts downstream of GSK3β activation.
Fig 8. Effect of HK II over-expression on GSK3β activation after metabolic stress.
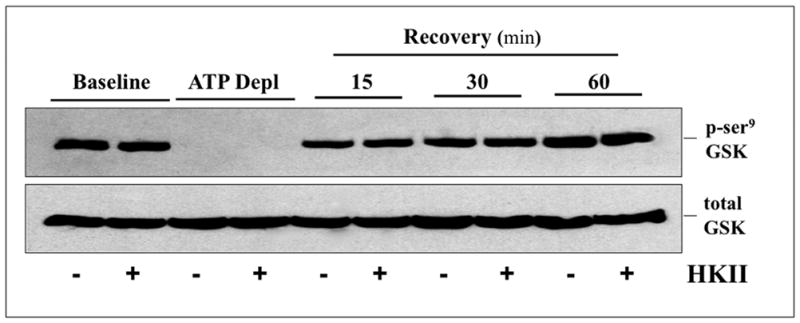
Inactive (p-ser9) GSK3β content in cell lysates by immunoblot analysis at baseline, after 60 minutes ATP depletion (ATP Depl), and following 15, 30, and 60 min recovery in cells treated with either empty vector (−) or HK II adenovirus (+). Total GSK3β loading control (lower panel).
To confirm the pathogenic role of Bax in our model, Bax content was reduced with a specific siRNA, resulting in a 50–70% decrease in immunoreactive Bax content (Fig 9A; top panel – lane 3 vs. 1). In contrast, non-specific RNA did not alter Bax content (lane 2). In ATP depleted cells, Bax knockdown markedly decreased the release of both AIF (Fig 9B, top panel – lane 4 vs. lanes 2 & 3) and cytochrome c (middle panel– lane 4 vs. lanes 2 & 3), confirming that Bax contributes to organelle injury after metabolic stress. To determine whether HK II regulates Bax activation, cell lysates were probed for the 6A7 epitope, a region exposed only after Bax activation (15). The steady state content of active Bax increased to a similar degree after ATP depletion and recovery in both control (Fig 10, top panel – lanes 1–3) and HK II over-expressing (top panel – lanes 4–6) cells, showing that HK II does not prevent cell death by inhibiting Bax activation. To the contrary, these results suggest that HK II limits active Bax-mediated injury at the level of the outer mitochondrial membrane.
Fig 9. Bax knockdown prevents mitochondrial leak of pro-apoptotic effector proteins after metabolic stress.
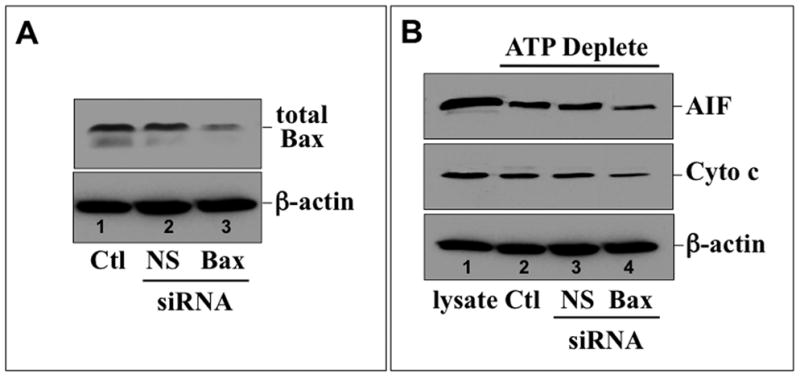
(A) Total Bax content (upper panel) in control (Ctl) and cells treated with either non-specific (NS) or Bax-specific (Bax) siRNA by immunoblot; β-actin loading control (lower panel); (B) AIF (upper panel) and cytochrome c (Cyto c, middle panel) measured by immunoblot in the digitonin-permeabilized control (Ctl), or cells exposed to non-specific (NS) or Bax-specific (Bax) siRNA following 60 min ATP depletion; s-actin loading control (lower panel).
Fig 10. Effect of HK II over-expression on stress-induced Bax activation.
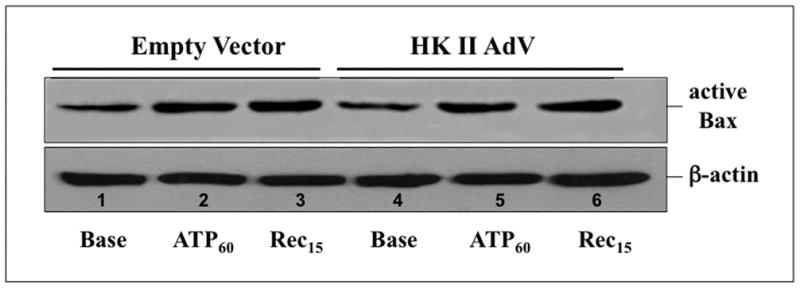
Active Bax (upper panel) detected by immunoblot with an anti-6A7 epitope-specific antibody in cells exposed to either empty vector (empty vector) or HK II (HK II AdV) adenovirus at baseline (Base), after 60 min ATP depletion (ATP60) and 15 min recovery (Rec15); β-actin loading control (lower panel).
Effect of metabolic stress on HK II-Bax interaction
To evaluate a potential mechanism of HK-mediated Bax antagonism and cytoprotection, interaction between HK II and Bax was examined in isolated renal epithelial cells before and after ATP depletion. Although total Bax was readily immunoprecipitated at baseline and after stress, no HK II could be detected before or after stress despite HK II over-expression (data not shown).
HK II prevents mitochondrial Bax accumulation during metabolic stress
Mitochondrial Bax translocation and accumulation are required to cause membrane injury and the release of pro-apoptotic mediators. During recovery from ATP depletion (Fig 11) HK II content decreased (upper panel), whereas Bax content increased in control mitochondria (middle panel). Compared to control, selective HK II expression increased mitochondrial HK II content and markedly reduced Bax accumulation content. Interestingly, the residual amount of mitochondrial-associated HK II after stress in HK II over-expressing cells was comparable to that noted in control at baseline (lane 3 vs. lane 2). These results suggest that HK II expression inhibits mitochondrial Bax translocation and/or accumulation and is important for preventing Bax-mediated organelle injury.
Fig 11. Effect of HK II over-expression on mitochondrial HK II and Bax content.
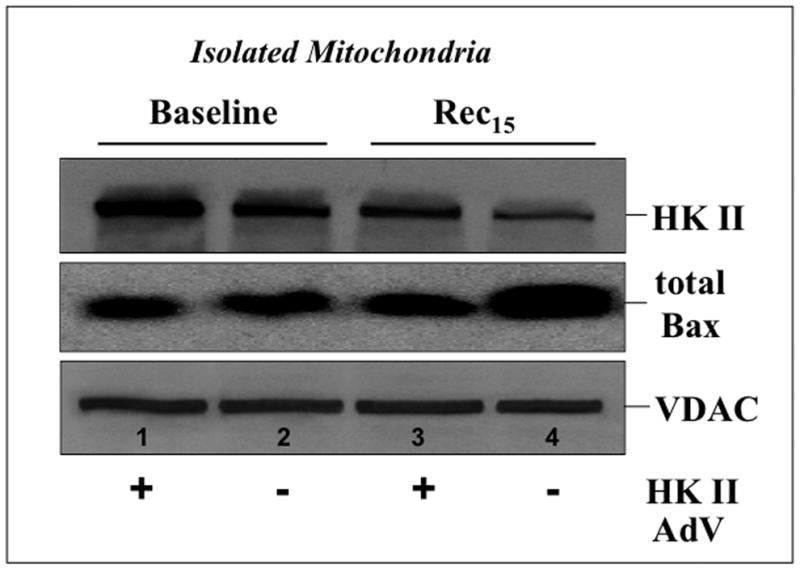
HK II (upper panel) and total Bax (middle panel) assessed in isolated mitochondria harvested from cells exposed to empty vector (−) or HK II (+) containing adenovirus at baseline and 15 min recovery following ATP depletion (Rec15). VDAC, an outer mitochondrial membrane protein, serves as loading control (lower panel).
DISCUSSION
The present study shows that HK is a key regulator of susceptibility to Bax-mediated mitochondrial membrane permeabilization in renal epithelial cells. HK II is abundant only in the proximal tubules, indicating for the first time that this isoform is relevant to proximal tubule injury. After ATP depletion in vitro (fig 1), HK II is displaced from mitochondria, cytosolic HK II content transiently increases, and total HK II content markedly falls (fig 2). Ischemia/re-perfusion in vivo causes similar displacement of mitochondrial HK II (fig 3B) and a decrement in overall intracellular HK II content (fig 4A,B). The later could be due to HK II degradation and/or to loss of HK to the extracellular milieu. In a contemporaneous manner, mitochondrial HK II content decreases (i.e., 15 min recovery after ATP depletion) and both GSK3β and Bax are activated (figs 8,10). Once activated, Bax accumulates in mitochondria (fig 11), caspase-dependent and independent mediators (cytochrome C and AIF, respectively) are released (figs 5B and 9B), pro-caspase 3 is consumed (fig 5C), and cell death follows (fig 5D). Although other BCL2 proteins (including Bcl2, BclXL and Bak and Bid) also mediate apoptosis, Bax is critical in our model, since RNAi-mediated Bax knockdown is sufficient to markedly reduce both mitochondrial AIF and cytochrome C leakage (fig 9B). The time course of these events is consistent with prior reports in which apoptotic signaling occurs within minutes of recovery from metabolic stress (15, 20, 22). Specifically, Akt inactivation combined with GSK3β activation lead to site-specific serine phosphorylation that promotes Bax activation and cell death (14, 18, 19, 23). Furthermore, activated GSK3β promotes mitochondrial HK II displacement and apoptosis (14), whereas GSK3β inhibition prevents HK II displacement and suppresses apoptosis (24). Once displaced, HK II is susceptible to ubiquitination and proteasomal degradation (25), likely contributing to the rapid loss of total cell HK II after ATP depletion (fig 2) or renal ischemia (fig 4).
In our study, loss of mitochondrial HK II is clearly associated with mitochondrial Bax accumulation (fig 11), AIF release (fig 5B), caspase 3 activation (fig 5C) and cell death (fig 5D). HK II over-expression increased mitochondrial HK II content and reduced each of these apoptotic parameters without altering HK I content (and vice versa). Expression of either HK I or II, the two isoforms with a mitochondrial targeting sequence, is sufficient to prevent ATP depletion-induced mitochondrial injury (fig 5B). After ATP depletion, HK II over-expression was equivalent (but did not exceed) the level of mitochondrial-associated HK II detected in control cells at baseline (fig 11; lane 3 vs. 2). This shows that maintaining mitochondrial associated HK II within the physiologic range is sufficient to inhibit Bax accumulation during stress (fig 11), an important cause of mitochondrial membrane permeabilization in renal ((15, 17) and fig 9) and non-renal cells (14). Although it is conceivable that HK II decreases susceptibility of the outer membrane to “Bax attack” by regulating mitochondrial dynamics during stress (26), HK II over-expression failed to prevent the dramatic shift from filamentous to punctate mitochondria (fig 7). This observation is consistent with the absence of any effect on HK II over-expression on net ATP content (fig 6), since intact mitochondria and HK are both prerequisites for metabolic coupling between the two. In addition, neither HK I or HK II over-expression altered the mitochondrial content of the other isoform (fig 5A). This observation excludes the possibility that HK I or II protect mitochondria by reciprocally increasing organelle binding of the other isoform. Furthermore, HK II failed to inhibit either stress-induced GSK3β (fig 8) or Bax activation (fig 10) and does not interact with Bax (data not shown). These findings indicate that HK II does not normally saturate the outer mitochondrial membrane and potentially interferes with membrane attack by competing with active Bax for binding at its target site(s) (27, 28). Although truncated HK I and II mutants that lack mitochondrial targeting partially protect cardiac myocytes against oxidant stress (29), this cell type and the model of injury differ from the present study. Admittedly, the kinetics of mitochondrial HK binding are complex and may include both regulated and unregulated components for each HK isoform. The physical basis and functional importance of individual subsets of bound HK are incompletely understood (2, 11) and are not addressed in the present work.
How mitochondrial HK II antagonizes “Bax attack” is presently debated. However, it is clear that the loss of mitochondrial-bound HK promotes apoptosis (11, 14). Recently, Chiara, et. al., confirmed that mitochondrial HK inhibits apoptosis. These investigators used an HK II amino-terminal peptide to release mitochondrial HK II, resulting in dose-dependent cell death (30). The present study is consistent with this prior report by showing that displacement of HK from mitochondria promotes cytochrome c release and apoptosis, whereas increasing mitochondrial-associated HK I or II limits mitochondrial membrane injury and enhances renal cell survival. Loss of mitochondrial HK could expose putative outer mitochondrial membrane sites for attack by Bax or alter binding kinetics for targets such as the voltage dependent anion channel (VDAC; (2, 3, 13, 27, 31–35). In either scenario, Bax interaction with VDAC (or another proximate site) appears to open a membrane pore to induce apoptosis, whereas maneuvers that increase HK II-VDAC interaction decrease outer membrane susceptibility to Bax (36, 37).
Although HK II failed to inhibit the activation of GSK3β (fig 8), a key Bax regulatory enzyme (18)) or limit Bax activation after stress (fig 10), it is possible that HK binds and sequesters active Bax in the cytosol. However, no interaction between active Bax and HK II was detected by IP even in stressed renal cells that over-express HK II, suggesting that this is unlikely to be its primary mode of cytoprotection. The multi-modal effects of HK II on cell survival are illustrated by the fact that HK protects cells with double Bax and Bak knockout against apoptogenic stress (38). As well, HK has two major binding sites: phosphohexose-dissociable (Type A) and non-dissociable (Type B). Therefore, relatively small changes in HK expression, compartmentalization and/or binding in our model, mediated by a distinct subset of HK binding sites, could exert major effects on cytoprotection (39).
Whether or not VDAC is the only HK binding site is controversial. On one hand, VDAC-1 over-expression causes apoptosis in diverse cell lines (27, 32, 40–42), whereas VDAC-1 knockdown suppresses apoptosis (42, 43), suggesting that this site plays a critical role in outer mitochondrial membrane permeabilization. On the other hand, ionomycin, staurosporine, H2O2, and TNFα induce mitochondrial membrane permeabilization and apoptosis even in cells with complete VDAC-1 and 3 knockout and RNAi-mediated partial knockdown of VDAC-2 (12). Similarly, an HK-related peptide that displaces endogenous HK caused apoptosis in cells that lacked both VDAC-1 and 3 (30). In these studies in which VDAC 1 and 3 were absent some VDAC-2 remained, since the complete absence of VDAC-2 is lethal (12, 30). Unfortunately in these reports, the amount of mitochondrial HK (perhaps more relevant than total HK content) was not assessed. This may be an important omission, since HK could bind multiple mitochondrial target proteins in a hierarchical fashion (e.g., VDAC 1, 2, 3) or bind to other mitochondrial proteins present on the outer membrane that interact with Bax. For example, Bax appears to bind TOM22, a component of the translocase of the outer mitochondrial membrane (44–46), an alternative HK II binding site that has not been investigated.
Despite this controversy regarding its binding site, the present study shows that mitochondrial bound HK II exerts anti-apoptotic effects that are potentially independent of its effect on ATP content or organelle morphology either during or after cell injury. Our results differ from those reported in some cancer cell lines in which the metabolic role of HK was believed to afford cytoprotection (7, 8). In these cancer cells, abundant HK resulted in higher levels of ATP, glucose-6-phosphate and lactic acid than found in non-cancer cells (6–8, 47). In renal epithelial cells, a single study suggests that endogenous and exogenous HK reduces oxidant injury via an ATP-dependent mechanism (1). Although ATP content is unchanged in HK over-expressing cell in the presence of metabolic inhibitors, we cannot exclude the possibility that ATP flux or other pathways including the generation of reactive oxygen species or VDAC phosphorylation, are altered and contribute to hexokinase-mediated protection. Others have shown that HK mutants with defective mitochondrial binding capacity inhibited oxidant induced cell injury, confirming that HK also has non-mitochondrial protective effects (29). Additionally, HK II could afford cytoprotection by altering proteolysis and/or autophagy. The present study does suggest that either directly or indirectly, HK alters mitochondrial susceptibility to Bax-mediated membrane injury downstream of either GSK3β or Bax activation (figs 8,10). Since mitochondrial HK II binding is not normally saturated and is further reduced by stress (fig 11), we suggest that mitochondrial hexokinase is a potential target for preventing organelle injury and for sustaining cellular viability after an apoptogenic insult.
METHODS
Cell Culture
Opossum kidney proximal tubule (OK) cells were cultured in Dulbecco’s Modified Eagles Medium (DMEM; Mediatech, Manassas, VA; 4.5 g/L of glucose, 584 mg/L L-glutamine, without pyruvate) supplemented with 1% penicillin/streptomycin (Mediatech) and 10% fetal bovine serum (Equitech-Bio, Kerrville, TX) at 37°C in 5% CO2.
Metabolic Stress
ATP depletion was initiated by exposing confluent cells to glucose free DMEM (Invitrogen, Carlsbad, CA; 584 mg/L L-glutamine, without pyruvate) containing sodium cyanide (5 mM) and 2-deoxy-D-glucose (5 mM), a competitive glucose inhibitor, as previously described by our laboratory (15). Cells were washed three times with PBS and incubated with the above ATP depletion media for 60–120 minutes at 37°C in the presence of 5% CO2. Recovery was initiated by washing twice with PBS followed by incubation in standard DMEM containing 4.5g/L glucose but without metabolic inhibitors or serum. This insult consistently causes cell death by apoptosis using established biochemical and morphologic criteria (22, 48) and is associated with <5% plasma membrane injury, an important hallmark of cellular necrosis (21).
Renal Ischemia & Reperfusion
Six-week old mice weighing 20–22gm were used to perform ischemia reperfusion studies under standardized conditions as recently described (18). After intraperitoneal injection of tribromoethanol (250mg/kg BW), both kidneys were exposed by a midline incision. For HK II degradation studies, the left renal pedicle was ligated and the kidney removed to serve as ‘baseline’. An occlusive non-traumatic microaneurysm clamp was placed on the right renal pedicle for 40 minutes. After clamp removal, restoration of blood flow was visually confirmed for 1 min and the right kidney harvested after 3 hrs re-perfusion. For renal histology studies, ischemia/re-perfusion was performed on both kidneys using two clamps. Sham-operated mice received identical surgical procedures with the exception that clamps were not applied. Procedures were performed under conditions in accordance with guidelines set by the National Institutes of Health and the Institutional Animal Care and Use Committee of Boston University.
Immunoblot analysis
Lysates were harvested from cells in RIPA buffer with EDTA (Boston BioProducts) containing a protease inhibitor cocktail (Set I; Calbiochem, San Diego, CA). Cytosolic fractions were collected by rocking cell monolayers on ice in a permeabilizing buffer (in mM; KCl, 120; KH2PO4, 5; HEPES; 10; EGTA, 2 and digitonin, 15 μg/mL at pH 7.4) for 15 minutes. Digitonin selectively permeabilizes the plasma membrane (22). The supernatant was harvested as the “cytosolic” cell fraction. Samples were sonicated, spun at 10,000 × g, and supernatants collected. Mitochondria were harvested using a “Mitochondrial Isolation Kit for Mammalian Cells” (Pierce, Rockford, IL) following the manufacturer’s protocol. The degree of purification of each cell fraction was assessed using standard markers (49). Protein levels were measured using the BCA assay (Pierce) and equal amounts of protein were separated on 7.5–12% tris-glycine polyacrylamide gels. Separated proteins were transferred to nitrocellulose membranes and antigens detected with primary antibodies and chemiluminescent reagents described below.
Immunoprecipitation
IP was performed as recently described by our laboratory (50). In brief, 7.5x106 cells were harvested in buffer containing (in mM): NaCl, 150; Tris HCl, 10; EDTA, 5; EGTA, 1; 1% Triton X-100, 0.5% NP-40, with protease inhibitors at pH 7.4. Lysates were centrifuged at 10,000 × g for 10 min at 4 °C. Samples containing 200μg total protein were incubated overnight at 4°C with 2μg of antibody directed against total Bax (Cell Signaling, Danvers, MA, cat #2772, and were then probed with antibodies directed against total Bax or HK II (Cell Signaling).
Antibodies
Antibodies to HK II (C-14), VDAC, AIF, and total Bax (N-20) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The active (6A7) Bax antibody was purchased from Trevigen (Gaithersburg, MD). Antibodies directed against phospho-ser9 GSK3β (inactive), total GSK3β, pro-caspase 3 and an alternate HK II–C64G5 antibody (used for immunohistochemistry) were obtained from Cell Signaling. βactin was purchased from Sigma (St. Louis, MO), β-tubulin from ICN (Costa Mesa, CA) and mitochondrial-specific F1F0 ATPase (complex V β subunit) from Invitrogen. Secondary antibodies conjugated to horseradish peroxidase (Jackson Immunoresearch Laboratories, West Grove, PA) were used in conjunction with chemiluminescent reagents from Boston BioProducts (Worcester, MA). Cy3 and FITC-conjugated secondary antibodies were used to localize HK II in cells (Jackson; described below). Primary antibodies in tissue were detected with AlexaFluor 488 and 594-conjugated secondary antibodies (Invitrogen; described below).
Immunofluorescence
Cells were seeded on glass coverslips in 24 well plates for 24 hr. After stress, cells were washed in ice-cold 1x PBS, fixed in methanol for 30 minutes, and stored overnight in 75% ethanol at 4°C. After washing 3 times in PBS, cells were permeabilized for 5 min in 0.2% saponin, 0.03M sucrose, 1% BSA in PBS, washed three more times for 5 minutes each in PBS, and blocked for 30 min in 5% BSA in PBS. Cells were then incubated in anti-HK II (1:50 dilution; 1% BSA in PBS) and anti-Cox IV antibodies (1:50 dilution; 1% BSA in PBS) overnight at 4°C. After four washes in PBS, cells were incubated in cy3-conjugated secondary antibody (1:200; 1% BSA in PBS) for 1 hr, washed 3 times in PBS, and mounted onto glass slides with gelvatol. Slides were imaged by confocal microscopy (Perkin Elmer Ultraview Scanning Disc Microscope, Wellesley, MA).
Mitochondrial morphology was examined in live cells grown on MatTek culture dishes (MatTek Corp, Ashland, MA) and incubated with Mitotracker green FM (100nM; Invitrogen) in standard culture media for 1hr as previously described (17). Prior to visual inspection by confocal microscopy, the MitoTracker was removed and the medium replaced.
Tissue
After intraperitoneal injection of tribromoethanol (250mg/kg BW), the heart was exposed by bilateral thoracic incision. A catheter was placed in the left ventricle and 10 ml of 4% paraformaldehyde (PFA) was infused at a rate of 6 ml/min. The kidney was removed, cut into 4 pieces and placed in 4% PFA overnight. After washing twice in PBS, the kidney was placed in 15% sucrose for 1 hour, followed by 30% sucrose at 4°C until the sample no longer floated. The samples were then imbedded Tissue-Tek O.C.T. compound (MatTek Corp) and cut into 5 micron sections using a cryostat.
For tissue staining, sections were washed 3 times for 5 minutes each in PBS followed by 5 minutes in PBS containing 1% SDS. After 3 additional washes, tissue sections were blocked for 1 hr in PBS containing 5% normal goat serum and 0.3% Triton X-100. Following 3 washes, sections were incubated for 90 minutes with antibodies against F1F0 ATPase (2μg/μl) and HK II (1:400 dilution) in PBS with 1% BSA and 0.3% Triton X-100. After 3 PBS washes, sections were incubated in AlexaFluor 488 (1:250 dilution) and AlexaFluor 594 (1:500 dilution) conjugated secondary antibodies (1% BSA, 0.3% Triton X-100 in PBS) for 1 hr, washed 3 final times in PBS, mounted with gelvatol and examined by confocal microscopy as described above. Slides were also imaged by phase contrast and wide-field epifluorescence microscopy followed by deconvolution using Image J software.
Cell Viability
Survival after ATP depletion was measured by MTT (Promega, Madison, WI) assay, a technique based on the ability of cells to convert 3-(4,5dimethylthiazol)-2,5-diphenyl tetrazolium bromide into purple formazan crystals that can be solubilized in 10% SDS, 0.01 N HCl. Samples (200μl each) were loaded onto a 96-well plate and read at 595nm on a plate reader. The number of surviving cells is expressed as the percentage of viable control cells at baseline.
HK II Over-expression
Cells were incubated in DMEM containing 2% FBS and adenovirus containing a full-length human HK II construct (50 moi for 6 hr). The media was replaced with 10% FBS in DMEM and the cells allowed to incubate overnight before the experiments were performed. HK II over-expression was confirmed by immunoblot analysis.
ATP Content
A luciferase-based ATP kit (Invitrogen) was used to determine cellular ATP levels before, during and after stress. Following each experiment, cells were washed twice with ice-cold PBS followed by 2.5% (w/v) trichloroacetic acid. Cells were then scraped from the dish and centrifuged at 10,000 × g for 5 min at 4°C. The supernatant was diluted 10 times and neutralized with tris acetate buffer (pH 7.75). ATP content was determined in triplicate in each sample following the manufacturers protocol and was compared to a freshly prepared standard curve derived from samples with known ATP content.
Acknowledgments
This work was supported by National Institutes of Health research grants (RO-1) DK-52898 (JHS), DK-53387 (SCB), a James A. Scherbenske Award from the American Society of Nephrology (SCB), a Ruth L. Kirschstein National Research Service Award DK-082150 (JMG), and an ARC award from the Evans Center for Interdisciplinary Research at Boston University. None of the authors has any significant primary financial arrangements with commercial companies that produce or sell products that are the subject of the studies contained in this manuscript or with competitors of such companies.
Footnotes
DISCLOSURE
The authors have no competing financial interests.
References
- 1.Bryson JM, Coy PE, Gottlob K, Hay N, et al. Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J Biol Chem. 2002;277:11392–11400. doi: 10.1074/jbc.M110927200. [DOI] [PubMed] [Google Scholar]
- 2.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 3.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377:347–355. doi: 10.1042/BJ20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballatori N, Cohen JJ. Intracellular distribution of hexokinase in the tissue zones of rat kidney. Biochim Biophys Acta. 1981;657:448–456. doi: 10.1016/0005-2744(81)90330-2. [DOI] [PubMed] [Google Scholar]
- 5.Vandewalle A, Wirthensohn G, Heidrich HG, Guder WG. Distribution of hexokinase and phosphoenolpyruvate carboxykinase along the rabbit nephron. Am J Physiol. 1981;240:F492–500. doi: 10.1152/ajprenal.1981.240.6.F492. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e. elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 7.Mathupala SP, Rempel A, Pedersen PL. Aberrant glycolytic metabolism of cancer cells: a remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J Bioenerg Biomembr. 1997;29:339–343. doi: 10.1023/a:1022494613613. [DOI] [PubMed] [Google Scholar]
- 8.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 10.Vyssokikh M, Brdiczka D. VDAC and peripheral channeling complexes in health and disease. Mol Cell Biochem. 2004;256–257:117–126. doi: 10.1023/b:mcbi.0000009863.69249.d9. [DOI] [PubMed] [Google Scholar]
- 11.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008;40:171–182. doi: 10.1007/s10863-008-9148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, et al. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shoshan-Barmatz V, Zakar M, Rosenthal K, Abu-Hamad S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim Biophys Acta. 2009;1787:421–430. doi: 10.1016/j.bbabio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 15.Ruchalski K, Mao H, Li Z, Wang Z, et al. Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J Biol Chem. 2006;281:7873–7880. doi: 10.1074/jbc.M513728200. [DOI] [PubMed] [Google Scholar]
- 16.Havasi A, Li Z, Wang Z, Martin JL, et al. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem. 2008;283:12305–12313. doi: 10.1074/jbc.M801291200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Havasi A, Gall JM, Mao H, et al. Beta-catenin promotes survival of renal epithelial cells by inhibiting Bax. J Am Soc Nephrol. 2009;20:1919–1928. doi: 10.1681/ASN.2009030253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Havasi A, Gall J, Bonegio R, et al. GSK3beta promotes apoptosis after renal ischemic injury. J Am Soc Nephrol. 2010;21:284–294. doi: 10.1681/ASN.2009080828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linseman DA, Butts BD, Precht TA, Phelps RA, et al. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Havasi A, Gall JM, Bonegio R, et al. GSK3β, A molecular switch, regulates apoptosis and renal function after ischemic stress. J Am Soc Nephrol. 2009 [Google Scholar]
- 21.Wang YH, Borkan SC. Prior heat stress enhances survival of renal epithelial cells after ATP depletion. Am J Physiol. 1996;270:F1057–1065. doi: 10.1152/ajprenal.1996.270.6.F1057. [DOI] [PubMed] [Google Scholar]
- 22.Ruchalski K, Mao H, Singh SK, Wang Y, et al. HSP72 inhibits apoptosis-inducing factor release in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol. 2003;285:C1483–1493. doi: 10.1152/ajpcell.00049.2003. [DOI] [PubMed] [Google Scholar]
- 23.Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, et al. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem. 2004;279:21085–21095. doi: 10.1074/jbc.M400063200. [DOI] [PubMed] [Google Scholar]
- 24.Gimenez-Cassina A, Lim F, Cerrato T, Palomo GM, et al. Mitochondrial hexokinase II promotes neuronal survival and acts downstream of glycogen synthase kinase-3. J Biol Chem. 2009;284:3001–3011. doi: 10.1074/jbc.M808698200. [DOI] [PubMed] [Google Scholar]
- 25.Magnani M, Crinelli R, Antonelli A, Casabianca A, et al. The soluble but not mitochondrially bound hexokinase is a substrate for the ATP- and ubiquitin-dependent proteolytic system. Biochim Biophys Acta. 1994;1206:180–190. doi: 10.1016/0167-4838(94)90206-2. [DOI] [PubMed] [Google Scholar]
- 26.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abu-Hamad S, Zaid H, Israelson A, Nahon E, et al. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem. 2008;283:13482–13490. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- 28.Machida K, Ohta Y, Osada H. Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J Biol Chem. 2006 doi: 10.1074/jbc.M513297200. [DOI] [PubMed] [Google Scholar]
- 29.Sun L, Shukair S, Naik TJ, Moazed F, et al. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008;28:1007–1017. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiara F, Castellaro D, Marin O, Petronilli V, et al. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastorino JG, Hoek JB. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem. 2003;10:1535–1551. doi: 10.2174/0929867033457269. [DOI] [PubMed] [Google Scholar]
- 32.Zaid H, Abu-Hamad S, Israelson A, Nathan I, et al. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005;12:751–760. doi: 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
- 33.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006;25:4683–4696. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- 34.Shoshan-Barmatz V, Keinan N, Zaid H. Uncovering the role of VDAC in the regulation of cell life and death. J Bioenerg Biomembr. 2008;40:183–191. doi: 10.1007/s10863-008-9147-9. [DOI] [PubMed] [Google Scholar]
- 35.Abu-Hamad S, Arbel N, Calo D, Arzoine L, et al. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci. 2009;122:1906–1916. doi: 10.1242/jcs.040188. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, et al. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol. 2001;152:237–250. doi: 10.1083/jcb.152.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 38.Majewski N, Nogueira V, Bhaskar P, Coy PE, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 39.Hutny J, Wilson JE. Further studies on the role of phospholipids in determining the characteristics of mitochondrial binding sites for type I hexokinase. Acta Biochim Pol. 2000;47:1045–1060. [PubMed] [Google Scholar]
- 40.Godbole A, Varghese J, Sarin A, Mathew MK. VDAC is a conserved element of death pathways in plant and animal systems. Biochim Biophys Acta. 2003;1642:87–96. doi: 10.1016/s0167-4889(03)00102-2. [DOI] [PubMed] [Google Scholar]
- 41.Lu AJ, Dong CW, Du CS, Zhang QY. Characterization and expression analysis of Paralichthys olivaceus voltage-dependent anion channel (VDAC) gene in response to virus infection. Fish Shellfish Immunol. 2007;23:601–613. doi: 10.1016/j.fsi.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Yuan S, Fu Y, Wang X, Shi H, et al. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. Faseb J. 2008;22:2809–2820. doi: 10.1096/fj.08-107417. [DOI] [PubMed] [Google Scholar]
- 43.Tajeddine N, Galluzzi L, Kepp O, Hangen E, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene. 2008;27:4221–4232. doi: 10.1038/onc.2008.63. [DOI] [PubMed] [Google Scholar]
- 44.Bellot G, Cartron PF, Er E, Oliver L, et al. TOM22, a core component of the mitochondria outer membrane protein translocation pore, is a mitochondrial receptor for the proapoptotic protein Bax. Cell Death Differ. 2007;14:785–794. doi: 10.1038/sj.cdd.4402055. [DOI] [PubMed] [Google Scholar]
- 45.Cartron PF, Bellot G, Oliver L, Grandier-Vazeille X, et al. Bax inserts into the mitochondrial outer membrane by different mechanisms. FEBS Lett. 2008;582:3045–3051. doi: 10.1016/j.febslet.2008.07.047. [DOI] [PubMed] [Google Scholar]
- 46.Colin J, Garibal J, Mignotte B, Guenal I. The mitochondrial TOM complex modulates bax-induced apoptosis in Drosophila. Biochem Biophys Res Commun. 2009;379:939–943. doi: 10.1016/j.bbrc.2008.12.176. [DOI] [PubMed] [Google Scholar]
- 47.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, et al. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta. 2002;1555:14–20. doi: 10.1016/s0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Knowlton AA, Christensen TG, Shih T, et al. Prior heat stress inhibits apoptosis in adenosine triphosphate-depleted renal tubular cells. Kidney Int. 1999;55:2224–2235. doi: 10.1046/j.1523-1755.1999.00476.x. [DOI] [PubMed] [Google Scholar]
- 49.Borkan SC, Schwartz JH. Role of oxygen free radical species in in vitro models of proximal tubular ischemia. Am J Physiol. 1989;257:F114–125. doi: 10.1152/ajprenal.1989.257.1.F114. [DOI] [PubMed] [Google Scholar]
- 50.Havasi A, Wang Z, Gall JM, Spaderna M, et al. Hsp27 inhibits sublethal, Src-mediated renal epithelial cell injury. Am J Physiol Renal Physiol. 2009;297:F760–768. doi: 10.1152/ajprenal.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]